

Abstract
Listeria monocytogenes, a food-borne pathogen that infects immunocompromised patients, enters and proliferates within mammalian cells by taking advantage of host cell machinery. While entry into macrophages and other phagocytic cells occurs constitutively, intracellular invasion of nonphagocytic cells, such as epithelial and endothelial cells, occurs through induced phagocytosis. Invasion of these nonphagocytic cell types is under the control of the secreted L. monocytogenes protein internalin B (InlB), which directly associates with and activates the receptor tyrosine kinase Met. Activation of Met by InlB has previously been shown to be potentiated by binding of glycosaminoglycans to the GW domains of this protein. We studied the interaction between heparin and full-length InlB as well as a truncated, functional form of InlB to understand the mode of interaction between these two molecules. InlB preferred long-chain (≥dp14) heparin oligosaccharides, and the interaction with heparin fit a complicated binding model with a dissociation constant in the nanomolar range. While there are various explanations for this complicated binding model, one supported by our data involves binding and rebinding of InlB to multiple binding sites on heparin in a positive and weakly cooperative manner. This mode is consistent with enhancement of interaction of InlB with glycosaminoglycans for activation of Met.
Listeria monocytogenes is a food-borne microbial pathogen that causes listeriosis. The disease primarily affects pregnant women, newborns, and adults with compromised immune systems. Symptoms can include severe gastroenteritis, meningitis, septicaemia, and abortions (1). The diversity of symptoms is due to the capacity of L. monocytogenes to traverse three tight human barriers: the intestinal, the bloodbrain, and the feto-placental barriers. Making a major contribution to the virulence of L. monocytogenes is its capacity to trigger uptake through induced phagocytosis into nonphagocytic mammalian cells. For example, intestinal epithelial cells are triggered to phagocytose the pathogen through signaling pathways triggered by the interaction of the bacterial cell wall-attached protein internalin A (InlA)1 with host cell E-cadherin (2-4). Invasion of many other cell types, including epithelial, endothelial, hepatocyte, and fibroblast-like cells, is dependent on a second internalin, InlB, which appears to be important for dissemination from the intestine and infection of other tissues (5). InlB directly binds and activates the host cell receptor tyrosine kinase Met (hepatocyte growth factor/scatter factor receptor) (6), resulting in stimulation of host cell signaling pathways that control the actin cytoskeleton and effect intracellular invasion.
InlB is an ∼69 kDa elongated L-shaped molecule (Figure 1) consisting of at least two functional domains, the N-terminal receptor binding domain (RBD) and the C-terminal GW domains. The RBD binds Met (6, 7) and contains three structural components common to the internalin family of Listeria proteins. The distinguishing characteristic shared by the 24 proteins in this family is a β-loophelix-loop motif constructed of tandemly arranged 22-amino acid leucine-rich repeat units (LRRs) (1, 8). Approximately 7.5 LRR motifs comprise the LRR domain of InlB, forming a curved, tubelike structure, with a concave surface that mediates protein-protein interactions (8-10). The LRR is flanked at one end by an N-terminal cap and at the other by an immunoglobulin (Ig)-like domain (11). The N-terminal cap (residues 36-76) and LRR region (residues 77-240)form the shorter arm of the L shape and span ∼60 Å in length, and the Ig-like domain projects at a nearly right angle from the base of the LRR. The RBD is connected to the GW domains by a segment known as the B-repeat region. The 72 residues encompassing this region are highly flexible and span ∼30 Å.
FIGURE 1.
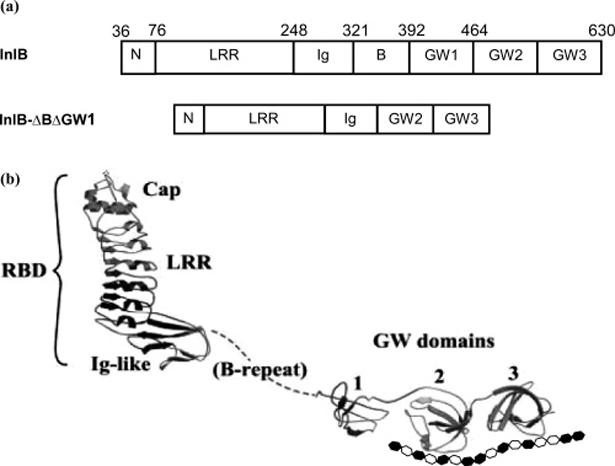
(a) Structural domains of InlB and InlB-ΔBΔGW1. (b) The tertiary structure of InlB is shown in ribbon representation complexed with a schematic illustration of heparin. Reprinted with permission from ref 8. Copyright 2002 Macmillan Publishers Ltd.
The C-terminal domain of InlB is comprised of three ∼80-residue tandem GW repeats, named for a conserved Gly-Trp (GW) dipeptide motif. The first GW repeat, GW1, is unpaired and flexible, which accounts for its susceptibility to proteolytic cleavage. Along with the B-repeat region, GW1 is dispensable for Met activation and bacterial invasion (12). Pairwise association between the second and third GW domains (GW2 and GW3), mediated through conserved hydrophobic residues and a conserved hydrogen bond, stabilizes these small protein domains through an enhanced secondary structure that is not susceptible to proteolysis. The surfaces of all three domains are entirely basic (pI ∼ 10) and enriched with arginine residues, which are known to mediate interactions with heparan sulfate (HS) or heparin through electrostatic attraction (13, 14). The GW domains promote attachment of InlB to the bacterial surface; specifically, the GW domains associate with negatively charged lipoteichoic acids (LTA) which are partially buried in the peptidoglycan layer of the bacterial cell wall (15). This association is noncovalent and dissociable. Although InlB partitions nearly equally between bacterial surface-attached and released forms (16), burial of InlB in the bacterial cell wall potentially hinders its ability to interact with eukaryotic cell ligands (8). In agreement with this, a model in which the released rather than surface-attached form of InlB activates Met is supported by several lines of evidence (12). The functional purpose of dissociable InlB attachment may be to coordinate host membrane dynamics with bacterial proximity during invasion (8).
Physiologically relevant activation of Met has been shown to require coordinated action in cis of the InlB N-terminal RBD in binding Met and the C-terminal GW domains in binding glycosaminoglycans (12). Interaction of the InlB GW domains with the glycosaminoglycan heparin increases the potency of InlB approximately 10-fold for Met activation (12). Furthermore, addition of soluble heparin to the glycosaminoglycan-deficient CHO-S745 cell line has been shown to make it permissive to InlB-mediated invasion (12). The glycosaminoglycan HS has been suggested to be the physiologically relevant mediator for InlB (16), but since more highly sulfated sequences of HS generally interact with proteins, heparin is often used as a model for HS (17). HS and heparin are important mediators in various physiologic and pathophysiologic processes, including cell adhesion, cell-cell communication, and cell growth (18-22). Despite structural differences between LTA and heparin, the charge density and flexibility of heparin are sufficient for competitive displacement of InlB from the bacterial surface (15).
