

The amide bond is one of the most important linkages in chemistry. Although the majority of amide bonds are formed by the condensation of an amine and a carboxylic acid equivalent, alternative approaches to amide synthesis are of continuing interest.1 In particular, aldehydes are attractive precursors to amides due to their ready availability and reactivity. A number of ways of accomplishing this conversion have been reported.2,3
Another way of preparing amides is through rearrangement.4 In this regard, the Schmidt reaction has long been known to provide amides from aldehydes or ketones, with nitriles as commonly observed side products when aldehydes are used as the carbonyl component.5 Although the Schmidt reaction classically uses hydrazoic acid as a nitrogen source, it is now known that alkyl azides can be used in Schmidt chemistry as well.6,7
Our own work in this area has focused on the acid-promoted reactions of alkyl azide with ketones.8 In contrast, only a few examples of useful azide/aldehyde reactions to afford amides have been reported. In the 1950’s, Boyer and coworkers showed that certain aromatic aldehydes could react with azides to afford amides under the influence of H2SO4, albeit in poor yield.9 Much later, we reported the intramolecular reactions of one γ-azido aldehyde and one δ-azido aldehyde, the latter using a variety of protic and Lewis acid conditions.8d
In work described herein, we reinvestigated the reactions of azides with aldehydes to determine whether this potentially useful transformation could be realized using modern Lewis acid conditions. In the intermolecular framework, we focused both on possible Lewis acids as well as the issue of alkyl group versus hydride migration in the Schmidt reaction step. It has been amply established that the intramolecular reactions of azides with ketones are much more facile than their intermolecular counterparts.8 Accordingly, we began the present study by examining the intramolecular reactions of ω-azido alkyl aldehydes in greater detail than previously reported,8d again focusing on the matter of regioselectivity.
Intramolecular Reactions
A series of azidoalkyl aldehydes containing a 2–5 carbon chain connecting the two functional group were prepared and examined under protic (TFA) and Lewis acid (TiCl4) promotion. Electronically neutral substituents were added α to the aldehyde to facilitate product isolation and identification. Except for the case of β-azidoalkyl aldehyde (3g), which was prepared via the Michael addition of hydrazoic acid to acrylaldehyde (Table 1),10 the azidoalkyl aldehydes were synthesized from commercially available bromoalkyl esters. Thus, the bromide was first displaced by sodium azide, followed by α-alkylation of the ester11 and reduction using DIBAL-H.12 As suggested by precedent,12 the azides typically survived the conditions required to carry out the reduction of the ester group.
Table 1.
Synthesis of azidoalkyl aldehydes.
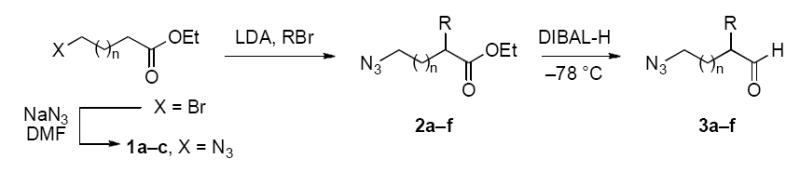
| entry | compd | n | 1 (yield, %) | compd | R | 2 (yield, %) | compd | 3 (yield, %) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1a | 1 | 96 | 2a | allyl | 84 | 3a | 59 |
| 2 | 1a | 1 | 96 | 2b | benzyl | 79 | 3b | 41 |
| 3 | 1b | 2 | 99 | 2c | allyl | 44 | 3c | 66 |
| 4 | 1b | 2 | 99 | 2d | benzyl | 65 | 3d | 83 |
| 5 | 1c | 3 | 99 | 2e | allyl | 83 | 3e | 81 |
| 6 | 1c | 3 | 99 | 2f | benzyl | 54 | 3f | 78 |
Treatment of the azido aldehydes 3a–g with acid can in principle afford two kinds of products, depending on mechanism. First, azide adds to the aldehyde to form an azidohydrin intermediate, which can lead to formamide 4 or lactam 5 by carbon–carbon bond migration or direct 1,2-hydride shift (followed by tautomerization), respectively (Scheme 2).
Scheme 2.
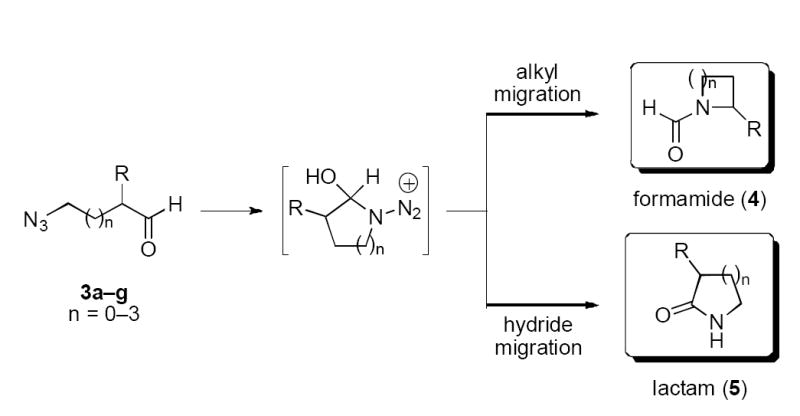
When the azido aldehydes were exposed to either protic or Lewis acid at room temperature, gas evolution was observed and products isolated following workup as summarized in Table 2. A successful Schmidt reaction of β-azido aldehyde would in principle afford a β-lactam; however, no tractable products were detected upon protic or Lewis acid treatment of 3g (entries 1 and 2). Increasing tether length by one carbon led to the exclusive formation of lactams 5a or 5b in good yields (entries 3–6), while further increases in chain length switched the product profile such that only formamides were observed (entries 7–14). In general, all of these reactions were efficient, although higher yields were usually observed when TiCl4 was used. In addition, the yields obtained in this study were generally improved over the two examples we reported previously.8d
Table 2.
Intramolecular reactions of azides with aldehydes.
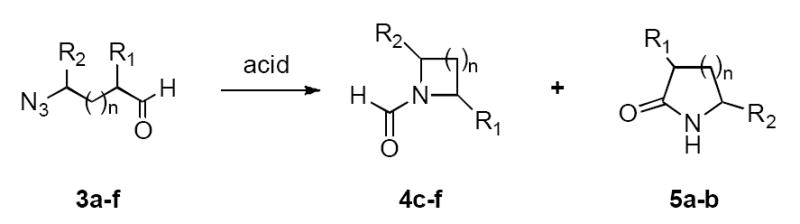
| entry | compd | n | R1 | R2 | acid | Product (yield, %)
|
|
|---|---|---|---|---|---|---|---|
| 4 | 5 | ||||||
| 1 | 3g | 0 | H | C6H13 | TFA | - | - |
| 2 | 3g | 0 | H | C6H13 | TiCl4 | - | - |
| 3 | 3a | 1 | allyl | H | TFA | - | 63 |
| 4 | 3a | 1 | allyl | H | TiCl4 | - | 90 |
| 5 | 3b | 1 | benzyl | H | TFA | - | 77 |
| 6 | 3b | 1 | benzyl | H | TiCl4 | - | 94 |
| 7 | 3c | 2 | allyl | H | TFA | 60 | - |
| 8 | 3c | 2 | allyl | H | TiCl4 | 96 | - |
| 9 | 3d | 2 | benzyl | H | TFA | 68 | - |
| 10 | 3d | 2 | benzyl | H | TiCl4 | 85 | - |
| 11 | 3e | 3 | allyl | H | TFA | 53 | - |
| 12 | 3e | 3 | allyl | H | TiCl4 | 41 | - |
| 13 | 3f | 3 | benzyl | H | TFA | 71 | - |
| 14 | 3f | 3 | benzyl | H | TiCl4 | 49 | - |
In general, these results can be rationalized by strain considerations (Scheme 3). The lack of β-lactam formation (entries 1 and 2) is ascribed to the non-formation of a necessary 4-membered cyclic azidohydrin intermediate from the β-azido aldehyde. In contrast, a γ-azidoalkyl aldehyde readily forms a 5-membered cyclic azidohydrin that can in principle lead to migration of either an endocyclic bond (affording formamide 4) or hydride (affording lactam 5). Two of the stereoisomeric possibilities for this intermediate are shown where X = H (protic acid promotion) or complexed TiCl4 (two epimers at the aminol carbon are also possible but in this case do not affect the regiochemical outcome of the Schmidt reaction). Assuming antiperiplanar migration in the azidohydrin (darkened bonds indicate the bond antiperiplanar to the N2+ leaving group), either alkyl or hydride migration is possible on stereoelectronic grounds, but it appears in the present case that developing strain in the 4-membered azetidine product disfavors alkyl migration and so valerolactam is exclusively formed (entries 3–6).
