

Abstract
The Mexican axolotl, Ambystoma mexicanum, has been a useful animal model to study heart development and cardiac myofibrillogenesis. A naturally-occurring recessive mutant, gene “c”, for cardiac non-function in the Mexican axolotl causes a failure of myofibrillogenesis due to a lack of tropomyosin expression in homozygous mutant (c/c) embryonic hearts.. Myofibril-Inducing RNA (MIR) rescues mutant hearts in vitro by promoting tropomyosin expression and myofibril formation thereafter. We have studied the effect of MIR on the expression of various isoforms of cardiac Troponin-T (cTnT), a component of the thin filament that binds with tropomyosin. Four alternatively spliced cTnT isoforms have been characterized from developing axolotl heart. The expression of various cTnT isoforms in normal, mutant, and mutant hearts corrected with MIR, is evaluated by real-time RT-PCR using isoform specific primer pairs; MIR affects the total transcription as well as the splicing of the cTnT in axolotl heart
Keywords: cardiac Troponin T, axolotl, myofibril, alternative splicing
Introduction
Troponin T (TnT) is a component of the troponin complex (troponin T, troponin I and troponin C) that anchors the complex to the thin filaments in vertebrate striated muscle. The troponin complex functions as a regulatory system for muscle contraction, responding to the cytosolic calcium concentration changes and other signalling [1]. Three muscle type-specific TnT isoform genes, cardiac TnT (cTnT or TNNT2), slow skeletal TnT (ssTnT or TNNT1) and fast skeletal TnT (fsTnT or TNNT3) have been cloned from higher vertebrates [2,3]. In addition, alternative RNA splicing adds another dimension of generating more protein isoforms for TnT [3,4]. The human cardiac Troponin-T gene contains 17 exons, some of which can be alternatively spliced, leading to the creation of different isoforms [5–7].
The differential splicing for some cTnT exons is developmentally regulated. Thus, some isoforms are present only in specific stages of development [3,6–7]. Different cTnT isoforms resulting from alternative splicing respond differently to cytosolic Ca2+ concentrations within the cell. The different isoforms also show differing actomyosin ATPase activity within the sarcomeres. Exclusion of the 10 amino acids in adult cTnT, encoded by exon 5, results in significant changes in activation of the actomyosin ATPase when compared to the embryonic isoforms [8,9]; not surprisingly, much smaller functional differences are observed from alternate splicing of exon 4 in bovine cardiac troponin T [10].
Our current studies are on a salamander model, the Mexican axolotl, Ambystoma mexicanum, in which a naturally occurring genetic recessive cardiac lethal mutation--gene c-- was discovered [11]. Homozygous mutant (c/c) embryos are first distinguishable from their normal siblings (+/+ or +/c) at stage 34, when the mutant embryos develop hearts that fail to beat and eventually die from lack of circulation. Subsequent studies have demonstrated the defects in myofibrillogenesis and significantly decreased tropomyosin expression in the mutant embryonic hearts [12–17]. As a tropomyosin-interacting protein, cardiac troponin T has also been studied in this system in our laboratory. It has been demonstrated that the expression of TnT protein is significantly decreased in the mutant hearts [18]. One possible explanation for this observation is that splicing abnormalities could exist in the mutant embryonic hearts. To test this hypothesis, we cloned four different isoforms of the cTnT cDNA (TNNT2-1, -2, -3 and -4), resulting from an alternative splicing process, from the developing axolotl heart. Similar to some, but not all the mammalian systems [19], the embryonic isoforms of cTnT (TNNT2-1) expression persists into adulthood in the axolotl system. We conclude that the aberrant splicing of cTnT occurs in mutant embryonic axolotl hearts in addition to the significantly decreased total RNA level compared to normal hearts. Interestingly, the aberrant splicing pattern of the cTnT in mutant embryonic hearts could be compensated by supplying a small RNA molecule exogenously, the Myofibril-inducing RNA (MIR). This observation is in good agreement with our previous studies that MIR is able to rescue the mutant hearts by promoting myofibrillogenesis and cardiomyocyte differentiation [15].
Materials and Methods
Procurement and Maintenance of Axolotls
A colony of homozygous normal (+/+) and heterozygous (+/c) adult animals were maintained at Florida Atlantic University for these studies. In a temperature and light controlled room, they were kept within individual aquaria in Holtfreter's solution and fed salmon fish pellets and brine shrimp. Gene c is maintained in the colony for production of mutant spawnings with occasional supplementation of embryos from the Indiana University Axolotl Colony. The cardiac lethal mutation designated by gene c is a simple recessive mutation. With each mating, 25% of the embryos are c/c mutants.