While the importance of glycosaminoglycans in InlB-mediated intracellular invasion is clear (12, 16), the precise mode of interaction remains to be elucidated. In the study present here, we used surface plasmon resonance (SPR) to study the interaction of surface-immobilized heparin with InlB and a truncated, functional form of InlB lacking the B-repeat and GW1 (InlB-ΔBΔGW1) (12).
EXPERIMENTAL PROCEDURES
Materials
Biotinylated albumin-heparin conjugate and biotinylated albumin were purchased from Sigma (St. Louis, MO). Streptavidin covalently linked to a carboxymethylated dextran matrix sensor chip and HBS-EP buffer [10 mM HEPES, 150 mM NaCl, 1.5 mM EDTA, and 0.005% P20 (pH 7.4)] were from BIAcore (Uppsala, Sweden). Porcine intestinal heparin (average molecular weight MWavg of 12 500) and low-molecular weight heparin (LMWH, MWavg of 5000) were purchased from Celsus (Cincinnati, OH).
Methods
Preparation of InlB and InlB-ΔBΔGW1
Previously reported procedures were used for expression and purification of InlB and InlB-ΔBΔGW1 (8, 12).
Preparation of Biosensor Surfaces
Biotinylated albumin-heparin conjugate was immobilized to sensor chips comprised of streptavidin covalently linked to a carboxymethylated dextran matrix. The conjugate was immobilized in two of the four flow cells of each sensor at a flow rate of 20 μL/min. Successful immobilization was confirmed by the observation of an ∼190-250 response unit (RU) increase. A control flow cell was coated with ∼400 RU of biotinylated albumin. The final flow cell was unmodified, and it served as the reference surface for detection of changes in the bulk refractive index, injection noise, baseline drift, and nonspecific binding. Multiple sensor chips were prepared in an identical fashion and used for different aspects of this study.
Data Acquisition and Analysis
Interaction data were acquired with a Biacore 3000 biosensor, which was operated with Biacore Control 3.2. All experiments were performed at 25 °C. Buffers were filtered and degassed prior to use in each experiment. Concentrated solutions of InlB or InlB-ΔBΔGW1 were serially diluted with HBS-EP buffer, yielding concentrations ranging from 0.3 to 62.5 nM. Samples (250 μL) were injected over the sensor surface at a flow rate of 50 μL/min. The sensor surface was regenerated with a 10 μL pulse of 0.5% SDS, which was followed by a 50μL injection of 1 M NaCl in 25 mM NaOH, and a 50 μL injection of 2 M NaCl. All experiments, except for the competition and solution affinity measurements, were repeated to ensure reproducibility. Each experiment was independently evaluated for kinetic and equilibrium binding parameters due to a loss in binding capacity, which may be attributed to harsh regeneration conditions. For each protein, three independent sets of data were included for analysis. Data obtained from four other binding studies on different sensor chips were analyzed to confirm the proper selection of a kinetic binding model.
The binding of InlB and InlB-ΔBΔGW1 to heparin was assessed through a procedure based on double referencing (23, 24). The procedure involved subtraction of the data for the reference flow cell from the data obtained for the albumin- and heparin-coated surfaces, correction of the heparin binding data for nonspecific binding to albumin, and then subtraction of a blank injection. Nonspecific binding was more prevalent at the higher solution concentrations of protein but represented, at most, 10% of the total measured binding to the heparin-coated surfaces that were selected for analysis. Rate and affinity constants were determined by globally fitting the experimental data with BIAevaluation, version 3.1.
Analysis of the Steady-State Binding Levels
The steady-state binding of InlB or InlB-ΔBΔGW1 to heparin was approximated using nonlinear least-squares curve fitting to the equation
where A is InlB or InlB-ΔBΔGW1, B heparin, ABmax the maximum surface binding capacity (RU), and KD the equilibrium dissociation constant. Both interactions were tested for mass transfer limitations and heterogeneity prior to kinetic analyses of selected data sets. The test for mass transfer limitations between the bulk solution phase and the surface layer involved a variation in the flow rate (15, 50, and 100 μL/min). The test for heterogeneous binding consisted of variation in the sample volume (50, 200, and 325 μL). Binding levels measured in the last 100 s of the association phase were averaged for each injected concentration. Curves that demonstrated a change greater than 5 RU over this interval were interpreted as not having achieved steady state. Curves in which biosensor-related effects were predominant (solution concentrations above 10 nM for InlB or 25 nM for InlB-ΔBΔGW1) were not included in the analyses.
Oligosaccharide and Polysaccharide Competition Experiments
The preparation of bovine lung heparin-derived oligosaccharides of defined and uniform structure (dp2-dp14) has been previously described (25). Oligosaccharides, polysaccharides, LMWH, and porcine intestinal heparin were mixed with InlB or InlB-ΔBΔGW1 at a ratio of 1000:1 (final concentrations of 12.5 nM InlB or InlB-ΔBΔGW1 and 12.5 μM oligosaccharide or polysaccharide) and incubated for 5 min prior to injection over the biosensor surfaces. Steady-state binding levels were determined by subtracting the maximum response for just the 12.5 μM oligosaccharide or polysaccharide samples from the measured maximum responses for the protein/oligosaccharide or protein/polysaccharide mixtures. This difference in response was used to compare the affinity of InlB and InlB-ΔBΔGW1 for each oligosaccharide and polysaccharide.
Solution Affinity Experiments
A known amount of InlB-ΔBΔGW1 was incubated with a range of oligosaccharide concentrations prior to injection over the sensor surface. The measured response represented the fraction of InlB-ΔBΔGW1 available to bind heparin, and the InlB-ΔBΔGW1 solution concentration was determined by graphing the steady-state responses of known concentrations of InlB-ΔBΔGW1 as a function of concentration and fitting the data with a polynomial to the fourth power using nonlinear regression. The use of the polynomial equation to estimate the InlB-ΔBΔGW1 solution concentrations was ascertained by comparison to estimates obtained with the four-parameter equation (BIAevaluation, version 3.1). InlB-ΔBΔGW1 (12.5 nM) was mixed and incubated with varying concentrations of tetradecasaccharide (dp14), LMWH, or heparin for 5 min. The solution affinity data were normalized according to the response measured for InlB-ΔBΔGW1 or InlB in the absence of polysaccharide. A similar experiment was also performed using InlB and LMWH.
Kinetic Analysis of the Interaction between InlB and Heparin
Kinetic binding parameters were estimated using a two-step mass transfer-limited reaction model for the interaction of the soluble analyte with a surface-bound heterogeneous ligand. The following equations were used to model the kinetic binding data:
where Ao represents the concentration of InlB or InlB-ΔBΔGW1 in the bulk solution phase, [A] is the concentration of InlB or InlB-ΔBΔGW1 at the sensor surface, and kM is the coefficient of mass transfer for the transport of InlB or InlB-ΔBΔGW1 between these solution phases. For the purpose of unit consistency, the modeling software converts kM (M s-1) to kt (RU M-1 s-1) with the approximation kt ≈ 109 × MW × kM (26). Using the ProtParam tool (http://us.expasy.org/tools/protparam.html), the theoretical molecular masses of InlB and InlB-ΔBΔGW1 were calculated to be 68.5 and 53.2 kDa, respectively. B and C represent the two different types of binding sites, where Rmax1 is the maximum surface binding capacity for site B and Rmax2 is the maximum surface binding capacity for site C. Both Rmax parameters are measured in terms of relative response (RU). The association and dissociation rate constants for site B are ka1 and kd1, respectively. The association and dissociation rate constants for site C are ka2 and kd2, respectively. The concentration of InlB or InlB-ΔBΔGW1 in the bulk flow (Ao) was assumed to be constant during the association phase. The measured response ([AB] + [AC]) was attributed to the simultaneous binding of InlB or InlB-ΔBΔGW1 to both classes of binding sites; terms were modeled in RUs. An additional term was included to adjust for biosensor-related effects (e.g., baseline offset).