Scheme 3.
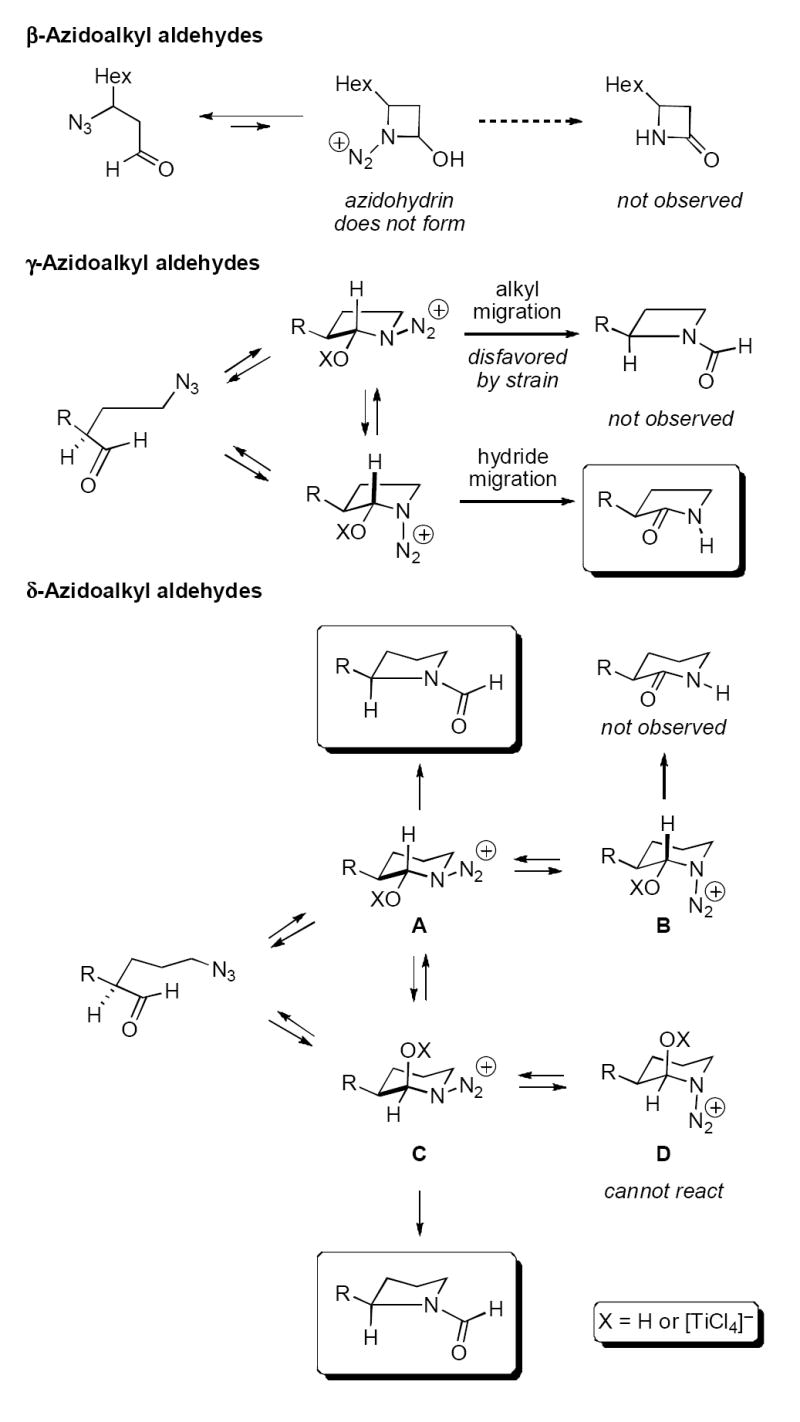
In the larger ring sizes, ring strain is less of a consideration (entries 7–14). Of four possible azidohydrins, antiperiplanar alkyl migration from either A or C affords the formamide products that are solely observed in these series. It is not clear if the reason for this is (1) intrinsic preference for or greater reactivity of A or C (equatorial N2+) vs. B or D (axial N2+), (2) greater migratory aptitude of an alkyl group over a hydride, or (3) preferred formation of C or D (axial OX) over A or B (equatorial OX) coupled with the lack of a mechanism for hydride migration from D (oxaziridines or products therefrom have never been observed in the azido-Schmidt reaction8). Although not shown, similar considerations apply for the ε-azidoaldehyde series. The relatively modest yields in this series suggest that either cyclization or orbital alignment for the migration step is less ideal for the 7-membered azidohydrin intermediate. In none of the cases examined were products of intermolecular reactions identified.
Intermolecular Reactions
We sought to expand the reaction to its intermolecular variant using different aldehydes and alkyl azides as depicted in Scheme 4. As above, it was expected that an acid-activated aldehyde would react with the alkyl azide to form an azidohydrin intermediate that would ultimately afford either formamide resulting from alkyl migration or simple amide resulting from hydride migration/tautomerization.
Scheme 4.
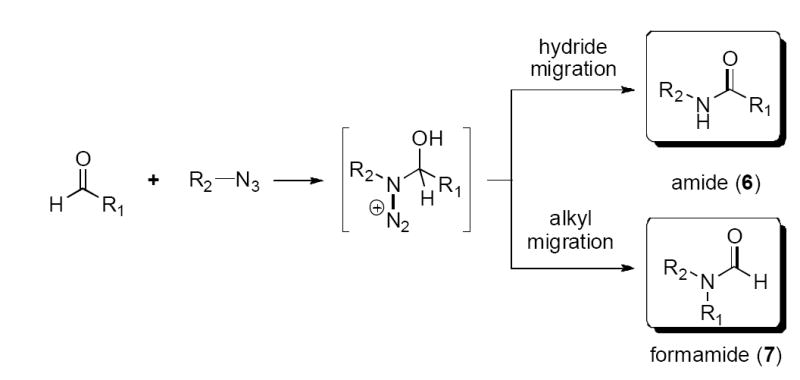
Although low yields of amides had been reported to result from treatment of aromatic aldehydes with H2SO4,9 we had hoped to achieve superior results using Lewis acid promotion. To this end, a variety of Lewis acid conditions were initially tried for the reaction of n-hexyl azide with either heptanal or phenyl acetaldehyde (SnBr4, BF3•OEt2, Ti(O-i-Pr)3, Sc(OTf)3, CF3SO3H and 1N HCl). Unfortunately, none of these conditions provided useful quantities of amide. Only two acid promoters, TFA and TiCl4, provided sufficient yields of products that they were further examined with a series of reactants (Table 3). In general, the reactions proceeded best when TiCl4 was used to promote them.
Table 3.
Intermolecular reactions of alkyl azides with aldehydes.
| entry | R1 | R2 | acid | compds 6 and 7 | yield, %a |
|
|---|---|---|---|---|---|---|
| 6 | 7 | |||||
| 1 | Ph | PhCH2 | TFA | a | trace | trace |
| 2 | Ph | PhCH2 | TiCl4 | a | 21 | 10 |
| 3 | Ph | n-hex | TFA | b | trace | trace |
| 4 | Ph | n-hex | TiCl4 | b | 16 | 10 |
| 5 | Ph | Ph(CH3)CH | TFA | c | trace | trace |
| 6 | Ph | Ph(CH3)CH | TiCl4 | c | 33b | 11b |
| 7 | PhCH2 | PhCH2 | TFA | d | 16 | 16 |
| 8 | PhCH2 | PhCH2 | TiCl4 | d | 22 | 19 |
| 9 | PhCH2 | n-hex | TFA | e | 9 | 9 |
| 10 | PhCH2 | n-hex | TiCl4 | e | 45 | 39 |
| 11 | PhCH2 | Ph(CH3)CH | TFA | f | 13 | 5 |
| 12 | PhCH2 | Ph(CH3)CH | TiCl4 | f | 20 | 30 |
| 13 | CH3(CH2)3CH2 | PhCH2 | TFA | g | 19 | 13 |
| 14 | CH3(CH2)3CH2 | PhCH2 | TiCl4 | g | 29 | 26 |
| 15 | CH3(CH2)3CH2 | n-hex | TFA | h | 28 | 12 |
| 16 | CH3(CH2)3CH2 | n-hex | TiCl4 | h | 37 | 26 |
| 17 | CH3(CH2)3CH2 | Ph(CH3)CH | TFA | i | 23 | 8 |
| 18 | CH3(CH2)3CH2 | Ph(CH3)CH | TiCl4 | i | 35 | 27 |
Isolated products, unless otherwise noted.