Cloning of cTnT Isoforms
Cardiac RNA was extracted from the embryonic axolotl hearts (stages 34-42) using TRI Reagent (Sigma, St. Louis). Reverse Transcription was done using the Thermoscript RT system (Invitrogen, CA). The cDNA synthesized was then used to carry out PCR using Pfx DNA polymerase (Invitrogen, CA). Primers to amplify cTnT isoforms were designed in 5′UTR (forward primer) and 3′UTR (reverse primer) sequences.
Forward primer: 5′-CCTTCCTGGATCTGCAAGCCTCCTC.
Reverse primer: 5′-TGCTGCTAGGTCTTCTTGGCAGAGGTG
PCR bands with different sizes were isolated from agarose gels and cloned into a T-vector using the pGEM–T Easy Vector System (Promega, WI). The plasmids were purified after transformation and sequenced by Davis sequencing, LLC (San Diego, CA) after digestion with EcoRI enzyme and confirmed with insertion.
Organ cultures for embryonic hearts
Stages 37-38 embryos were anesthetized using a 1:5000 dilution of MS – 222 and hearts were dissected in Steinberg’s solution. Mutant hearts were separated from normal based on the presence or absence of beating activity. All dissections were carried out under a dissecting microscope. Hearts were then placed in 96 U-well shaped plates containing Steinberg’s solution for liposome-mediated RNA transfection following our published methods [15–18]. Mutant hearts were divided into two groups: those treated with sense MIR and those treated with antisense MIR. RNA molecules were in vitro synthesized following our published methods (Zhang et al., 2003). Control hearts were treated with Lipofectamine 2000 (Invitrogen, CA) and Steinberg’s solution only. Hearts were then collected and processed for total RNA extraction after RNA transfection for 36 hours and 72 hours.
Quantitative RT-PCR
Total RNA was extracted from normal and mutant hearts at stages 37-38 after the conuses were removed. cDNA was synthesized from 2μg of total RNA using Invitrogen’s Thermoscript RT-PCR System. Real-time RT-PCR experiments were performed on a Capillary Lightcycler (Roche) machine using a Roche Fast Start SYBR Green I Kit following the manufacturer’s instructions. Specific amplification of desired genes was confirmed by calculating melting temperatures (Tm) for the products from the melting peak curve (−dF/dT vs. temperature). All the amplicons were collected and confirmed again by agarose gel electrophoresis and sequencing. A standard curve of cross-point vs. Log concentration (copies) was created using one of the cDNA samples with serial dilutions or with known concentrations of plasmid DNA with a cTnT insert. Negative controls were included using cDNAs synthesized the same way as above but with no reverse transcriptase added. Each cDNA sample was run in triplicate. The data were averaged and standard deviations were calculated. The beta-actin gene was used as a standard control. The sizes of the PCR products were further confirmed by polyacrylamide gel electrophoresis. Primers for total cTnT amplification were designed based on the 3′ UTR sequence that is common for all the isoforms. Primers for TNNT2-2, 2-3 and 2-4 were designed to span the neighboring exons for specific amplification of individual isoforms. All primer pairs designed to amplify shorter isoforms have been pre-tested and their specificity has been verified by not being able to amplify the full length cTnT insert in a plasmid (data not shown). Primer sequences used in these studies are as follows:
-
TNNT2-2-for:
5′-TGGAGGAATACGAATCGTGATGATG
-
TNNT2-2-rev:
5′-CAAAGTGCGCCTCGATCAGC
-
TNNT2-3-for:
5′-ACCCAAACCAAAGAGAAGCGTCGG
-
TNNT2-3-rev:
5′-TTCTTCCCGCCTCTGCCAGTC
-
TNNT2-4-for:
5′-CCAGGCACGCATGGCAGAAG
-
TNNT2-4-rev:
5′-GGACATTGATTTCAGCTCGTCCTCC
-
cTnT-3UTR-for:
5′-CCAAGGGCTTCACCGGGCTCAA
-
cTnT-3UTR-rev:
5′-TGGCAGAGGTGGAATGGATCACAGG
-
Beta-actin-for:
5′-TCCATGAAGGCTGCCCAACT
-
Beta-actin-rev:
5′-TGGCGCCACATCTGATTGAT
Results and Discussion
We have previously cloned the full-length sequence of the cardiac troponin T cDNA from the heartbeat initiation stages of embryonic axolotls [18]. In our current studies, we have designed PCR primers based on both 5′- and 3′- UTR sequences of the full-length cTnT and tried amplifying all possible isoforms of this gene. RT-PCR was performed by using the primer pairs on total RNA extracted from normal (+/+) stage 34 to 37 embryonic hearts. After ligation of the amplified PCR bands from RT-PCR into T-vectors, more than seventy clones were randomly picked from the culture plates. Plasmid DNA from the individual clones were purified and digested by a restrictive enzyme for confirmation. Agarose gel electrophoresis results have shown successful amplifications of three different isoforms of axolotl cTnT (Figure 1A). Clones with different sized amplicons were sequenced and the sequences of the three different cTnT isoforms were compared. The three fragments of sequences that are alternatively spliced are indicated by the underlined sequences shown in Figure 1B. Each shorter isoform of cTnT (TNNT2-2, TNNT2-3 and TNNT2-4) is generated by splicing out only one fragment. No shorter cTnT isoform is detected with more than one fragment simultaneously spliced out. The full-length cTnT contains all of the otherwise alternatively spliced exons, which in our studies, proved to be the dominant form in the embryonic hearts. The isoform-specific primer pairs and a primer pair amplifying the common sequences have been designed as shown in Figure 1B. The predicted peptide organization pattern for all four different isoforms (TNNT2-1 (full length), -2, -3, and -4) is shown in Figure 1C. Interestingly, the axolotl TNNT2-2, a potential adult isoform, is generated by splicing out a much larger 5′ exon(s) sequence than many of the other vertebrates such as human, mouse, rat or zebrafish (not shown). The resultant shorter isoform TNNT2-2 in axolotl has a very high pI (isoelectric point) of 9.57 predicted by the ProtParam program from ExPASy, which is significantly different from the other animal systems. TNNT2-3 and TNNT2-4 isoforms isolated from embryonic axolotl hearts are generated from an unusual splicing pattern when compared to other animal systems although the exon-intron boundaries are quite conserved among the species for both TNNT2-3 and TNNT2-4 (Figure 2A). Due to differential splicing, the predicted protein sequence of TNNT2-3 is missing a cardiac specific exon that includes a unique epitope (SFMPNLVPPKIPDGERVDFD) in cardiac troponin T compared to skeletal muscle troponin Ts [6]. Compared to the full-length cardiac troponin T (TNNT2-1), TNNT2-4 has a truncated C-terminal end due to the exon alternative splicing.