RESULTS
SPR Measurements of InlB-Heparin Interaction
Sensor chips containing immobilized heparin prepared using several different chemistries (27-29) were evaluated to reduce the level of nonspecific binding of InlB. Minimal nonspecific interaction was achieved using biotinylated albumin-heparin conjugate bound to a streptavidin-coated surface (data not shown). The level of nonspecific interaction could be further decreased using a truncated, fully functional version of InlB, InlB-ΔBΔGW1, missing the B-repeat region and GW1 domain (12). On the basis of these preliminary experiments, all further studies utilized albumin-heparin chips and both InlB and InlB-ΔBΔGW1.
Varying Contact Time for the Examination of Binding Complexities
Preliminary examination of the interaction between InlB and heparin using SPR suggested a complex binding mechanism that did not follow a single pseudo-first-order reaction. The contact time between the InlB analyte and immobilized heparin ligand was examined. The dissociation phase profile of an analyte, binding to one or more classes of sites within a ligand on the sensor surface, should remain unaffected by the duration of time over which binding occurs (30, 31). An InlB-ΔBΔGW1 solution at high concentrations (100-500 nM) obscured the dissociation phase kinetics but yielded binding progress curves closely resembling those obtained for binding of InlB to heparin (data not shown). Lower concentrations (10 nM) of InlB-ΔBΔGW1 afforded interpretable results at contact times of 1, 4, and 6.5 min (data not shown). The dissociation rate of InlB-ΔBΔGW1 was faster when the association phase was limited to 1 min, suggesting that this interaction involves a level of complexity beyond a simple bimolecular interaction.
Steady-State Analysis To Resolve Three Binding Mechanism Possibilities
The three binding mechanisms, known to result in an alteration in dissociation phase kinetics, can often be resolved by the evaluation of steady-state binding levels. If the mechanism is consistent with either a change in conformation or a heterogeneous analyte, then the steady-state binding levels will conform to a 1:1 binding model. A multivalent analyte (e.g., a bivalent antibody) that binds to a monovalent class of receptors will not conform to the case of 1:1 binding, and Scatchard plot analysis will reveal a curvature consistent with negative cooperative binding.
The measured response (Req) was plotted as a function of the solution concentration (Figure 2a). Each set of data was fit to a 1:1 binding model using nonlinear regression, and the modeled parameters are reported in Table 1. Both InlB and InlB-ΔBΔGW1 bind heparin with nanomolar affinity. Scatchard analysis (Figure 2b) reveals curvature suggestive of positive cooperative binding, providing additional insight into the binding mechanism. Extension of the linear portion of the Scatchard plots predicts approximately 2 mol of InlB (or InlB-ΔBΔGW1) binds to each mole of heparin. However, the Scatchard analysis lacks a degree of certainty since the interactions were limited to a narrow range of concentrations.
FIGURE 2.
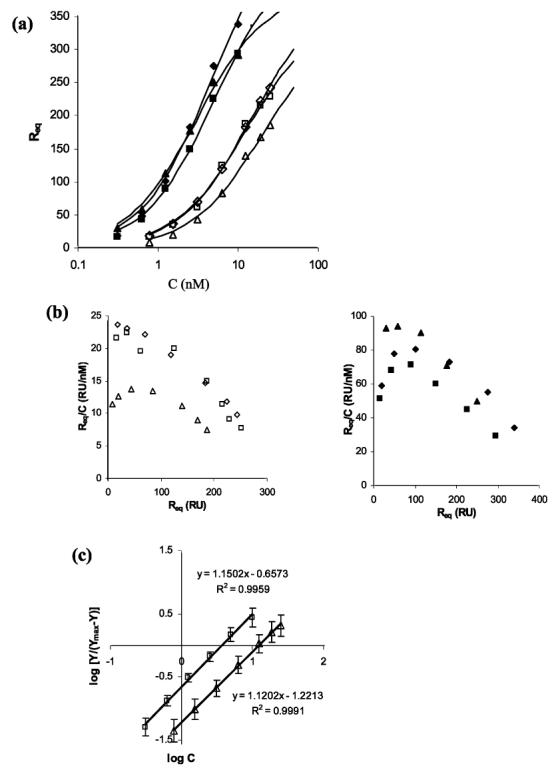
(a) Three different sets of levels of steady-state binding of InlB (filled symbols) and InlB-ΔBΔGW1 (empty symbols) to immobilized heparin. Each set of data was independently fit to the 1:1 binding model. (b) Steady-state binding levels of InlB (left) and InlB-ΔBΔGW1 (right) in a Scatchard plot. The x-axis intercept provides a theoretical estimate for the total number of binding sites. Extension of the linear portion of these data, which includes the last four (InlB) or five (InlB-ΔBΔGW1) data points, predicts approximately two protein binding sites for each heparin chain. (c) Hill representation of the steady-state binding levels for InlB (□) and InlB-ΔBΔGW1 (△). A function of the bound protein [log Y/(Ymax - Y)] was plotted vs the logarithmic concentration of the amount of protein injected, where Y represents the molar ratio of InlB (or InlB-ΔBΔGW1) bound to heparin ([InlB]/[heparin]). The maximum binding level for each protein was based on two binding sites per heparin chain.
Table 1.
Parameter Estimates for 1:1 Binding at the Steady State for Three Independent Sets of Data
| InlB 1 | InlB 2 | InlB 3 | InlB-ΔBΔGW1 1 | InlB-ΔBΔGW1 2 | InlB-ΔBΔGW1 3 | |
|---|---|---|---|---|---|---|
| KA (M) | 211 × 106 ± 34 | 178 × 106 ± 18 | 324 × 106 ± 32 | 71.9 × 106 ± 3.5 | 82.4 × 106 ± 12.9 | 52.6 × 106 ± 7.7 |
| KD (M-1) | 4.75 × 10-9 | 5.62 × 10-9 | 3.09 × 10-9 | 13.9 × 10-9 | 12.1 × 10-9 | 19 × 10-9 |
| Rmax (RU) | 512 ± 40 | 493 ± 24 | 391 ± 16 | 383 ± 9 | 351 ± 25 | 335 ± 26 |
| T (KA1) | 6.12 | 9.78 | 10.1 | 20.3 | 6.39 | 6.85 |
| χ2 | 150 | 40.4 | 52.3 | 6.3 | 65.5 | 25.3 |
| Rmax1 | 339 | 249 | 293 | 243 | 237 | 186 |
The Hill coefficient was calculated from the steady-state data to investigate the possibility of positive cooperative reactions (32-34). Qualitative interpretation of the Hill coefficient gives a measure of the number of binding sites that interact cooperatively (33) and is often considered a diagnostic measure of binding cooperativity (32). A coefficient of >1 is considered positively cooperative and a coefficient of <1 negatively cooperative, and at a value of 1, the Hill equation reduces to the Langmuir equation corresponding to an interaction that is noncooperative. The Hill coefficient for each independent set of data was close to the one obtained for the averaged data (Figure 2c), and error bars represent one standard deviation from the averaged value determined subsequent to the transformation of the independent estimates for Y. In the interaction of InlB with heparin, the estimated Hill coefficient was 1.15, and the half-saturation point was 3.7 nM. A slightly lower Hill coefficient of 1.12 was determined for the interaction between InlB-ΔBΔGW1 and heparin, with half of the sites occupied at a concentration of 12.3 nM. Thus, the Hill coefficients are consistent with a mechanism of dimerization on a single heparin chain. However, the values are considerably lower than the theoretical maximum of 2, indicating only a weak tendency for dimerization.