Inseparable mixture. Yields estimated from 1H NMR.
Benzyl azide gave the lowest yields among those examined in these reactions (entries 1 to 6). This was not surprising as benzylic azides are known to decompose under acidic conditions.13 In contrast, use of azidoethylbenzene or azidohexane afforded increased yields. Although the combined yields of products were in some cases rather high (i.e., entries 10, 12, 14, 16 and 18), selectivities of products resulting from hydride and alkyl migration were low throughout. This suggests that the intrinsic migratory preference for alkyl and hydride are comparable in this system, which in turn suggests that the considerably better selectivities noted for the intramolecular examples in Table 2 could well arise from the stereoelectronic effects imposed in those reactions.
Summary
The intramolecular variant of the reaction, like the version using azidoketone substrates, is a useful tool for the synthesis of nitrogenous heterocycles. In those reactions, the product profile was shown to entirely depend on the length of the tether between the azide and aldehyde. Although in some cases, the intermolecular reaction of azides and aldehydes proceeds with useful efficiency, especially in the presence of TiCl4, the lack of regiochemical control in the migration step is a drawback.
Experimental Section
Unless otherwise stated, all reactions were performed under a argon atmosphere in flame-dried glassware. All chromatography was performed using Sorbent Technologies silica gel (230-400 mesh) with the indicated solvent mixtures. Infrared spectra were recorded on a Perkin-Elmer 1420 spectrometer or a Nicolet Fourier transform infrared spectrometer and are expressed in wavenumbers (cm−1). Proton nuclear magnetic resonance (1H NMR) and carbon nuclear magnetic resonance (13C NMR) spectra were recorded in deuterochloroform using a Bruker AV-400 spectrometer. Chemical shifts are reported in parts per million (ppm) and are referenced to the centreline of deuterochloroform (δ 7.24 ppm 1H NMR, 77.0 ppm 13C NMR). Coupling constants (J values) are given in Hertz (Hz). Low resolution mass spectra were obtained using a Ribermag R10-10 quadrupole instrument. High resolution mass spectra (HRMS) were obtained using a VG Analytical ZAG double focusing spectrometer.
Tetrahydrofuran, dichloromethane, and ether (when used as reaction solvents) were purchased from Fisher Scientific and purified using an Innovative Technologies solvent purification system. All other reagents were commercially available and were used without further purification. The azido esters 1a-c were prepared according to a modified procedure of Khoukhi, Vaultier and Carrié.14
General Procedure for the Synthesis of Azido Esters 2a-f
Diisopropylamine (1.30 equiv) was dissolved in THF (50 mL) at 0 °C, followed by the addition of n-butyllithium (1.10 equiv). The mixture was allowed to stir for 40 min. The temperature was decreased to -78 °C and the azido ester 1 (1.00 equiv) was added. After 30 min of stirring, allyl bromide (1.10 equiv) was added into the reaction mixture. Then, the temperature was allowed to slowly increase to rt over 3.5 h before the reaction mixture was diluted with Et2O (50.0 mL) and water (20.0 mL). The organic layer was separated and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic layers were washed with brine (1 × 30 mL), dried (NaSO4) and concentrated to give the crude product, which was purified by flash chromatography to afford the azido ester 2.
Ethyl 2-(2′-azidoethyl)pent-4-enoate (2a)
(1.25 g, 84%) was isolated after chromatography (5% EtOAc/hexanes) as a pale yellow oil. Rf = 0.50 (30% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 5.73 (m, 1H), 5.07 (m, 2H), 4.16 (q, J = 7.2 Hz, 2H), 3.32 (m, 2H), 2.56 (m, 1H), 2.63 (m, 1H), 2.41 (m, 1H), 1.93 (m, 1H), 1.75 (m, 1H), 1.27 (t, J = 3.9 Hz, 3H); 13C NMR (100.6 MHz, CDCl3) δ 174.6, 134.7, 117.4, 60.6, 49.4, 42.3, 36.4, 30.5, 14.3; IR (NaCl) 3000, 2100, 1740; MS (ES+), m/z 198.2 (M++H); HRMS calcd for C9H16N3O2 (M++H) 198.1242, found 198.1516.
Ethyl 4-azido-2-benzylbutanoate (2b)
(1.62 g, 79%) was isolated after chromatography (10% EtOAc/hexanes) as a pale yellow oil. Rf = 0.50 (30% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.15 (m, 5H), 4.07 (q, J = 7.12 Hz, 2H), 3.30 (m, 2H), 2.95 (m, 1H), 2.75 (m, 2H), 1.94 (m, 1H), 1.75 (m, 1H), 1.55 (t, J = 7.12 Hz, 3H); 13C NMR (100.6 MHz, CDCl3) δ 174.7, 138.5, 130.1, 128.9, 128.5, 126.6, 60.6, 49.5, 44.8, 38.5, 30.8, 14.1; IR (NaCl) 3100, 3000, 2100, 1730; MS (ES+), m/z 248.2 (M++H); HRMS calcd for C13H18N3O2 248.1399 (M++H): found 248.1682.
Ethyl 2-(3′-azidopropyl)pent-4-enoate (2c)
(0.38 g, 44%) was isolated after chromatography (10% EtOAc/hexanes) as a pale yellow oil. Rf = 0.50 (30% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 5.75 (m, 1H), 5.06 (m, 2H), 4.16 (q, J = 7.1 Hz, 2H), 3.29 (t, J = 6.5 Hz, 2H), 2.41 (m, 2H), 2.26 (m, 1H), 1.60 (m, 4H), 1.28 (t, J = 7.1 Hz, 3H); 13C NMR (100.6 MHz, CDCl3) δ 175.1, 135.1, 117.1, 60.4, 51.2, 44.8, 36.5, 28.7, 26.7, 14.3; IR (NaCl) 2020, 1660, 1400, 1120; MS (ES+), m/z 184.1 (M+-2N+H); HRMS calcd for C10H18NO2 (M+-2N+H) 184.1338, found 184.1337.
Ethyl 5-azido-2-benzylpentanoate (2d)
(0.69 g, 65%) was isolated after chromatography (10% EtOAc/hexanes) as a pale yellow oil. Rf = 0.50 (30% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.23 (m, 5H), 4.09 (q, J = 7.1 Hz, 2H), 3.27 (t, J = 6.6 Hz, 2H), 2.97 (m, 1H), 2.76 (dd, J = 6.8 Hz, 6.7 Hz, 1H), 2.68 (m, 1H), 1.65 (m, 4H), 1.18 (t, J = 7.2 Hz, 3H); 13C NMR (100.6 MHz, CDCl3) δ 175.1, 139.0, 128.9, 128.4, 126.4, 60.4, 51.2, 47.2, 38.6, 29.0, 26.7, 14.2; IR (NaCl) 2000, 1680, 1450; MS (ES+), m/z 262.2 (M++H); HRMS calcd for C14H20N3O2 (M++H) 262.1555 found 262.1553.
Ethyl 2-allyl-6-azidohexanoate (2e)
(0.50 g, 83%) was isolated after chromatography (10% EtOAc/hexanes) as a pale yellow oil. Rf = 0.50 (30% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 5.66 (m, 1H), 4.96 (m, 2H), 4.07 (q, J = 4.9 Hz, 2H), 3.19 (t, J = 6.8 Hz, 2H), 2.33 (m, 2H), 2.16 (m, 1H), 1.51 (m, 4H), 1.32 (m, 2H), 1.19 (t, J = 4.9 Hz, 3H); 13C NMR (100.6 MHz, CDCl3) δ 175.1, 135.3, 116.7, 60.1, 51.1, 45.0, 36.4, 31.1, 28.7, 34.4, 14.2; IR (NaCl) 2950, 2060, 1710; MS (ES+), m/z 200.2 (M+-2N+3H); HRMS calcd for C11H22NO2 (M+-2N+3H) 200.1650, found 200.1644.