Figure 1.

A. Agarose gel electrophoresis of the digestion of clones containing the PCR products of cTnT cDNAs subcloned into the pGEM-T-Easy vector system. Notice that bands 2 and 4 (from left to right) are smaller than expected, corresponding to TNNT2-2 and TNNT2-3, respectively. Clones containing TNNT2-4 are not shown here. A majority of the clones contain full-length cTnT (TNNT2-1). B. Location of the 3 missing exon sequences (underlined) on each of the cTnT isoforms: the first underlined region is missing in TNNT2-2; the second underlined region is missing in TNNT2-3; and the third underlined sequence is missing in TNNT2-4. The primers used to amplify all of the cTnT isoforms are indicated by the shaded sequences at each end. The two ATGs in the boxes indicate start codons for TNNT2-1 (first ATG) and TNNT2-2 (second ATG) respectively. The TGA in the box indicates the shared stop codon for both TNNT2-1 and TNNT2-2. C. Gene structure comparisons for all of the cTnT isoforms. The exons excluded from TNNT2-2, 3 and 4 are shown by the bridging lines between common exons. TNNT2-1, 3 and 4 share the same start codon while TNNT2-3 and 4 have truncated C-terminal ends compared to TNNT2-1 and 2.
Figure 2.
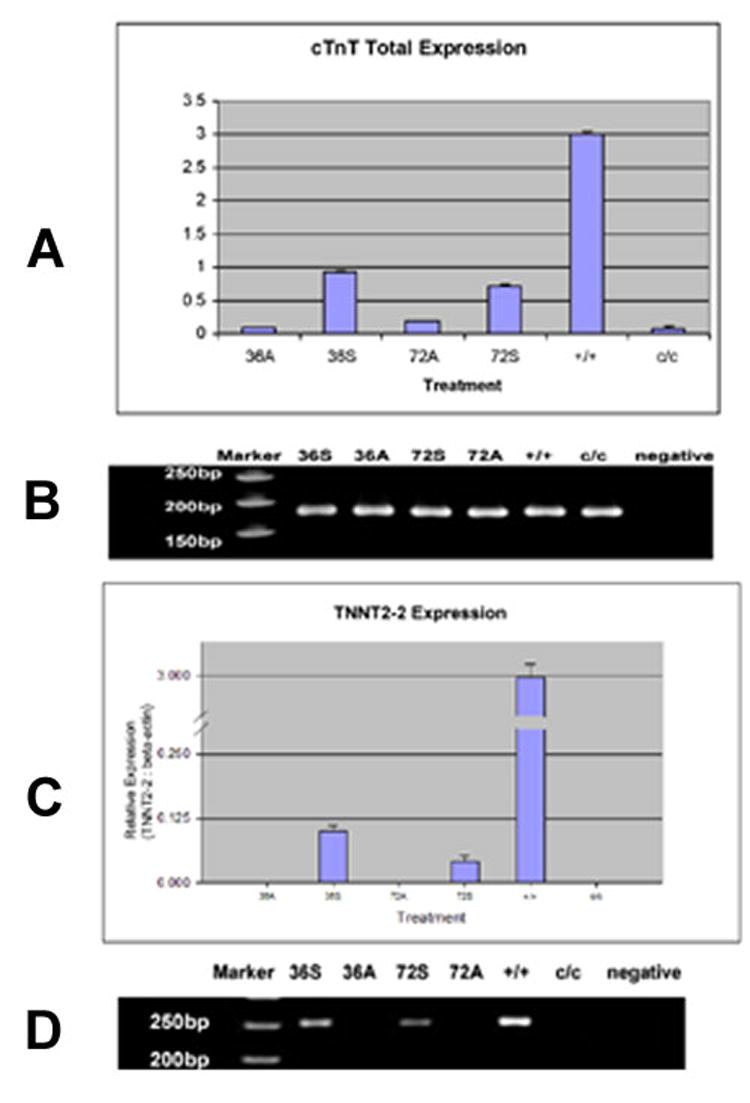
Expression of cTnT and TNNT-2 expression in normal and mutant axolotl hearts A. Real-time RT-PCR experiments showing down regulation of cTnT total mRNA expression in the mutant embryonic hearts compared to normal and increased expression of total cTnT in the mutant embryonic hearts by exogenously supplying the sense Myofibril-inducing RNA (MIR). +/+: homozygous normal hearts; c/c: homozygous mutant hearts; 36A: c/c mutant hearts treated by anti-sense MIR for 36 hours; 36S: c/c mutant hearts treated by sense MIR for 36 hours; 72A: c/c mutant hearts treated by anti-sense MIR for 72 hours; 72S: c/c mutant hearts treated by sense MIR for 72 hours. The cTnT expression levels are normalized by beta-actin expression levels in each sample. All hearts are at stages 37-38. B. PAGE of the amplicons from RT-PCR shows bands of expected size. The PCR products were collected after the reactions reached a plateau. The densities of the DNA bands no longer reflect the concentrations of total cTnT cDNA in the samples. C. Real-time RT-PCR experiments showing the missing of TNNT2-2 mRNA in the mutant embryonic heart compared to normal and increased expression of TNNT2-2 mRNA in the mutant embryonic hearts by exogenously supplying the sense myofibril-inducing RNA (MIR). All the labels are the same as in Figure 2. cTnT expression levels are normalized by beta-actin expression levels in each sample. All hearts are at stages 37-38. D. PAGE of the amplicons from RT-PCR shows bands with expected size. The PCR products were collected after the reactions reached a plateau. No amplicons of TNNT2-2 were detected in untreated c/c mutant hearts or antisense MIR-treated mutant hearts at either 36 hours or 72 hours interval.