Competition Binding Assay for Investigating the Size of the Heparin Binding Site in InlB
Crystallographic data of InlB afford the dimensions for each of the GW domains, approximately 27 Å × 25 Å × 15 Å (8). On the basis of these dimensions, it seemed reasonable that an octasaccharide, which is 34 Å in length (35), could easily span one of the two GW domains (Figure 1b).
Structurally defined heparin oligosaccharides (dp2-dp14), LMWH, and heparin in a large molar excess with respect to InlB or InlB-ΔBΔGW1 (1000:1) were injected over the heparin surface. The measured response is proportional to the amount of unbound protein in the solution phase (Figure 3). Disaccharide (dp2), tetrasaccharide (dp4), and hexasaccharide (dp6) did not alter binding of InlB or InlB-ΔBΔGW1 to immobilized heparin. The octasaccharide (dp8) resulted in a modest decrease in the level of binding of InlB and InlB-ΔBΔGW1 to immobilized heparin. dp14 was markedly more potent in reducing the level of binding to immobilized heparin by more than 50% for both InlB and InlB-ΔBΔGW1. LMWH, a mixture of oligosaccharides ranging from dp8 to dp20 (average dp14), showed an even greater reduction. Heparin (average dp38) at the concentration tested effectively blocked measurable levels of binding. The enhanced binding affinity of dp14 for both InlB and InlB-ΔBΔGW1, compared to that of dp8-dp12, suggests that the heparin binding site in InlB might span the GW2 and GW3 domains (Figure 1b). Alternatively, the high binding affinity might require multivalent interactions, which could involve multiple binding sites between InlB and heparin or various states of InlB oligomerization (e.g., dimerization). The smaller-sized oligosaccharides might simply be unable to accommodate the additional interactions for high-affinity binding.
FIGURE 3.
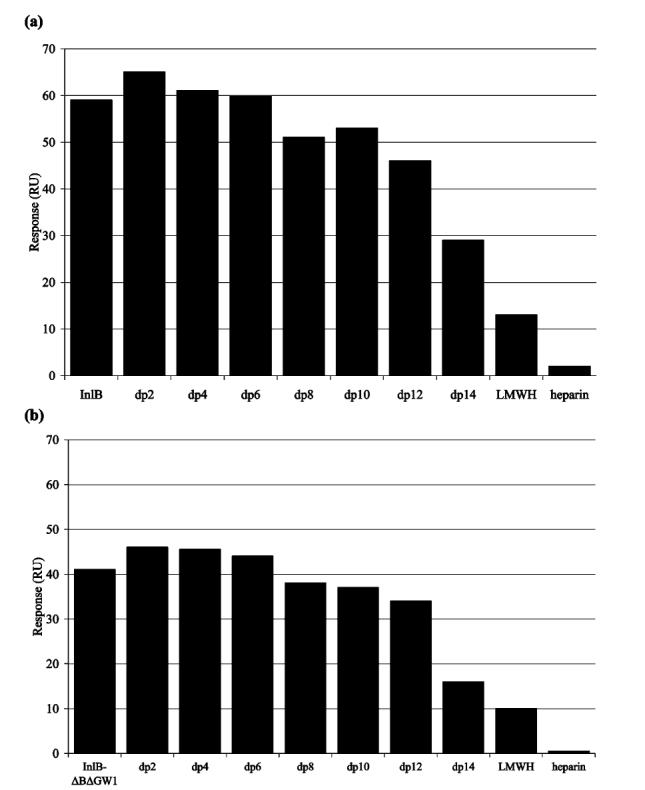
Oligosaccharide competition experiments for (a) InlB and (b) InlB-ΔBΔGW1.
Solution Affinity Studies for Determination of IC50 Values
Solution affinity experiments were next undertaken to determine IC50 values for the competition of dp14, LMWH, and heparin with binding of InlB-ΔBΔGW1 to immobilized heparin. In these experiments, a known amount of InlB-ΔBΔGW1 was incubated with a range of oligosaccharide concentrations prior to injection over the sensor surface. The measured response represented the fraction of InlB-ΔBΔGW1 available to bind heparin, and the InlB-ΔBΔGW1 solution concentration was determined from a calibration curve (Figure 4a). Construction of the calibration curve involved graphing the steady-state responses of known concentrations of InlB-ΔBΔGW1 as a function of concentration, and fitting the data with a polynomial to the fourth power using nonlinear regression.
FIGURE 4.
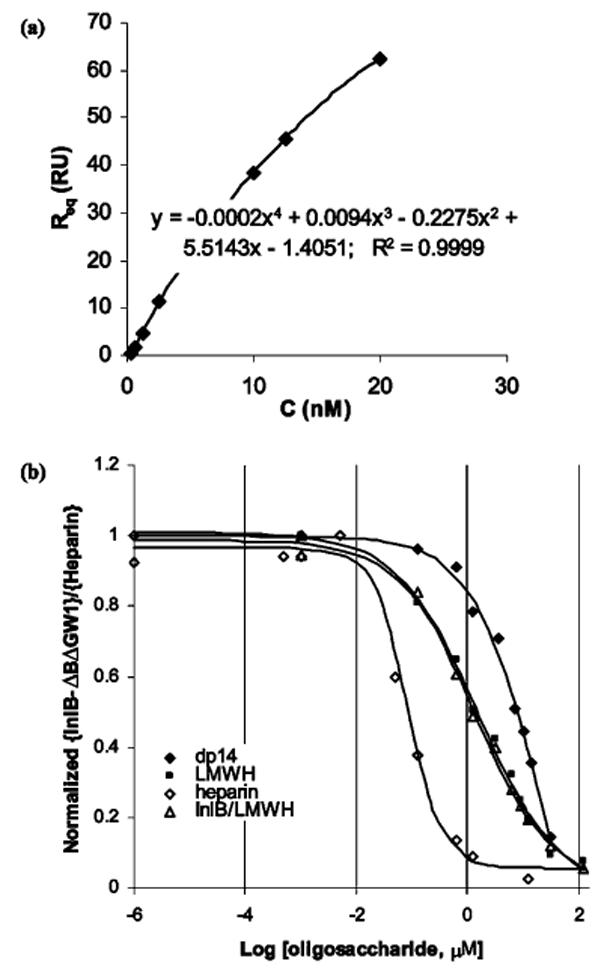
Solution affinity data for the interactions between InlB-ΔBΔGW1 and dp14, LMWH, or heparin, combined with solution affinity data for InlB and LMWH. (a) Calibration curve used to determine the free solution concentration of InlB-ΔBΔGW1 in the solution affinity experiments. (b) Steady-state binding levels corresponded to the free concentration of protein in the solution mixtures and were normalized according to the response measured for InlB-ΔBΔGW1 or InlB in the absence of polysaccharide.