Ethyl 6-azido-2-benzylhexanoate (2f)
(0.80 g, 54%) was isolated after chromatography (20% EtOAc/hexanes) as a pale yellow oil. Rf = 0.60 (50% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.21 (m, 5H), 4.06 (q, J = 7.1 Hz, 2H), 3.17 (t, J = 6.8 Hz, 2H), 2.95 (m, 1H), 2.70 (m, 2H), 1.67 (m, 1H), 1.51 (m, 3H), 1.35 (m, 2H), 1.13 (t, J = 7.1 Hz, 3H); 13C NMR (100.6 MHz, CDCl3) δ 175.1, 139.3, 128.9, 128.3, 126.3, 60.1, 51.1, 47.4, 38.6, 31.5, 28.7, 24.5, 14.2; IR (NaCl) 2880, 2020, 1700; MS (ES+), m/z 298.2 (M++Na); HRMS calcd for C15H21N3O2Na (M++Na) 298.1531, found 298.1532.
General Procedure for the Synthesis of Azido Aldehydes 3a-f
DIBAL-H (1.08 equiv) was slowly added to a solution of the azido ester 2 (1.00 equiv) in anhydrous Et2O (10.0 mL) at -70 °C. The reaction mixture was stirred for 1 h and quenched with MeOH (0.50 mL) followed by the addition of saturated aqueous potassium sodium tartrate solution (10 mL). The reaction mixture was then passed through a thin layer of celite. After the separation of organic layer, the aqueous layer was extracted with Et2O (3 × 5 mL). The combined organic layers were washed with brine (1 × 30 mL), dried (NaSO4) and concentrated to give the crude product, which was purified by flash chromatography to afford the azido ester 3.
2-(2′-Azidoethyl)pent-4-enal (3a)
(0.32 g, 41%) was isolated after chromatography (20% EtOAc/hexanes) as a pale yellow oil. Rf = 0.40 (20% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 9.68 (d, J = 1.5 Hz, 1H), 5.74 (m, 1H), 5.13 (m, 2H), 3.35 (m, 2H), 2.50 (m, 2H), 2.28 (m, 1H), 1.97 (m, 1H), 1.71 (m, 1H); 13C NMR (100.6 MHz, CDCl3) δ 203.1, 134.1, 118.1, 49.1, 48.3, 32.9, 27.3; IR (NaCl) 2960, 2140, 1730; MS (ES+), m/z 126.1 (M+-2N+H); HRMS calcd for C7H12NO (M+-2N+H) 126.0919, found 126.0911.
4-Azido-2-benzylbutanal (3b)
(0.73 g, 59%) was isolated after chromatography (5% EtOAc/hexanes) as a pale yellow oil. Rf = 0.35 (20% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 9.73 (d, J = 1.5 Hz, 1H), 7.27 (m, 5H), 3.32 (m, 2H), 3.06 (dd, J = 5.5 Hz, 7.0 Hz, 1H), 2.77 (m, 2H), 1.96 (m, 1H), 1.70 (m, 1H); 13C NMR (100.6 MHz, CDCl3) δ 203.1, 137.9, 128.9, 128.8, 126.8, 50.5, 49.1, 35.1, 27.6; IR (NaCl) 2960, 2120, 1740; MS (ES+), m/z 178.1 (M+-2N+3H); HRMS calcd for C11H16NO (M+-2N+3H) 178.1232, found 178.1227.
2-(3′-Azidopropyl)pent-4-enal (3c)
(0.25 g, 66%) was isolated as a pale yellow oil. Rf = 0.60 (30% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 9.66 (s, 1H), 5.76 (m, 1H), 5.13 (m, 2H), 3.33 (t, J = 6.0 Hz, 2H), 2.47 (m, 2H), 2.29 (m, 1H), 1.68 (m, 4H); 13C NMR (100.6 MHz, CDCl3) δ 204.0, 134.4, 117.7, 51.3, 50.7, 33.1, 26.3, 25.3; IR (NaCl) 2880, 2010, 1660, 1600; MS (ES+), m/z 140.1 (M+-2N+H); HRMS calcd for C8H14NO (M+−2N+H) 140.1075, found 140.1078.
5-Azido-2-benzylpentanal (3d)
(0.54 g, 83%) was isolated as a pale yellow oil. Rf = 0.50 (30% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 9.71 (d, J = 2.2 Hz, 1H), 7.26 (m, 5H), 3.28 (t, J = 6.5 Hz, 2H), 3.04 (q, J = 7.2 Hz, 6.7 Hz, 1H), 2.76 (dd, J = 7.2 Hz, 6.7 Hz, 1H), 2.68 (m, 1H), 1.65 (m, 4H); 13C NMR (100.6 MHz, CDCl3) δ 203.9, 138.3, 128.9, 128.7, 126.6, 52.9, 51.3, 35.2, 26.4, 25.5; IR (NaCl) 3000, 2010, 1700, 1650; MS (ES+), m/z 217.1 (M+); HRMS calcd for C12H15N3O (M+) 217.1215, found 217.1056.
2-Allyl-6-azidohexanal (3e)
(97 mg, 81%) was isolated as a pale yellow oil. Rf = 0.50 (30% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 9.57 (d, J = 2.2 Hz, 1H), 5.69 (m, 1H), 5.04 (m, 2H), 3.23 (t, J = 6.8 Hz, 2H), 2.35 (m, 2H), 2.23 (m, 1H), 1.59 (m, 3H), 1.40 (m, 3H); 13C NMR (100.6 MHz, CDCl3) δ 204.3, 134.7, 117.3, 51.1, 50.9, 32.9, 29.0, 27.6, 24.0; IR (NaCl) 2850, 2040, 1680; MS (ES+), m/z 154.1 (M+-2N+H); HRMS calcd for C9H16NO (M+-2N+H) 154.1232, found 154.1236.
6-Azido-2-benzylhexanal (3f)
(0.52 g, 78%) was isolated as a pale yellow oil. Rf = 0.50 (30% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 9.66 (d, J = 2.1 Hz, 1H), 7.24 (m, 5H), 3.21 (t, J = 6.7 Hz, 2H), 3.00 (dd, J = 7.0 Hz, 6.9 Hz, 1H), 2.73 (dd, J = 7.0 Hz, 6.9 Hz, 1H), 2.62 (m, 1H), 1.67 (m, 1H), 1.38 (m, 5H); 13C NMR (100.6 MHz, CDCl3) δ 204.1, 138.8, 138.2, 129.3, 129.0, 128.8, 128.6, 128.4, 127.6, 126.8, 126.5, 53.2, 51.1, 35.0, 28.8, 27.9, 24.1; IR (NaCl) 2960, 2020, 1680, 1660; MS (ES+), m/z 204.1 (M+); HRMS calcd for C13H18NO (M+-2N+H) 204.1388, found 204.1389.
3-Azidononanal (3g)
To a solution of NaN3 (1.46 g, 22.5mmol) in aqueous acetic acid (21.6 mmol in 2.0 mL of H2O) was added nonenal (1.20 mL, 7.23 mmol) at 0 °C. After 1 h of consistent stirring, the reaction mixture was diluted with Et2O (20 mL) before washing with NaHCO3 (3 × 5 mL) and brine (10 mL). The organic layer was dried (NaSO4) and concentrated to give the crude product, which was purified by flash chromatography (2% EtOAc/hexanes) to afford 3g (1.14 g, 87%). Rf = 0.30 (10% EtOAc/hexanes) as a pale yellow oil.). 1H NMR (400 MHz, CDCl3) δ 9.78 (m, 1H), 3.87 (m, 1H), 2.62 (m, 2H), 1.41 (m, 10H), 0.88 (m, 3H); 13C NMR (100.6 MHz, CDCl3) δ 199.9, 57.4, 48.6, 34.9, 32.0, 29.3, 26.2, 22.9, 14.4; IR (NaCl) 2115, 1731; MS (FAB+), m/z 184 (M++H); HRMS calcd for C9H18N3O (M++H) 185.1450, found 185.0824.