The hearts of normal embryos (+/+) have a much higher expression level of the cardiac troponin-T gene than mutants (c/c) (Figure 2). Mutant hearts express approximately one twelfth of the mRNA expressed in normal hearts at stage 37-38, the post-heartbeat stages. Real-time RT PCR detected all three of the cTnT shorter isoforms (TNNT2-2, 3 and 4) in normal embryonic axolotl hearts (+/+). However, TNNT2-2 could not be amplified from the mutant embryonic hearts (c/c) using RT-PCR (Figure 3B). In contrast, TNNT2-3 was detected in mutant hearts at stages 37-38 at the same expression levels as in normal hearts (Figure 3); TNNT2-4 also was amplified from mutant hearts but at a much lower expression level than in normals (Figure 4).
Figure 3.
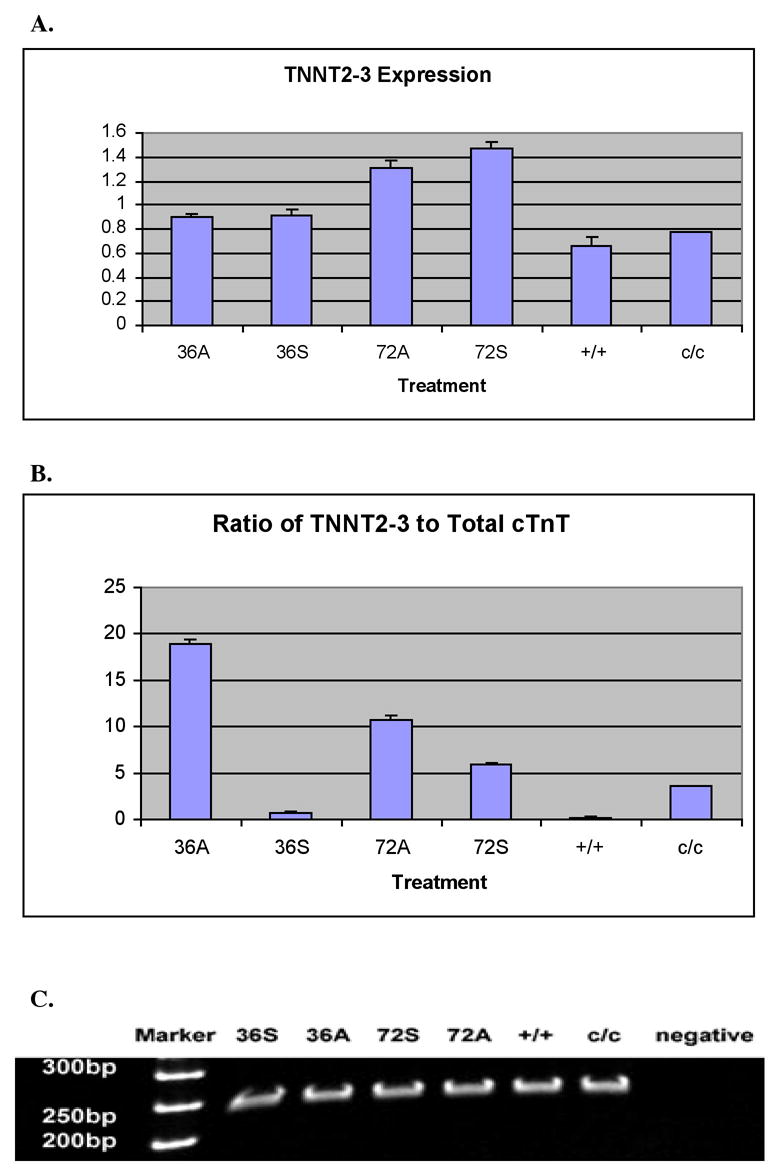
A. Real-time RT-PCR experiments showing equal absolute amounts of TNNT2-3 mRNA in the mutant embryonic heart when compared to normal. The expression of TNNT2-3 mRNA in the mutant embryonic hearts is not altered by exogenously supplying the Myofibril-inducing RNA (MIR). All of the labels are the same as in Figure 3. β-actin expression levels in each sample normalize TNNT2-3 expression levels. All hearts used are at stages 37-38. B. Comparisons of the ratios of TNNT2-3 to total cTnT between different heart samples indicate sense MIR treatments on the mutant hearts for 36 hours decrease the ratio of this isoform, which does not contain a cardiac specific sequence, to total cTnT, resembling the ratio in normal hearts. C. PAGE of the amplicons from RT-PCR shows bands with expected size. The PCR products were collected after the reactions reached a plateau. The densities of the DNA bands no longer represent the concentrations of total cTnT cDNA in the samples.
Figure 4.
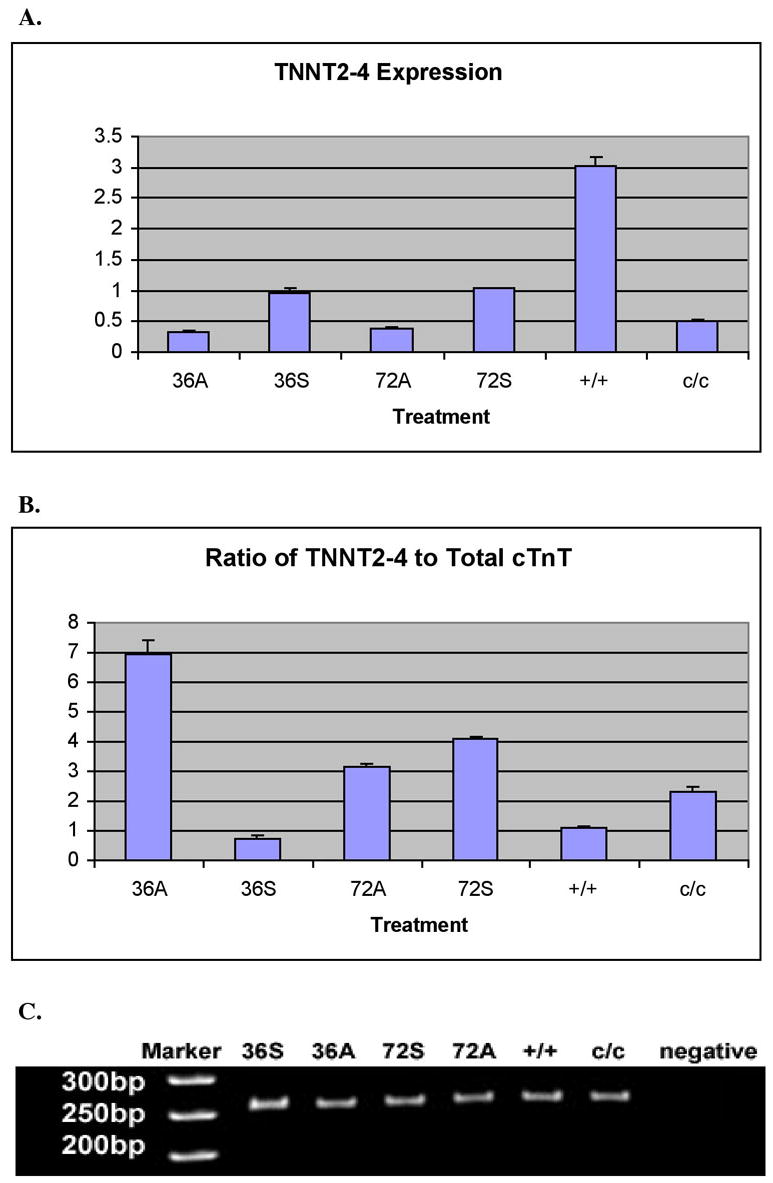
Real-time RT-PCR experiments showing up-regulation of TNNT2-4 mRNA expression in mutant embryonic hearts compared to normal and increased expression of TNNT2-4 in the mutant embryonic hearts by exogenously supplying the sense Myofibril-inducing RNA (MIR). All the labels are the same as in Figure 2. β-actin expression levels in each sample normalize TNNT2-4 expression levels. All hearts used are at stages 37-38. B. Comparisons of the ratios of TNNT2-4 to total cTnT between different heart samples indicate sense MIR treatments on the mutant hearts for 36 hours increase the ratio of this isoform, which has a truncated c-terminus, to total cTnT, resembling the ratio in normal hearts. C. PAGE of the amplicons from RT-PCR shows bands with expected size. The PCR products were collected after the reactions reached plateau. The densities of the DNA bands no longer represent the concentrations of TNNT2-4 cDNA in the samples.