Equilibrated mixtures of InlB-ΔBΔGW1 and InlB with polysaccharides were injected over the heparin-coated surface. Steady-state binding levels corresponded to the free concentration of protein in the solution mixtures and were normalized according to the response measured for InlB-ΔBΔGW1 or InlB in the absence of polysaccharide, and the results are combined in Figure 4b. As expected, an increased chain length led to a reduction in the apparent IC50. The concentration of dp14 required to inhibit 50% of the binding was 7.1 μM; for LMWH, the approximate IC50 decreased to 1.3 μM, whereas the IC50 for heparin was around 60 nM. The solution affinity of full-length InlB for LMWH yielded an IC50 identical to the one determined for InlB-ΔBΔGW1 and LMWH.
Kinetic Characterization Studies of the Interaction between InlB and Heparin
The binding of InlB to heparin was further examined by globally fitting the kinetic binding data. Global analysis, which involves fitting the association and dissociation phase data for a series of concentrations simultaneously, provides a stringent test for the binding model and better parameter estimates (23, 24, 36). The results obtained showed that InlB exhibited mass transfer limitations (Figure 5), where the rate at which the analyte reaches the surfaces from the bulk solution is slower than the rate of binding to the ligand, creating a shortage of analyte to the surface, thus interfering with kinetics. The presence of mass transfer limitations was confirmed by varying the flow rate used in the SPR experiments. Since the kinetics varied with flow rate, mass transfer limitation was likely (37). Thus, a term accounting for mass transfer was included in the models that were considered. Representative fits using a heterogeneous ligand model for InlB and InlB-ΔBΔGW1 and corresponding residuals are shown in panels a and b of Figure 6, respectively, and the parameter estimates summarized in Table 2. Although the numerical estimates are somewhat different for the two proteins, both interactions are similar, and neither appears to support a mechanism involving dimerization. The model predicts that a large proportion of the measured response is from the fast association of InlB with lower-affinity sites, while the remainder is attributed to higher-affinity sites with a slower association rate. The binding parameters for the higher-affinity site (Table 2, site 1) were consistent on the different sensor chips and at different heparin surface concentrations. The solution concentrations eligible for kinetic evaluation (≤20 nM for InlB and ≤50 nM for InlB-ΔBΔGW1) reduced the accuracy of these parameter estimates, especially for the site with the lower binding affinity (Table 2, site 2). While the overall affinity of the second (lower-affinity) binding site was maintained, the estimates for the kinetic rate constants varied nearly 10-fold. The ratio of the Rmax for the high-affinity site to the maximum response measured in the modeled data is slightly less than 20%. Successful deconvolution of biosensor data for a heterogeneous ligand typically requires a low-affinity site accounting for 20% of the binding (38). Thus, while reasonable fits were obtained, the two-site model may not be adequate for rigorous quantitative interpretation of the binding kinetics, and the values presented in Table 2 can be considered only apparent binding constants.
FIGURE 5.
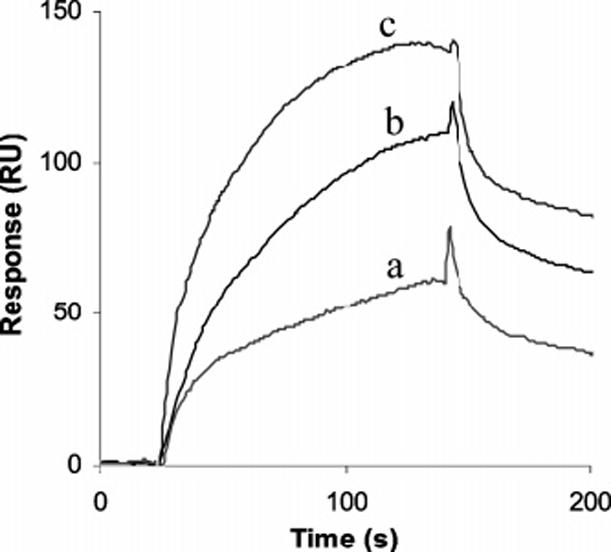
Binding progress curves for the interaction between InlB and heparin with a variation in flow rate. A solution consisting of 15 nM InlB was injected over a heparin-coated sensor surface at (a) 15, (b) 50, and (c) 100 μL/min.
FIGURE 6.
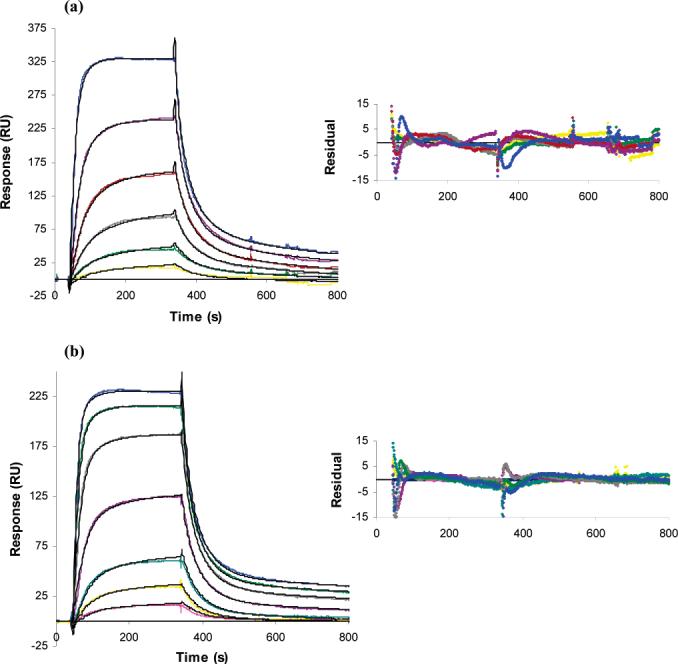
Kinetic analysis of the interaction between InlB and heparin. (a) Independent sets of curves were globally fit to a model for two binding sites, or heterogeneous ligand. InlB solution concentrations were 0.313, 0.625, 1.25, 2.5, 5, 10, 15, and 20 nM. (b) Kinetic analysis of the interaction between InlB-ΔBΔGW1 and heparin. One representative set of fitted curves is shown using solution concentrations of 0.78, 1.56, 3.13, 6.25, 12.5, 18.8, 25, 32.2, and 50 nM.
Table 2.