General Procedure for Intramolecular Schmidt Reactions Using TiCl4
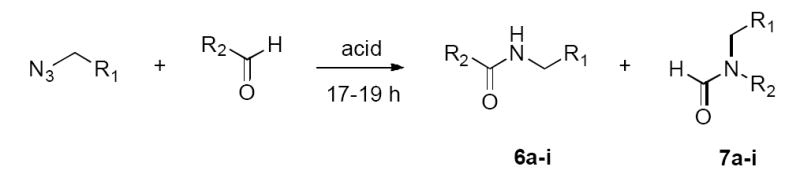
The azido aldehyde (1.0 equiv) was activated by TiCl4 (1.3 equiv) in CH2Cl2 (2.0 mL) at room temperature. After 17–19 h of stirring, the mixture was quenched with saturated NaHCO3 and diluted with CH2Cl2. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were washed with brine, dried (NaSO4) and concentrated to give crude product, which was purified by chromatography.
General Procedure for Intramolecular Schmidt Reactions Using TFA
The azido aldehyde was dissolved in TFA (1.0 mL) at room temperature and quenched after 17–19 h of stirring. Workup as described above afforded crude product, which was purified by chromatography.
3-Allylpyrrolidin-2-one (5a)
(81 mg, 90% with TiCl4 and 57 mg, 63% with TFA) was afforded after chromatography (75% EtOAc/hexanes) as a pale yellow oil. Rf = 0.20 (100% EtOAc). 1H NMR (400 MHz, CDCl3) δ 7.33 (s, 1H), 5.75 (m, 1H), 5.07 (m, 2H), 3.31 (m, 2H), 2.55 (m, 1H), 2.46 (m, 1H), 2.20 (m, 2H), 1.81 (m, 1H); 13C NMR (100.6 MHz, CDCl3) δ 1180.3, 135.6, 116.8, 40.6, 40.5, 35.0, 26.6; IR (NaCl) 3000, 1710, 1660, 1290, 1080; MS (ES+), m/z 126.1 (M++H); HRMS calcd for C7H12NO (M++H) 126.0919, found 126.0917.
3-Benzylpyrrolidin-2-one (5b)
(89 mg, 94% with TiCl4 and 100 mg, 77% with TFA) was afforded after chromatography (70% EtOAc/hexanes) as a pale yellow oil. Rf = 0.20 (100% EtOAc). 1H NMR (400 MHz, CDCl3) δ 7.47 (s, 1H), 7.30 (m, 5H), 3.23 (m, 3H), 2.65 (m, 2H), 2.09 (m, 1H), 1.80 (m, 1H); 13C NMR (100.6 MHz, CDCl3) δ 180.2, 139.6, 129.0, 128.8, 128.5, 126.3, 43.0, 40.5, 36.7, 21.1; IR (NaCl) 3240, 3020, 1660; MS (ES+), m/z 176.1 (M++H); HRMS calcd for C11H14NO (M++H) 176.1075, found 176.1071.
2-Allylpyrrolidine-1-carbaldehyde (4c)
(80 mg, 96% with TiCl4 and 50 mg, 60% with TFA) was isolated as a mixture of rotamers in a 6:4 ratio after chromatography (50% EtOAc/hexanes) as a pale yellow oil. Rf = 0.10 (50% EtOAc/hexanes). IR (NaCl) 2900, 1620, 1350; MS (ES+), m/z 140.1 (M++H); HRMS calcd for C8H14NO (M++H) 140.1075, found 140.1070. Major rotamer: 1H NMR (400 MHz, CDCl3) δ 8.26 (s, 1H), 5.76 (m, 1H), 5.09 (m, 2H), 3.55 (m, 1H), 3.84 (m, 1H), 3.39 (m, 1H), 2.28 (m, 2H), 1.90 (m, 3H), 1.77 (m, 1H); 13C NMR (100.6 MHz, CDCl3) δ 161.1, 134.3, 118.7, 57.1, 46.7, 40.5, 30.2, 23.7. Minor rotamer: 1H NMR (400 MHz, CDCl3) δ 8.26 (s, 1H), 5.76 (m, 1H), 5.09 (m, 2H), 3.84 H NMR (400 MHz, CDCl3) δ (m, 1H), 3.55 (m, 1H), 3.39 (m, 1H), 2.28 (m, 2H), 1.90 (m, 3H), 1.77 (m, 1H); 13C NMR (100.6 MHz, CDCl3) δ 161.0, 133.6, 117.6, 54.7, 43.5, 37.5, 29.5, 22.4.
2-Benzylpyrrolidine-1-carbaldehyde (4d)
(74 mg, 85% with TiCl4 and 59 mg, 68% with TFA) was isolated as rotamers in a 2:1 ratio after purified by flash chromatography (50% EtOAc/hexanes) as a pale yellow oil. Rf = 0.10 (50% EtOAc/hexanes). IR (NaCl) 2950, 1650, 750, 690; MS (ES+), m/z 190.1 (M++H); HRMS calcd for C12H16NO (M++H) 190.1232, found 190.1227. Major rotamer: 1H NMR (400 MHz, CDCl3) δ 7.87 (s, 1H), 7.22 (m, 5H), 4.02 (m, 1H), 3.52 (m, 1H), 3.40 (m, 1H), 2.74 (m, 2H), 1.85 (m, 4H); 13C NMR (100.6 MHz, CDCl3) δ 161.1, 137.5, 129.6, 129.4, 128.8, 128.4, 126.8, 56.5, 46.8, 38.7, 30.3, 22.3; Minor rotamer: 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 7.22 (m, 5H), 4.24 (m, 1H), 3.52 (m, 1H), 3.30 (m, 1H), 2.74 (m, 2H), 1.85 (m, 4H); 13C NMR (100.6 MHz, CDCl3) δ 161.1, 138.3, 129.6, 129.4, 128.8, 128.4, 126.3, 59.1, 43.4, 42.4, 29.2, 23.5.
2-Allylpiperidine-1-carbaldehyde (4e)
(35 mg, 41% with TiCl4 and 45 mg, 53% with TFA) was isolated as rotamers in a 1:1 ratio after purified by flash chromatography (40% EtOAc/hexanes) as a pale yellow oil. Rf = 0.15 (50% EtOAc/hexanes). IR (NaCl) 3000, 2860, 1620, 970, 880; MS (ES+), m/z 154.1 (M++H); HRMS calcd for C9H16NO (M++H) 154.1232, found 154.1230. Rotamer 1: 1H NMR (400 MHz, CDCl3) δ 8.00 (s, 1H), 5.66 (m, 1H), 5.05 (m, 2H), 4.21 (dd, J = 3.4 Hz, 9.6 Hz, 1H), 3.60 (m, 1H), 2.70 (t, J = 3.0 Hz, 10.4 Hz, 1H), 2.52 (m, 1H), 2.27 (m, 1H), 1.69 (m, 5H), 1.39 (m, 1H); 13C NMR (100.6 MHz, CDCl3) δ 161.3, 134.9, 118.2, 54.5, 42.5, 34.9, 29.4, 26.3, 19.9. Rotamer 2: 1H NMR (400 MHz, CDCl3) δ 8.00 (s, 1H), 5.66 (m, 1H), 5.05 (m, 2H), 4.60 (q, J = 6.4 Hz, 1H), 3.36 (dd, J = 4.2 Hz, 9.2 Hz, 1H), 3.14 (t, J = 2.8 Hz, 10.5 Hz, 1H), 2.41 (m, 1H), 2.27 (m, 1H), 1.69 (m, 5H), 1.39 (m, 1H); 13C NMR (100.6 MHz, CDCl3) δ 161.2, 134.1, 117.1, 46.8, 36.3, 34.1, 27.1, 25.0, 19.5.
2-Benzylpiperidine-1-carbaldehyde (4f)
(43 mg, 49% with TiCl4 and 62 mg, 71% with TFA) was isolated as rotamers in a 3:2 ratio after chromatography (40% EtOAc/hexanes) as a pale yellow oil. Rf = 0.20 (50% EtOAc/hexanes). IR (NaCl) 2880, 1620, 980, 700; MS (ES+), m/z 226.1 (M++Na); HRMS calcd for C13H17NONa (M++Na) 226.1208, found 226.1204. Major rotamer: 1H NMR (400 MHz, CDCl3) δ 7.64 (s, 1H), 7.23 (m, 5H), 4.29 (dd, J = 4.3 Hz, 9.2 Hz, 1H), 3.76 (m, 1H), 3.07 (q, J = 4.3 Hz, 1H), 2.85 (m, 2H), 1.69 (m, 6H); 13C NMR (100.6 MHz, CDCl3) δ 161.2, 138.2, 138.0, 129.2, 129.0, 128.8, 126.8, 56.6, 42.7, 37.0, 29.5, 26.4, 20.0. Minor rotamer: 1H NMR (400 MHz, CDCl3) δ 7.98 (s, 1H), 7.23 (m, 5H), 4.81 (q, J = 7.2 Hz, 1H), 3.43 (dd, J = 4.9 Hz, 8.4 Hz, 1H), 3.28 (t, J = 2.7 Hz, 10.3 Hz, 1H), 2.85 (m, 2H), 1.69 (m, 6H); 13C NMR (100.6 MHz, CDCl3) δ 161.2, 138.2, 138.0, 129.2, 129.0, 128.5, 126.4, 48.6, 36.3, 35.7, 26.1, 25.1, 19.5.