We have previously reported that treating the mutant hearts with the normal sense strand RNA of Myofibril-Inducing RNA (MIR) rescued the mutant phenotypes by promoting tropomyosin production followed by myofibril formation in mutant hearts in vitro [17,18]. In this study, we have shown for the first time that MIR, a small regulatory RNA, also is capable of promoting cTnT expression at the mRNA level as well as regulating the splicing patterns of cTnT gene in mutant hearts making them more like normal. The mutant hearts were incubated with sense or anti-sense MIR for either 36 hours or 72 hours before the total RNA was collected. Real-time RT-PCR experiments were performed on these RNA samples using isoform-specific primer pairs.
As depicted in Figure 3, after treatment of sense MIR for 36 hours or 72 hours, mutant hearts showed significantly increased amounts of cTnT mRNA in the cells compared to anti-sense RNA treated groups. Our Real-time RT-PCR results suggest that TNNT2-2 is not present in mutant hearts (Figure 2C). Results have been confirmed by agarose gel electrophoresis of the PCR products; no corresponding band was detected from samples of untreated mutant hearts or mutant hearts incubated with anti-sense MIR for 36 or 72 hours (Figure 2D). Interestingly, upon treatment with sense MIR for 36 hours, the appearance of TNNT2-2 can be readily seen. The inducing effects of sense MIR on TNNT2-2 expression in mutant hearts is a maximum at 36 hours of treatment (Figure 3A). We have not detected significant differences in TNNT2-3 expression in untreated normal and mutant hearts (Figure 3A). No differences have been shown between mutant heart groups with sense or anti-sense MIR treatment for 36 hours. Neither are there significant differences for mutant hearts treated with sense or anti-sense MIR for 72 hours. Considering both the facts of the equal absolute mRNA concentrations of TNNT2-3 in normal and mutant hearts, and the dramatically increased total cTnT mRNA levels in the sense MIR treated mutant hearts, the ratio of TNNT2-3 to the total cTnT mRNA changes after MIR treatment, with the 36-hour sense MIR treated mutant heart group most closely mimicking normal hearts (Figure 3B). The TNNT2-4 mRNA levels in the sense MIR-treated mutant hearts were increased approximately two-fold over that of anti-sense MIR-treated mutant hearts incubated for 36 or 72 hours. In terms of the ratio of TNNT2-4 to total cTnT, mutant hearts treated with sense MIR for 36 hours exhibited the closest number to that from untreated normal hearts (Figure 4).
Our RT-PCR studies with adult heart RNA failed to detect any splicing isoforms of cTnT at the N-terminal sequence other than TNNT2-1 and TNNT2-2 (data not shown). Thus, the dominant isoform in both adult and embryonic hearts is TNNT2-1. The ratio of TNNT2-2 to the total cTnT transcripts remains at a very low level. Meanwhile, the percentage of TNNT2-3 in the total cTnT transcripts decreases to a very insignificant level (less than 0.1%) while TNNT2-4 persists at about 10% in the adult heart, even higher than in the embryonic axolotl hearts (~2.6%) (results not shown).