Kinetic Parameter Estimates for Binding of InlB and InlB-ΔBΔGW1 to Heparin with the Two-Site Heterogeneous Ligand Model for Each Set of Dataa
| average InlB | average InlB-ΔBΔGW1 | |
|---|---|---|
| kt (M-1 s-1) | 1.5 × 109 ± 0.3 | 5 × 108 ± 1 |
| site 1 | ||
| ka1 (M-1 s-1) | 3.2 × 106 ± 0.6 | 1.2 × 106 ± 0.5 |
| kd1 (s-1) | 2 × 10-3 ± 0.6 | 1.2 × 10-3 ± 0.2 |
| Rmax1 (RU) | 46 ± 5 | 29 ± 7 |
| KD1 (M-1) | 6.4 × 10-10 ± 0.8 | 1.1 × 10-9 ± 0.5 |
| site 2 | ||
| ka2 (M-1 s-1) | 1 × 108 ± 0.1 | 17 × 107 ± 24 |
| kd2 (s-1) | 0.5 ± 0.1 | 2 ± 2 |
| Rmax2 (RU) | 440 ± 41 | 350 ± 23 |
| KD2 (M-1) | 5 × 10-9 ± 1 | 1.5 × 10-8 ± 0.4 |
| χ2 | 4.9 ± 3.7 | 2.9 ± 0.7 |
The average was calculated on the basis of the kinetic data obtained from three independent sets of experiments (data not shown).
Another mechanism that was investigated was a rebinding event that could be taking place on the surface of the chip, where the protein, InlB, binds to the ligand, heparin, then comes off for a short amount of time and eventually reassociates back to the surface to bind to the ligand (39). The first suggestion of rebinding was that at higher flow rates a faster dissociation was observed (40) (Figure 5). Rebinding was tested by injecting soluble heparin during the dissociation of InlB-ΔBΔGW1 from the biotinylated albuminheparin conjugate immobilized to the chip (41). When soluble heparin was injected, a faster decrease in response was observed, suggesting that InlB-ΔBΔGW1 was taken away by soluble heparin when it dissociated from the surface, preventing it from binding back on the chip (data not shown). As the rebinding phenomenon leads to sensorgrams with a slower dissociation phase, the off rate and dissociation constants determined by curve fitting will be lower and higher, respectively, than the true values (41).
DISCUSSION
A detailed examination of the mechanism of binding between InlB and heparin was undertaken using SPR. In initial experiments, using standard SPR methods, InlB exhibited an unusually high degree of nonspecific binding to the surface of biosensor chips. The use of surfaces containing albumin-heparin conjugate and InlB-ΔBΔGW1, a simplified and fully functional version of the InlB protein, missing the B and GW1 domains (Figure 1a), reduced the level of nonspecific binding but retained the elements required for heparin binding, namely, the GW2 and GW3 domains. Thus, both InlB and InlB-ΔBΔG1 were used throughout the course of this study. Previous studies using biotinylated albumin-heparin conjugate-based chips (27) suggested that they might further reduce the level of nonspecific binding, facilitating the analysis of InlB-heparin interactions. Unfortunately, these chips have low stability that would require their replacement after the collection of each data set. Thus, appropriate controls and repetitive studies were undertaken in this study to ensure the accuracy and repeatability of our measurements. While nonspecific binding could not be completely eliminated, we were able to acquire interpretable data over a narrow range of InlB concentrations.
We first examined whether a simple bimolecular interaction was responsible for InlB-heparin binding by varying the association time. InlB has previously been reported to bind heparin as a 1:1 complex (8), and it exists only as monomers by sedimentation equilibrium ultracentrifugation (12). However, when the time allowed for the association phase was limited, InlB-ΔBΔGW1 was shown to have a faster dissociation rate. This deviation from ideal behavior suggests three possible interaction mechanisms. First, interaction of InlB with heparin could be accompanied by a change in conformation, leading to the formation of a more stable complex, and this increased association time allows a greater proportion of these stable complexes to form. Second, the binding event might involve multivalency of InlB, and the increase in contact time favors the formation of stabilized complexes composed of a single multivalent InlB cross-linking two or more binding sites within the immobilized heparin ligand. Third, the binding involves InlB being composed of multiple species that are capable of binding to a uniform heparin ligand.
Via evaluation of the steady-state binding levels, the three possible binding mechanisms could be narrowed down. Both InlB and InlB-ΔBΔGW1 were able to bind heparin with nanomolar affinity. On the basis of the steady-state binding data, Hill coefficients of 1.12 and 1.15 were calculated for InlB-ΔBΔGW1 and InlB, respectively. These values suggest a positive cooperativity; however, since these coefficients are significantly less than 2, this positive cooperativity is extremely weak. Scatchard analysis revealed curvature, also suggestive of positive cooperative binding. If the last group of data points in Figure 2b is extended, the plots suggest that there are two protein binding sites per heparin chain. The stoichiometry of binding appears to be consistent with the paradigm that dimers are capable of forming on a heparin chain but only with a weak level of positive cooperativity.
Next, we investigated the relative size(s) of the heparin binding site(s) in InlB and InlB-ΔBΔGW1. Only ≥dp14 oligosaccharides were capable of noticeably decreasing the level of binding of InlB to heparin. Comparable competition data (Figure 3) and IC50 values were observed for full-length InlB and InlB-ΔBΔGW1. These results suggest that the heparin-binding site spans both GW2 and GW3 domains.
SPR curves were next globally fit to a heterogeneous ligand model, in which one InlB binds independently to two sites within heparin. The SPR chip prepared using biotinylated albumin-heparin conjugate gave data that fit this model well with very few systematic residuals. Since variations of the flow rate indicated binding kinetics were prone to mass transfer limitations, this was taken into account when the data were analyzed. This model afforded an average KD of 0.64 nM for the first binding site and a KD of 4.57 nM for the second binding site for the interaction of full-length InlB and heparin. Similarly, the InlB-ΔBΔGW1-heparin interaction exhibited average KD values of 1.11 and 14.6 nM for the first and second binding sites, respectively. This kinetic analysis is limited, however, because of the narrow range of concentrations available to fit and because the low-affinity binding accounts for <20% of the interaction (34). A rebinding event might also be occurring along with other mechanisms of interaction. Rebinding was examined by injecting soluble heparin during the dissociation, revealing a drop in response and, thus, supporting a rebinding event.
These studies suggest that the interaction of InlB with heparin involves a complex mechanism of binding. The stoichiometry of the interaction appears to be 2:1 InlB (or InlB-ΔBΔGW1):heparin and shows weak positive cooperativity as indicated by the Hill relationships; however, a generalized mechanism for dimerization is not consistent with the kinetic interaction data. Comparison of the steady-state binding data, competition, and affinity experiments for InlB and InlB-ΔBΔGW1 show that InlB binds to heparin with a slightly higher affinity than does InlB-ΔBΔGW1, suggesting that the B and GW1 domains may play some small role in binding heparin. The studies also reveal that dp14 is the minimum heparin chain length required for strong binding, which likely involves interactions with both the GW2 and GW3 domains. The enhanced solution affinity for heparin compared to dp14 and LMWH suggests that heparin potentially accommodates multiple InlB-ΔBΔGW1 molecules. This would help explain the weak positive binding cooperativity indicated by a Hill coefficient of >1. An alternative explanation is that a lysine-rich putative heparin binding site in the LRR region of InlB participates as a secondary binding site, which requires full-length heparin to bridge the two binding sites to enhance the overall binding affinity. The kinetic interactions are reasonably consistent with nonspecific binding to a lattice or both specific and nonspecific binding to a lattice. These data suggest a significant proportion of the binding may be attributed to nonspecific electrostatic attractions. A smaller proportion of the binding appears to be attributed to specific binding and potentially dimerization in heterogeneous regions of heparin.