General Procedure for Intermolecular Schmidt Reactions Using TiCl4
The aldehyde (1.0 equiv) was activated by TiCl4 (1.3 equiv) in CH2Cl2 (2.0 mL) at room temperature, followed by slow addition of alkyl azide (1.1 equiv). After 17–19 h of consistent stirring, the mixture was quenched with saturated NaHCO3 and diluted with CH2Cl2. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were washed with brine, dried (NaSO4) and concentrated to give the crude product, which was purified by flash chromatography.
General Procedure for Intermolecular Schmidt Reactions Using TFA
The aldehyde was reacted with the alkyl azide in TFA (1.0 mL) at room temperature and quenched after 17–19 h of consistent stirring under the same workup condition described above to give crude product, which was purified by flash chromatography.
N-Benzyl-2-phenylacetamide (6a) (40 mg, 21%) and N, N-dibenzylformamide (7a) (20 mg, 10%) were isolated after chromatography (20% EtOAc/hexanes) as a pale yellow oil. Compound 6a: Rf = 0.35 (50% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.30 (m, 10H), 5.90 (s, 1H), 4.43 (d, J = 5.8 Hz, 2H), 3.64 (s, 2H); 13C NMR (100.6 MHz, CDCl3) δ 171.0, 138.2, 134.9, 129.5, 129.1, 128.7, 127.5, 127.4, 127.4, 60.4, 43.8, 43.6; IR (NaCl) 3320, 3100, 1660, 1070; MS (ES+), m/z 226.1 (M++H); HRMS calcd for C15H16NO (M++H) 226.1232, found 226.1245. Compound 7a: Rf = 0.30 (50% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H), 7.31 (m, 10H), 4.44 (s, 2H), 4.29 (s, 2H); 13C NMR (100.6 MHz, CDCl3) δ 162.9, 136.0, 135.6, 129.0, 128.7, 128.5, 128.2, 127.7, 127.7, 50.3, 44.7; IR (NaCl) 3080, 2980, 1680, 1210; MS (ES+), m/z 226.1 (M++H); HRMS calcd for C15H16NO (M++H) 226.1232, found 226.1245.
N-Benzylheptanamide (6b) (30 mg, 16%) and N-benzyl-N-hexylformamide (7b) (20 mg, 10%, rotamers in a 5:6 ratio) were isolated after chromatography (30% EtOAc/hexanes) as a pale yellow oil. Compound 6b: Rf = 0.30 (50% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.33 (m, 5H), 5.88 (s, 1H), 4.44 (d, J = 5.7 Hz, 2H), 2.22 (t, J = 7.4 Hz, 2H), 1.66 (t, J = 7.1 Hz, 2H), 1.32 (m, 6H), 0.896 (t, J = 6.8 Hz, 3H); 13C NMR (100.6 MHz, CDCl3) δ 173.1, 138.5, 128.7, 127.8, 127.5, 43.6, 36.8, 31.5, 29.0, 25.8, 22.5, 14.0; IR (NaCl) 3320, 3100, 1700, 1200; MS (ES+), m/z 220.2 (M++H); HRMS calcd for C14H22NO (M++H) 220.1701, found 220.1703. Compound 7b: Rf = 0.45 (50% EtOAc/hexanes). IR (NaCl) 3000, 2960, 2900, 1680; MS (ES+), m/z 220.2 (M++H); HRMS calcd for C14H22NO (M++H) 220.1701, found 220.1694. Rotamer 1: 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 7.31 (m, 5H), 4.41 (s, 2H), 3.24 (t, J = 7.5 Hz, 2H), 1.50 (m, 2H), 1.27 (m, 6H), 0.89 (q, J = 6.7 Hz, 3H); 13C NMR (100.6 MHz, CDCl3) δ 162.9, 136.3, 128.9, 128.6, 128.2, 128.1, 127.5, 51.2, 45.2, 31.5, 28.1, 26.8, 26.1, 14.0. Rotamer 2: 1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 7.31 (m, 5H), 4.55 (s, 2H), 3.14 (t, J = 7.1 Hz, 2H), 1.50 (m, 2H), 1.27 (m, 6H), 0.89 (q, J = 6.7 Hz, 3H); 13C NMR (100.6 MHz, CDCl3) δ 162.8, 136.3, 128.9, 128.6, 128.2, 128.1, 127.5, 46.8, 42.0, 31.3, 28.1, 26.5, 22.5, 14.0.
N-Benzyl-2-phenylpropanamide (6c) and N-benzyl-N-(1′-phenylethyl)-formamide (7c) (86 mg, 44%) were isolated as inseparable product mixture with a 1:3 ratio after chromatography (20% EtOAc/hexanes) as a pale yellow oil. Compound 7c: Exist as rotamer in a 3:1 ratio. Rf = 0.15 (25% EtOAc/hexanes). IR (NaCl) 3320, 3100, 1660; MS (ES+), m/z 240.1 (M++Na); HRMS calcd for C16H17NONa (M++Na) 262.1208, found 262.1204. Major rotamer: 1H NMR (400 MHz, CDCl3) δ 8.54 (s, 1H), 7.15 (m, 10H), 4.40 (m, 2H), 4.07 (m, 1H), 1.54 (m, 3H); 13C NMR (100.6 MHz, CDCl3) δ 163.6, 141.4, 139.7, 138.4, 137.7, 128.9, 128.6, 128.5, 128.0, 127.8, 127.7, 127.3, 126.8, 56.7, 48.3, 20.2. Minor rotamer: 1H NMR (400 MHz, CDCl3) δ 8.31 (d, J = 10.6 Hz, 1H), 7.15 (m, 10H), 4.40 (m, 2H), 3.61 (m, 1H), 1.54 (m, 3H); 13C NMR (100.6 MHz, CDCl3) δ 162.5, 140.4, 139.7, 138.4, 137.5, 128.7, 128.6, 128.5, 128.0, 127.8, 127.7, 127.3, 126.8, 50.6, 45.3, 17.0.
N-Phenethyl-2-phenylacetamide (6d) (40 mg, 22% with TiCl4 and 28 mg, 16% with TFA) and N-benzyl-N-phenethyl-formamide (7d) (33 mg, 19% with TiCl4 and 28 mg, 16% with TFA, rotamers in a 1:1 ratio) were isolated after chromatography (30% EtOAc/hexanes) as a pale yellow oil. Compound 6d: Rf = 0.10 (25% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.23 (m, 8H), 7.01 (m, 2H), 5.37 (s,1H), 3.52 (s, 2H), 3.44 (q, J = 6.8 Hz, 2H), 2.71 (t, J = 6.8, 2H); 13C NMR (100.6 MHz, CDCl3) δ 170.9, 138.7, 134.8, 129.5, 129.0, 128.7, 128.6, 127.3, 126.4, 43.9, 40.7, 35.5; IR (NaCl) 3260, 3000, 1620; MS (ES+), m/z 240.1 (M++H); HRMS calcd for C16H18NO (M++H) 240.1388, found 240.1380. Compound 7d: Rf = 0.15 (25% EtOAc/hexanes). IR (NaCl) 3000, 1650, 1180; MS (ES+), m/z 240.1 (M++H); HRMS calcd for C16H18NO (M++H) 240.1388, found 240.1377. Rotamer 1: 1H NMR (400 MHz, CDCl3) δ 8.26 (s, 1H), 7.18 (m, 10H), 4.55 (s, 2H), 3.44 (t, J = 7.4 Hz, 2H), 2.78 (m, 2H); 13C NMR (100.6 MHz, CDCl3) δ 162.9, 138.8, 136.4, 129.9, 129.2, 128.9, 128.7, 128.3, 127.7, 126.9, 126.5, 51.9, 45.5, 35.1. Rotamer 2: 1H NMR (400 MHz, CDCl3) δ 7.96 (s, 1H), 7.18 (m, 10H), 4.22 (s, 2H), 3.37 (t, J = 7.0 Hz, 2H), 2.78 (m, 2H); 13C NMR (100.6 MHz, CDCl3) δ 162.8, 137.8, 136.1, 129.4, 129.1, 128.8, 128.6, 128.1, 127.6, 126.9, 126.5, 48.4, 43.9, 33.5.