The expression of TNNT2-2 in early developmental stages 37-38 is very low (0.5% of all the cTnT transcripts). TNNT2-2 expression is augmented in the hearts of adult animals (2 years old). However, because of the dramatic increase in the total cTnT transcription level in adult hearts, the ratio of TNNT2-2 to the total cTnT transcripts in adult heart is increased very little (~6%) (not shown). The increase of total cTnT transcription level is probably due to much higher concentrations of the cTnT transcripts per adult heart cell or a much higher percentage of cardiomyocytes among all of the cell types in the adult hearts compared to embryonic. We believe that the remaining low ratio of TNNT2-2 to total cTnT expression, even in adult axolotl hearts, which is quite different from other vertebrates, is due to this species being neotenous.
In recent studies, numerous protein factors have been identified that regulate gene alternative splicing, including the cardiac troponin T gene [20]. This study suggests that MIR, a small regulatory RNA might function through participating in the spliceosome, a ribonucleoprotein complex, thus selectively promoting exclusion of the cardiac troponin T embryonic exon. The uniqueness of MIR is that in addition to being involved in cTnT splicing, it also can increase the total cTnT mRNA concentration in the cells. We have already shown that MIR is capable of binding with one or more proteins present in axolotl hearts [17]. Also, we have already reported that in mutant axolotl hearts MIR is expressed. However, in mutant axolotl heart there is a G93U mutation in MIR. The G93U mutant MIR fails to rescue mutant hearts in situ and also the mutant hearts do not bind with the protein(s) that bind with wild-type MIR [17].
Acknowledgments
This study was supported by NIH grants HL-58435 and HL-061246 and by a Christine E. Lynn American Heart Association Grant-in-Aid to L.F.L. Some of the embryos used for this study were from the Axolotl Colony at Indiana University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Gian-Franco Sferrazza, Department of Biomedical Science, Florida Atlantic University, Boca Raton, FL 33431.
Chi Zhang, Department of Biomedical Science, Florida Atlantic University, Boca Raton, FL 33431.
Pingping Jia, Department of Biomedical Science, Florida Atlantic University, Boca Raton, FL 33431.
Sharon L. Lemanski, Department of Biomedical Science, Florida Atlantic University, Boca Raton, FL 33431
Gagani Athauda, Department of Biomedical Science, Florida Atlantic University, Boca Raton, FL 33431.
Alyssa Stassi, Department of Biomedical Science, Florida Atlantic University, Boca Raton, FL 33431.
Kristine Halager, Department of Biomedical Science, Florida Atlantic University, Boca Raton, FL 33431.
Jennifer A. Maier, Department of Biomedical Science, Florida Atlantic University, Boca Raton, FL 33431
Elena Rueda-de-Leon, Department of Biomedical Science, Florida Atlantic University, Boca Raton, FL 33431.
Amit Gupta, Department of Biomedical Science, Florida Atlantic University, Boca Raton, FL 33431.
Syamalima Dube, Department of Medicine, SUNY Upstate Medical University, Syracuse, NY 13210.
Xupei Huang, Department of Biomedical Science, Florida Atlantic University, Boca Raton, FL 33431.
Howard M. Prentice, Department of Biomedical Science, Florida Atlantic University, Boca Raton, FL 33431
Dipak K. Dube, Department of Medicine, SUNY Upstate Medical University, Syracuse, NY 13210
Larry F. Lemanski, Department of Biomedical Science, Florida Atlantic University, Boca Raton, FL 33431
References
- 1.Perry SV. Troponin T: genetics, properties and function. J Muscle Res Cell Motil. 1998;19:575–602. doi: 10.1023/a:1005397501968. [DOI] [PubMed] [Google Scholar]
- 2.Breitbart RE, Nadal-Ginard B. Complete nucleotide sequence of the fast skeletal troponin T gene: alternatively spliced exons exhibit unusual interspecies divergence. J Mol Biol. 1986;188:313–324. doi: 10.1016/0022-2836(86)90157-9. [DOI] [PubMed] [Google Scholar]
- 3.Cooper TA, Ordahl CP. A single cardiac troponin-t gene generates embryonic and adult isoforms via developmentally regulated alternative splicing. J Biol Chem. 1985;260:11140–11148. [PubMed] [Google Scholar]
- 4.Jin JP, Lin J-C. Isolation and characterization of cDNA clones encoding embryonic and adult isoforms of rat cardiac troponin T. J Biol Chem. 1989;264:14471–14477. [PubMed] [Google Scholar]