The current study, while unable to definitively establish the mechanism of binding of InlB and heparin, suggests that multiple mechanisms are responsible. A rebinding event could be occurring that obscures the kinetic analysis (39-43). Such a mechanism is supported by faster dissociation at higher flow rates (Figure 5) (41) and faster dissociation after injection of free heparin. Such rebinding might facilitate microbial entry into host cell, by allowing InlB to interact with GAGs for a longer amount of time, allowing more efficient activation of Met and thereby promoting actin rearrangements that lead to internalization. Additional binding studies with complementary analytical techniques will be required to more fully elucidate the mechanism with which InlB binds glycosaminoglycans and potentiates activation of Met.
Footnotes
- InlA
- internalin A
- InlB
- internalin B
- RBD
- N-terminal receptor binding domain
- LRR
- leucine-rich repeat
- GW
- Gly-Trp
- HS
- heparan sulfate
- LTA
- lipoteichoic acid
- HEPES
- 1-piperazineethanesulfonic acid, 4-(2-hydroxyethyl)monosodium salt
- EDTA
- ethylenediaminetetraacetate tetrasodium salt
- HBS-EP
- HEPES-buffered saline containing EDTA and P20
- RU
- response units
- SPR
- surface plasmon resonance
- InlB-ΔBΔGW1
- InlB lacking the B repeat and GW1 domains
- dp
- degree of polymerization
- dp14
- tetradecasaccharide
- LMWH
- low-molecular weight heparin
- IC50
- concentration inhibiting 50% of binding
REFERENCES
- 1.Cossart P, Pizarro-Cerda J, Lecuit M. Invasion of mammalian cells by Listeria monocytogenes: Functional mimicry to subvert cellular functions. Trends Cell Biol. 2003;13:23–31. doi: 10.1016/s0962-8924(02)00006-5. [DOI] [PubMed] [Google Scholar]
- 2.Mengaud J, Ohayon H, Gounon P, Mege R, Cossart P. E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell. 1996;84:923–932. doi: 10.1016/s0092-8674(00)81070-3. [DOI] [PubMed] [Google Scholar]
- 3.Lecuit M, Vandormael-Pournin S, Lefort J, Huerre M, Gounon P, Dupuy C, Babinet C, Cossart P. A transgenic model for Listeriosis: Role of internalin in crossing the intestinal barrier. Science. 2001;292:1722–1725. doi: 10.1126/science.1059852. [DOI] [PubMed] [Google Scholar]
- 4.Pentecost M, Otto G, Theriot JA, Amieva MR. Listeria monocytogenes invades the epithelial junctions at sites of cell extrusion. PLoS Pathog. 2006;2:29–40. doi: 10.1371/journal.ppat.0020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaillard J, Jaubert F, Berche P. The InlAB locus mediates the entry of Listeria monocytogenes into hepatocytes in vivo. J. Exp. Med. 1996;183:359–369. doi: 10.1084/jem.183.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen Y, Naujokas M, Park M, Ireton K. InIBdependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell. 2000;103:501–510. doi: 10.1016/s0092-8674(00)00141-0. [DOI] [PubMed] [Google Scholar]
- 7.Machner MP, Frese S, Schubert W, Orian-Rousseau V, Gherardi E, Wehland J, Niemann HH, Heinz DW. Aromatic amino acids at the surface of InlB are essential for host cell invasion by Listeria monocytogenes. Mol. Microbiol. 2003;48:1525–1536. doi: 10.1046/j.1365-2958.2003.03532.x. [DOI] [PubMed] [Google Scholar]
- 8.Marino M, Banerjee M, Jonquieres R, Cossart P, Ghosh P. GW domains of the Listeria monocytogenes invasion protein InlB are SH3-like and mediate binding to host ligands. EMBO J. 2002;21:5623–5634. doi: 10.1093/emboj/cdf558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marino M, Braun L, Cossart P, Ghosh P. Structure of the InlB leucine-rich repeats, a domain that triggers host cell invasion by the bacterial pathogen L. monocytogenes. Mol. Cell. 1999;4:1063–1072. doi: 10.1016/s1097-2765(00)80234-8. [DOI] [PubMed] [Google Scholar]
- 10.Kobe B, Deisenhofer J. A structural basis of the interactions between leucine-rich repeats and protein ligands. Nature. 1995;374:183–186. doi: 10.1038/374183a0. [DOI] [PubMed] [Google Scholar]
- 11.Schubert WD, Gobel G, Diepholz M, Darji A, Kloer D, Hain T, Chakraborty T, Wehland J, Domann E, Heinz DW. Internalins from the human pathogen Listeria monocytogenes combine three distinct folds into a contiguous internalin domain. J. Mol. Biol. 2001;312:783–794. doi: 10.1006/jmbi.2001.4989. [DOI] [PubMed] [Google Scholar]
- 12.Banerjee M, Copp J, Vuga D, Marino M, Chapman T, van der Geer P, Ghosh P. GW domains of the Listeria monocytogenes invasion protein InlB are required for potentiation of Met activation. Mol. Microbiol. 2004;52:257–271. doi: 10.1111/j.1365-2958.2003.03968.x. [DOI] [PubMed] [Google Scholar]
- 13.Hileman RE, Fromm JR, Weiler JM, Linhardt RJ. Glycosaminoglycan-protein interactions: Definition of consensus sites in glycosaminoglycan binding proteins. BioEssays. 1998;20:156–167. doi: 10.1002/(SICI)1521-1878(199802)20:2<156::AID-BIES8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 14.Hileman RE, Jennings RN, Linhardt RJ. Thermodynamic analysis of the heparin interaction with a basic cyclic peptide using isothermal titration calorimetry. Biochemistry. 1998;37:15231–15237. doi: 10.1021/bi980212x. [DOI] [PubMed] [Google Scholar]
- 15.Jonquieres R, Bierne H, Fiedler F, Gounon P, Cossart P. Interaction between the protein InlB of Listeria monocytogenes and lipoteichoic acid: A novel mechanism of protein association at the surface of Gram-positive bacteria. Mol. Microbiol. 1999;34:902–914. doi: 10.1046/j.1365-2958.1999.01652.x. [DOI] [PubMed] [Google Scholar]
- 16.Jonquieres R, Pizarro-Cerda J, Cossart P. Synergy between the N- and C-terminal domains of InlB for efficient invasion of non-phagocytic cells by Listeria monocytogenes. Mol. Microbiol. 2001;42:955–965. doi: 10.1046/j.1365-2958.2001.02704.x. [DOI] [PubMed] [Google Scholar]
- 17.Mulloy B, Linhardt RJ. Order out of complexity: Protein structures that interact with heparin. Curr. Opin. Struct. Biol. 2001;11:623–628. doi: 10.1016/s0959-440x(00)00257-8. [DOI] [PubMed] [Google Scholar]