N-Phenethylheptanamide (6e) (77 mg, 45% with TiCl4 and 16 mg, 9% with TFA) and N-hexyl-N-phenethylformamide (7e) (68 mg, 39% with TiCl4 and 16 mg, 9% with TFA, rotamers in a 5:4 ratio) were isolated after chromatography (20% EtOAc/hexanes) as a pale yellow oil. Compound 6e: Rf = 0.10 (25% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.26 (m, 5H), 5.74 (s. 1H), 3.52 (m, 2H), 2.82 (t, J = 7.0 Hz, 2H), 2.13 (t, J = 7.4 Hz, 2H), 1.60 (m, 2H), 1.31 (m, 6H), 0.89 (t, J = 6.4 Hz, 3H); 13C NMR (100.6 MHz, CDCl3) δ 173.3, 139.0, 128.8, 128.6, 126.5, 40.5, 36.8, 35.7, 31.6, 29.0, 25.7, 22.5, 14.2, 14.0; IR (NaCl) 3350, 2880, 1620, 1180; MS (ES+), m/z 234.2 (M++H); HRMS calcd for C15H24NO (M++H) 234.1858, found 234.1843. Compound 7e: Rf = 0.20 (25% EtOAc/hexanes). IR (NaCl) 2880, 1650, 1620, 1430; MS (ES+), m/z 234.2 (M++H); HRMS calcd for C15H24NO (M++H) 234.1858, found 234.1847. Major rotamer: 1H NMR (400 MHz, CDCl3) δ 8.07 (s, 1H), 7.24 (m, 5H), 3.53 (t, J = 7.6 Hz, 2H), 3.11 (t, J = 7.2 Hz, 2H), 2.86 (m, 2H), 1.50 (m, 2H), 1.33 (m, 6H), 0.91 (m, 3H); 13C NMR (100.6 MHz, CDCl3) δ 162.9, 138.8, 137.8, 128.8, 128.7, 128.5, 126.8, 49.1, 44.2, 35.5, 31.5, 28.6, 26.6, 22.6, 14.0. Minor rotamer: 1H NMR (400 MHz, CDCl3) δ 7.85 (s, 1H), 7.24 (m, 5H), 3.46 (t, J = 7.2 Hz, 2H), 3.34 (t, J = 7.6 Hz, 2H), 2.86 (m, 2H), 1.50 (m, 2H), 1.33 (m, 6H), 0.91 (m, 3H); 13C NMR (100.6 MHz, CDCl3) δ 162.8, 138.8, 137.8, 128.8, 128.6, 128.5, 126.5, 48.0, 42.4, 33.7, 31.3, 27.3, 26.1, 22.5, 14.0.
N-Phenethyl-2-phenylpropanamide (6f) (38 mg, 20% with TiCl4 nd 24 mg, 13% with TFA) and N- phenethyl-N-(1′-phenylethyl)-formamide (7f) (57 mg, 30% with TiCl4 and 9 mg, 5% with TFA, rotamers in a 2:1 ratio) were isolated as pale yellow oil after chromatography (20% EtOAc/hexanes). Compound 6f: Rf = 0.15 (25% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.22 (m, 8H), 6.99 (d, J =6.3 Hz, 2H), 5.39 (s, 1H), 3.48 (m, 2H), 3.38 (m, 1H), 2.69 (t, J = 1.6 Hz, 5.1 Hz, 2H), 1.49 (d, J = 7.2, 3H); 13C NMR (100.6 MHz, CDCl3) δ 174.1, 141.3, 138.8, 128.9, 128.7, 128.5, 127.7, 127.2, 126.4, 47.1, 40.7, 35.5, 18.3; IR (NaCl) 3320, 3000, 1650, 1550; MS (ES+), m/z 254.1 (M++H); HRMS calcd for C17H20NO (M++H) 254.1545, found 254.1533. Compound 7f: Rf = 0.10 (25% EtOAc/hexanes). IR (NaCl) 3000, 2860, 1620, 1450; MS (ES+), m/z 254.2 (M++H); HRMS calcd for C17H20NO (M++H) 254.1545, found 254.1537. Major rotamer: 1H NMR (400 MHz, CDCl3) δ 8.39 (s, 1H), 7.25 (m, 10H), 4.67, (q, J = 7.0 Hz, 1H), 3.23 (m, 2H), 2.79 (m, 1H), 2.56 (m, 1H), 1.59 (dd, J = 2.2 Hz, 5.0 Hz, 3H); 13C NMR (100.6 MHz, CDCl3) δ 163.1, 141.3, 140.1, 138.9, 128.9, 128.7, 128.5, 128.3, 128.0, 127.8, 127.2, 126.7, 126.4, 51.1, 49.8, 44.2, 34.5, 16.8. Minor rotamer: 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H), 7.25 (m, 10H), 5.79 (q, J = 7.2 Hz, 1H), 3.36 (m, 2H), 2.69 (t, J = 1.3 Hz, 5.3 Hz, 1H), 2.42 (m, 1H), 1.49 (d, J = 7.2 Hz, 3H); 13C NMR (100.6 MHz, CDCl3) δ 162.5, 140.2, 139.1, 138.0, 128.8, 128.7, 128.5, 128.3, 128.0, 127.8, 127.2, 126.7, 126.4, 51.1, 46.0, 37.9, 19.3, 16.8.
N-Hexyl-2-phenylacetamide (6g) (51 mg, 29% with TiCl4 and 32 mg, 19% with TFA) and N-benzyl-N-hexylformamide (7g) (45 mg, 26% with TiCl4 and 22 mg, 13% with TFA rotamers in a 1:1 ratio) were isolated after chromatography (20-25% EtOAc/hexanes) as a pale yellow oil. Compound 6g: Rf = 0.50 (50% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.31 (m, 5H), 5.39 (s, 1H), 3.58 (s, 2H), 3.21 (q, J = 6.9 Hz, 2H), 1.42 (t, J = 7.0 Hz, 2H), 1.25 (m, 6H), 0.87 (t, J = 6.8 Hz, 3H); 13C NMR (100.6 MHz, CDCl3) δ 170.8, 135.1, 129.5, 129.0, 127.3, 43.9, 39.7, 31.4, 29.4, 26.4, 22.5, 14.0; IR (NaCl) 3300, 2980, 1640; MS (ES+), m/z 220.2 (M++H); HRMS calcd for C14H22NO 220.1701 (M++H): found 220.1692.
Hexylheptanamide (6h) (68 mg, 37% with TiCl4 and 51 mg, 28% with TFA) and N,N-dihexylformamide (7h)11 (48 mg, 26% with TiCl4 and 22 mg, 12% with TFA) were isolated after chromatography (20% EtOAc/hexanes) as a pale yellow oil. Compound 6h: Rf = 0.50 (50% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 5.66 (s. 1H), 3.22 (q, J = 6.9 Hz, 2H), 2.15 (t, J = 7.5 Hz, 2H), 1.60 (q, J = 6.5 Hz, 2H), 1.48 (q, J = 6.2 Hz, 2H), 1.32 (s, 12H), 0.88 (t, J = 5.0 Hz, 6H); 13C NMR (100.6 MHz, CDCl3) δ 173.1, 39.5, 36.9, 31.6, 31.5, 29.6, 29.0, 26.6, 25.8, 22.5, 22.5, 14.0, 14.0; IR (NaCl) 3300, 2960, 1650, 1570; MS (ES+), m/z 214.2 (M++H); HRMS calcd for C13H28NO (M++H) 214.2171, found 214.2161. Compound 7h: Rf = 0.25 (50% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 8.03 (s, 1H), 3.28 (t, J = 6.4 Hz, 2H), 3.18 (t, J = 6.9 Hz, 2H), 1.52 (s, 4H), 1.29 (s, 12H), 0.89 (m, 6H); 13C NMR (100.6 MHz, CDCl3) δ 162.7, 47.4, 42.1, 31.5, 31.4, 28.6, 27.3, 26.6, 26.1, 22.6, 22.5, 14.0, 14.0; IR (NaCl) 2980, 1660, 1480; MS (ES+), m/z 214.2 (M++H); HRMS calcd for C13H28NO (M++H) 214.2171, found 214.2147.