- 5.Townsend PJ, Farza H, MacGeoch C, Spurr C, Wade NK, Gahlmann R, Yacoub R, Barton PJ MH. Human cardiac troponin T: identification of fetal isoforms and assignment of the TNNT2 locus to chromosome 1q. Genomics. 1994;21:311–316. doi: 10.1006/geno.1994.1271. [DOI] [PubMed] [Google Scholar]
- 6.Anderson PAW, Greig A, Mark TM, Malouf NN, Oaklely AE, Ungerleider RM, Allen PD, Kay BK. Molecular Basis of Human Cardiac Troponin T Isoforms Expressed in the Developing, Adult and Failing Heart. Cir Res. 1995;4:681–686. doi: 10.1161/01.res.76.4.681. [DOI] [PubMed] [Google Scholar]
- 7.Farza H, Townsend PJ, Carrier L, Barton PJ, Mesnard L, Bahrend E, Forissier JF, Fiszman M, Yacoub MH, Schwartz K. Genomic organisation, alternative splicing and polymorphisms of the human cardiac troponin T gene. J Mol Cell Cardiol. 1998;30:1247–1253. doi: 10.1006/jmcc.1998.0698. [DOI] [PubMed] [Google Scholar]
- 8.Wang J, Jin J-P. Conformational modulation of troponin T by configuration of the NH2-terminal variable region and functional effects. Biochemistry. 1998;37:14519–14528. doi: 10.1021/bi9812322. [DOI] [PubMed] [Google Scholar]
- 9.Gomes AV, Guzman G, Zhao J, Potter JD. Cardiac troponin T isoforms affect the Ca2+ sensitivity and inhibition of force development. Insights into the role of troponin T isoforms in the heart. J Biol Chem. 2002;277:35341–35349. doi: 10.1074/jbc.M204118200. [DOI] [PubMed] [Google Scholar]
- 10.Tobacman LS. Structure-function studies of the amino-terminal region of bovine cardiac troponin T. J Biol Chem. 1988;263:2668–2672. [PubMed] [Google Scholar]
- 11.Humphrey RR. A genetically determined absence of heart function in embryos of the Mexican axolotl (Ambystoma mexicanum) Anat Rec. 1968;160:475. [Google Scholar]
- 12.Lemanski LF. Morphological and biochemical abnormalities in hearts of cardiac mutant salamanders (Ambystoma mexicanum) J Supramoic Struct. 1976;5:221–238. doi: 10.1002/jss.400050209. [DOI] [PubMed] [Google Scholar]
- 13.Luque EA, Lemanski LF, Dube DK. Molecular cloning, sequencing and expression of α tropomyosin from cardiac muscle of the Mexican axolotl, Ambystoma mexicanum. Biochem Biophys Res Com. 1994;203:319–325. doi: 10.1006/bbrc.1994.2184. [DOI] [PubMed] [Google Scholar]
- 14.Luque EA, Spinner BJ, Dube S, Dube DK, Lemanski LF. Differential expression of a novel isoform of alpha-tropomyosin in cardiac and skeletal muscle of the Mexican axolotl (Ambystoma mexicanum) Gene. 1997;185:175–180. doi: 10.1016/s0378-1119(96)00606-3. [DOI] [PubMed] [Google Scholar]
- 15.Lemanski LF, Nakatsugawa M, Bhatia R, Erginel-Unaltuna N, Spinner B, Dube DK. Characterization of an RNA which promotes myofibrillogenesis in embryonic cardiac mutant axolotl hearts. Biochem Biophys Res Comm. 1996;229:974–981. doi: 10.1006/bbrc.1996.1910. [DOI] [PubMed] [Google Scholar]
- 16.Zajdel RW, McLean MD, Lemanski SL, Muthuchamy M, Wieczorek DF, Lemanski LF, Dube DK. Ectopic expression of tropomyosin in cardiac mutant axolotl hearts promotes organized myofibril formation. Dev Dyn. 1998;213:412–420. doi: 10.1002/(SICI)1097-0177(199812)213:4<412::AID-AJA6>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 17.Zhang C, Dube DK, Huang X, Zajdel RW, Bhatia R, Foster D, Lemanski SL, Lemanski LF. A point mutation in bioactive RNA results in the failure of mutant heart correction in Mexican axolotls. Anat Embryol. 2003;206:495–506. doi: 10.1007/s00429-003-0315-8. [DOI] [PubMed] [Google Scholar]
- 18.Zhang C, Pietras CKM, Huang X, Lemanski S, Dube DK, Lemanski LF. Immunohistochemical and molecular analyses of cardiac troponin T gene during the cardiac development of Mexican axolotls, Ambystoma mexicanum. J Cell Biochem. 2006 doi: 10.1002/jcb.20918. In press. [DOI] [PubMed] [Google Scholar]
- 19.Biesiadecki BJ, Elder BD, Yu Z, Jin J. Cardiac Troponin T Variants Produced by Aberrant Splicing of Multiple Exons in Animals with High Instances of Dilated Cardiomyopathy. J Biol Chem. 2002;277:113–120. doi: 10.1074/jbc.M206369200. [DOI] [PubMed] [Google Scholar]
- 20.Ladd AN, Stenberg MG, Swanson MS, Cooper TA. Dynamic balance between activation and repression regulates pre-mRNA alternative splicing during heart development. Dev Dyn. 2005;233:783–793. doi: 10.1002/dvdy.20382. [DOI] [PubMed] [Google Scholar]