- 18.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 19.Iozzo RV. Matrix Proteoglycans: From molecular design to cellular function. Annu. Rev. Biochem. 1998;67:609–652. doi: 10.1146/annurev.biochem.67.1.609. [DOI] [PubMed] [Google Scholar]
- 20.Shworak NW, Liu J, Fritze LM, Schwartz JJ, Zhang L, Logeart D, Rosenberg RD. Molecular cloning and expression of mouse and human cDNAs encoding heparan sulfate D-glucosaminyl 3-O-sulfotransferase. J. Biol. Chem. 1997;272:28008–28019. doi: 10.1074/jbc.272.44.28008. [DOI] [PubMed] [Google Scholar]
- 21.Woods A, Couchman JR. Heparan sulfate proteoglycans and signalling in cell adhesion. Adv. Exp. Med. Biol. 1992;313:87–96. doi: 10.1007/978-1-4899-2444-5_9. [DOI] [PubMed] [Google Scholar]
- 22.Woods A, Couchman JR. Protein kinase C involvement in focal adhesion formation. J. Cell Sci. 1992;101:277–290. doi: 10.1242/jcs.101.2.277. [DOI] [PubMed] [Google Scholar]
- 23.Myszka DG. Kinetic, equilibrium, and thermodynamic analysis of macromolecular interactions with BIACORE. Methods Enzymol. 2000;323:325–340. doi: 10.1016/s0076-6879(00)23372-7. [DOI] [PubMed] [Google Scholar]
- 24.Myszka DG. Kinetic, equilibrium, and thermodynamic analysis of macromolecular interactions with BIACORE. Energetics of Biological Macromolecules, Part C. 2000;323:325. doi: 10.1016/s0076-6879(00)23372-7. [DOI] [PubMed] [Google Scholar]
- 25.Pervin A, Gallo C, Jandik KA, Han X, Linhardt RJ. Preparation and structural characterization of large heparinderived oligosaccharides. Glycobiology. 1995;5:83–95. doi: 10.1093/glycob/5.1.83. [DOI] [PubMed] [Google Scholar]
- 26.Myszka DG, Morton TA. Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Methods Enzymol. 1998;295:268–294. doi: 10.1016/s0076-6879(98)95044-3. [DOI] [PubMed] [Google Scholar]
- 27.Zhang F, Fath M, Marks R, Linhardt RJ. A highly stable covalent conjugated heparin biochip for heparin-protein interaction studies. Anal. Biochem. 2002;304:271–273. doi: 10.1006/abio.2002.5617. [DOI] [PubMed] [Google Scholar]
- 28.Muñoz EM, Yu H, Hallock J, Edens RE, Linhardt RJ. Poly(ethylene glycol)-based biosensor chip to study heparin-protein interactions. Anal. Biochem. 2005;343:176–178. doi: 10.1016/j.ab.2005.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mach H, Volkin DB, Burke CJ, Middaugh CR, Linhardt RJ, Fromm JR, Loganathan D, Mattsson L. Nature of the interaction of heparin with acidic fibroblast growth factor. Biochemistry. 1993;32:5480–5489. doi: 10.1021/bi00071a026. [DOI] [PubMed] [Google Scholar]
- 30.Karlsson R, Falt A. Experimental design for kinetic analysis of protein-protein interactions with surface plasmon resonance biosensors. J. Immunol. Methods. 1997;200:121–133. doi: 10.1016/s0022-1759(96)00195-0. [DOI] [PubMed] [Google Scholar]
- 31.Svitel J, Balbo A, Mariuzza RA, Gonzales NR, Schuck P. Combined affinity and rate constant distributions of ligand populations from experimental surface binding kinetics and equilibria. Biophys. J. 2003;84:4062–4077. doi: 10.1016/S0006-3495(03)75132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Limbird LE. Cell Surface Receptors: A Short Course on Theory and Methods. Kluwer Academic Publishers; Norwell, MA.: 1996. [Google Scholar]
- 33.Johnson ML. Mathematical modeling of cooperative interactions in hemoglobin. Methods Enzymol. 2000;323:124–155. doi: 10.1016/s0076-6879(00)23364-8. [DOI] [PubMed] [Google Scholar]
- 34.Johnson ML, Straume M. Deriving complex ligandbinding formulas. Methods Enzymol. 2000;323:155–167. doi: 10.1016/s0076-6879(00)23365-x. [DOI] [PubMed] [Google Scholar]
- 35.Faham S, Hileman RE, Fromm JR, Linhardt RJ, Rees DC. Heparin structure and interactions with basic fibroblast growth factor. Science. 1996;271:1116–1120. doi: 10.1126/science.271.5252.1116. [DOI] [PubMed] [Google Scholar]
- 36.Myszka DG. Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Curr. Opin. Biotechnol. 1997;8:50–57. doi: 10.1016/s0958-1669(97)80157-7. [DOI] [PubMed] [Google Scholar]
- 37.Karlsson R, Roos H, Fägerstam L, Persson B. Kinetic and concentration analysis using BIA technology. Methods. 1994;6:99–110. [Google Scholar]
- 38.Khalifa BM, Choulier L, Lortat-Jacob H, Altschuh D, Vernet T. BIACORE data processing: An evaluation of the global fitting procedure. Anal. Biochem. 2001;293:194–203. doi: 10.1006/abio.2001.5119. [DOI] [PubMed] [Google Scholar]
- 39.Gopalakrishnan M, Forsten-Williams K, Cassino TR, Padro L, Ryan TE, Täuber UC. Ligand rebinding: Selfconsistent mean-field theory and numerical simulations applied to surface plasmon resonance studies. Eur. Biophys. J. 2005;34:943–958. doi: 10.1007/s00249-005-0471-2. [DOI] [PubMed] [Google Scholar]
- 40.Nieba L, Nieba-Axmann SE, Persson A, Hämäläinen M, Edebratt F, Hansson A, Lidholm J, Magnusson K, Frostell Karlsson AÅ, Plückthun A. BIACORE analysis of histidine-tagged proteins using a chelating NTA sensor chip. Anal. Biochem. 1997;252:217–228. doi: 10.1006/abio.1997.2326. [DOI] [PubMed] [Google Scholar]
- 41.Lookene A, Chevreuil O, Østergaard P, Olivecrona G. Interaction of lipoprotein lipase with heparin fragments and with heparan sulfate: Stoichiometry, stabilization, and kinetics. Biochemistry. 1996;35:12155–12163. doi: 10.1021/bi960008e. [DOI] [PubMed] [Google Scholar]
- 42.Gopalakrishnan M, Forsten-Williams K, Nugent MA, Täuber UC. Effects of receptor clustering on ligand dissociation kinetics: Theory and simulations. Biophys. J. 2005;89:3686–3700. doi: 10.1529/biophysj.105.065300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shinohara Y, Hasegawa Y, Kaku H, Shibuya N. Elucidation of the mechanism enhancing the avidity of lectin with oligosaccharides on the solid phase surface. Glycobiology. 1997;7:1201–1208. doi: 10.1093/glycob/7.8.1201. [DOI] [PubMed] [Google Scholar]