N-Hexyl-2-phenylpropanamide (6i) (70 mg, 35%) and N-hexyl-N-(1-phenylethyl)-formamide (7i) (55 mg, 27%, rotamers in a 2:1 ratio) were isolated as a pale yellow oil after chromatography (20% EtOAc/hexanes). Compound 6i: Rf = 0.35 (25% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.37 (m, 5H), 5.51 (s, 1H), 3.56 (q, J = 7.2 Hz, 1H), 3.18 (m, 2H), 1.53 (d, J = 7.2 Hz, 3H), 1.40 (m, 2H), 1.24 (m, 6H), 0.86 (m, 3H); 13C NMR (100.6 MHz, CDCl3) δ 174.1, 141.6, 128.8, 128.4, 127.9, 127.5, 127.2, 126.8, 47.1, 39.6, 31.4, 29.4, 26.4, 22.5, 18.5, 14.0; IR (NaCl) 3500, 3300, 1650; MS (ES+), m/z 234.2 (M++H); HRMS calcd for C15H24NO (M++H) 234.1858, found 234.1847. Compound 7i: Rf = 0.30 (25% EtOAc/hexanes). IR (NaCl) 3500, 2980, 1670; MS (ES+), m/z 234.2 (M++H); HRMS calcd for C15H24NO (M++H) 234.1858, found 234.1847. Major rotamer: 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 7.30 (m, 5H), 4.72 (q, J = 7.1 Hz, 1H), 3.10 (t, J = 2.0 Hz, 5.0 Hz, 2H), 1.67 (d, J = 7.1 Hz, 3H), 1.42 (m, 2H), 1.20 (m, 6H), 0.85 (m, 3H); 13C NMR (100.6 MHz, CDCl3) δ 163.2, 140.5, 128.8, 128.4, 127.6, 127.5, 126.8, 56.8, 44.6, 31.4, 29.4, 26.8, 22.5, 19.6, 14.0. Minor rotamer: 1H NMR (400 MHz, CDCl3) δ 8.19, (s, 1H), 7.30 (m, 5H), 5.73 (q, J = 7.2 Hz, 1H), 3.00 (m, 2H), 1.57 (d, J = 7.2 Hz, 3H), 1.42 (m, 2H), 1.20 (m, 6H), 0.85 (m, 3H); 13C NMR (100.6 MHz, CDCl3) δ 162.4, 140.2, 128.7, 127.8, 127.5, 127.2, 126.8, 49.8, 44.6, 31.2, 28.3, 26.4, 22.4, 16.8, 13.9.
Compound 6i and 7i were also obtained using TFA (62 mg, 31%). The compounds were not separated in this experiment (6i:7i = 1:3 by NMR).
Scheme 1.
Acknowledgments
We thank the National Institute of General Medical Sciences (GM-49093) for financial support and Erik Fenster for assistance in preparing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Recent reviews of chemical methods for amide synthesis, see: Albericio F. Curr Opin Chem Biol. 2004;8:211–21. doi: 10.1016/j.cbpa.2004.03.002.Mahajan YR, Weinreb SM. Science of Synthesis. 2005;21:17–25.Ziegler T. Science of Synthesis. 2005;21:43–75.Bode JW. Curr Opin Drug Discovery Dev. 2006;9:765–775.
- 2.Review: Austin DJ, Miller SM. Science of Synthesis. 2005;21:77–109.
- 3.Some recent conversions of aldehydes to amides: Tillack A, Rudloff I, Beller M. Eur J Org Chem. 2001:523–528.Sharghi H, Sarvari H. Tetrahedron. 2002;58:10323–10328.Shie J-J, Fang J-M. J Org Chem. 2003;68:1158–1160. doi: 10.1021/jo026407z.Kashiwagi M, Fushuku K-I, Sugai T. J Mol Catal B: Enzym. 2004;29:249–258.Raj IVP, Sudalai A. Tetrahedron Lett. 2005;46:8303–8306.Nambu H, Hata K, Matsugi M, Kita Y. Eur J Org Chem. 2005;11:719–727. doi: 10.1002/chem.200400754.Yoo W-J, Li C-J. J Am Chem Soc. 2006;128:13064–13065. doi: 10.1021/ja064315b.
- 4.Review: Judd WR, Katz CE, Aubé J. Science of Synthesis. 2005;21:133–178.
- 5.Reviews: Wolff H. The Schmidt Reaction. Org React. 1946;3:307–336.Banthorpe DV. The Chemistry of the Azido Group. Vol. 1971. John Wiley & Sons; London: Rearrangements involving azido groups; pp. 397–440.
- 6.Reviews: Bräse S, Gil C, Knepper K, Zimmermann V. Angew Chem Int Ed. 2005;44:5188–5240. doi: 10.1002/anie.200400657.Lang S, Murphy JA. Chem Soc Rev. 2006;2:146–156. doi: 10.1039/b505080d.Nyfeler E, Renaud P. Chimia. 2006;60:276–284.
- 7.(a) Briggs LH, De Ath GC, Ellis SR. J Chem Soc. 1942:61–63. [Google Scholar]; (b) Smith PAS. J Am Chem Soc. 1948;70:320–323. [Google Scholar]; (c) Pearson WH, Schkeryantz JM. Tetrahedron Lett. 1992;33:5291–5294. [Google Scholar]; (d) Pearson WH, Walavalkar R, Schkeryantz JM, Fang W-K, Blickensdorf JD. J Am Chem Soc. 1993;115:10182–10194. [Google Scholar]; (e) Pearson WH, Fang W-K. J Org Chem. 1994;59:2682–2684. [Google Scholar]; (f) Pearson WH, Fang W-K. J Org Chem. 1995;60:4960–4961. [Google Scholar]; (g) Pearson WH. J Heterocycl Chem. 1996;33:1489–1496. [Google Scholar]; (h) Pearson WH, Fang W-K. Isr J Chem. 1997;37:39–46. [Google Scholar]
- 8.(a) Aubé J, Milligan GL. J Am Chem Soc. 1991;113:8965–8966. [Google Scholar]; (b) Aubé J, Milligan GL, Mossman CJ. J Org Chem. 1992;57:1635–1637. [Google Scholar]; (c) Aubé J, Rafferty PS, Milligan GL. Heterocycles. 1993;35:114–1147. [Google Scholar]; (d) Milligan GL, Mossman CJ, Aubé J. J Am Chem Soc. 1995;117:10449–11459. [Google Scholar]; (e) Mossman CJ, Aubé J. Tetrahedron. 1996;52:3403–3408. [Google Scholar]; (f) Forsee JE, Aubé J. J Org Chem. 1999;64:4381–4385. [Google Scholar]
- 9.(a) Boyer JH, Hamer J. J Am Chem Soc. 1955;77:951–954. [Google Scholar]; (b) Boyer JH, Canter FC, Hamer J, Putney RK. J Am Chem Soc. 1956;78:325–327. [Google Scholar]; (c) Boyer JH, Morgan LR., Jr J Org Chem. 1959;24:561–562. [Google Scholar]
- 10.Boyer JH. J Am Chem Soc. 1951;73:5248–5252. [Google Scholar]
- 11.Khoukhi N, Vaultier M, Carrié R. Tetrahedron. 1987;43:1811–1822. [Google Scholar]
- 12.Markidis T, Kokotos G. J Org Chem. 2001;66:1919–1923. doi: 10.1021/jo005677j. [DOI] [PubMed] [Google Scholar]
- 13.(a) Desai P, Schildknegt K, Agrios KA, Mossman CJ, Milligan GL, Aubé J. J Am Chem Soc. 2000;122:7226–7232. [Google Scholar]; (b) Wrobleski A, Aubé J. J Org Chem. 2001;66:886–889. doi: 10.1021/jo001367p. [DOI] [PubMed] [Google Scholar]
- 14.(a) Khoukhi N, Vaultier M, Carrié R. Tetrahedron. 1987;43:1811–1822. [Google Scholar]; (b) Kusumoto S, Sakai K, Shiba T. Bull Chem Soc Jpn. 1986;59:1296–1298. [Google Scholar]; (c) Keschmann E, Zbiral E, Schweng J. Justus Liebigs Annalen der Chemie. 1977;9:1508–1515. [Google Scholar]