

Abstract
Tobacco mosaic virus (TMV) coat protein (CP) in absence of RNA self-assembles into several different structures depending on pH and ionic strength. Transgenic plants that produce self-assembling CP are resistant to TMV infection, a phenomenon referred to as coat protein mediated resistance (CP-MR). The mutant CP Thr42Trp (CPT42W) produces enhanced CP-MR compared to wild type CP. To establish the relationship between the formation of 20S CP aggregates and CP-MR, virus-like particles (VLPs) produced by TMV variants that yield high levels of CP-MR, were characterized. We demonstrate that non-helical structures are found in VLPs formed in vivo by CPT42W but not by wild type CP and suggests that the mutation shifts the intracellular equilibrium of aggregates from low to higher proportions of non-helical 20S aggregates. A similar shift in equilibrium of aggregates was observed with CPD77R, another mutant that confers high level of CP-MR. The mutant CPD50R confers a level of CP-MR similar to wild type CP, and aggregates in a manner similar to wild type CP. We conclude that increased CP-MR is correlated with a shift in intracellular equilibrium of CP aggregates, including aggregates that interfere with virus replication.
Keywords: TMV, CP-MR mechanism, CP mutants, CP aggregates, stacked disk, 20S
Introduction
Tobacco mosaic virus (TMV) is a positive sense, ssRNA virus and the type species of the genus Tobamovirus. In the absence of vRNA, the TMV coat protein (CP) self-assembles into a wide range of different forms depending on pH and ionic strength of the medium. It was proposed that the 20S disk, with sedimentation coefficients from 17S to 19.5S, plays an important role in the encapsidation process (reviewed in Butler, 1999; Butler and Klug, 1978; Klug, 1999). The 20S disk is a two-layer cylindrical structure with 17 CP molecules in each ring. In contrast, there are 16 and 1/3 molecules in each helical layer of the mature virion.
The role of the 20S aggregates in encapsidation is somewhat controversial, with some authors proposing that the 20S form involved in encapsidation is a two layer helical disk (Caspar and Namba, 1990). It was also proposed that several different structures comprise the 20S form, depending on buffer conditions in which the aggregate is formed; i.e., some aggregates exhibit a helical arrangement and others are non-helical. The non-helical nature of the stacked disk structure was demonstrated by electron micrographs of stacked disks of CP produced in vitro, using a monoclonal antibody specific for one end of the virion; the antibody bound to both ends of the stacked disks but to a single end of helical disks (Dore et al., 1990). Later, it was shown that the structure of a disk pair that forms the stacked disk closely resembles the AA ring pair found in the crystal structure of the four-layer disk assembly (Diaz-Avalos and Caspar, 1998). It has also been demonstrated that interconversion between the 17S bilayer disk (thought to be non-helical) and other 20S structures are only possible through an intermediary 4S structure (Raghavendra et al., 1988). These authors therefore concluded that the non-helical bilayer is not involved in virus encapsidation.
Transgenic plants that produce TMV CP are resistant to TMV infection, a phenomenon referred to as coat protein mediated resistance (CP-MR; see reviews by, Beachy, 1999; Bendahmane and Beachy, 1999). When amino acid residue 42 (Thr) in the CP was mutated to Trp (CPT42W), CP-MR was greatly enhanced, while a mutant CP that did not assemble did not confer CP-MR (Bendahmane et al., 1997). In a subsequent report Bendahmane et al., (2002) reported that CPT42W reduces production of virus-encoded movement protein (MP) during TMV replication, thereby restricting cell-cell spread of infection. The CPT42W mutant also reduced the formation of virus replication complexes (VRCs), and we proposed that CP plays a regulatory role in establishing the VRCs (Asurmendi et al., 2004).
In an accompanying paper Bendahmane et al (this issue) reported that mutations of a.a. 50 and 77 caused different effects on encapsidation, CP-MR, and virus replication. To better understand the link between structures of CP aggregates and CP-MR, we characterized the relationship between the types of VLPs formed by several of the CP mutants (reported in Bendahmane et al; this issues) and the biological effects of the CP. Here we present evidence that, in vivo, CPT42W and other CP mutants that yield high levels of CP-MR produce VLPs that contain non-helical disks whereas wild type (w.t.) CP produces VLPs made up soley or primarily of helical discs. Velocity sedimentation studies show that CPT42W and other high-resistance mutations exhibit a shift in the intracellular equilibrium of CP aggregates toward a higher proportion of smaller 20S aggregates compared with w.t. CP. Further, CPs that are mutated at amino acid (a.a.) positions 50 and 77 and exhibit a similar shift in equilibrium of CP aggregates exhibit enhanced CP-MR similar to CPT42W. We propose that structural equilibrium of the aggregates of TMV-CP plays an important role both in the biological functions of CP and in CP-MR.
Results
Previous work from our laboratory demonstrated a correlation between assembly of TMV CP into VLPs and CP-MR against TMV (Bendahmane et al., 1997). CPT42W, a mutant CP that provides very high levels of CP-MR, forms long, rod-like protein aggregates (Bendahmane et al., 1997). While mechanisms for the increased efficacy of CPT42W in CP-MR are not experimentally established, we developed structural models of aggregates of CPT42W and conducted experiments to test the hypothesis that the state of aggregation of mutant CPs are correlated with high and low levels of CP-MR.
Structural model of CPT42W
CPT42W is known to form VLPs, some of which appeared to include helical and non-helical aggregates (Bendahmane et al, 1997; described in detail below). We therefore developed models that included the helical (Figure 1A–C) and non-helical (Figure 1E, F) configurations, and the structural environments of the 42W residue were analyzed (Tables 1, 2). In the helical structure, the subunits are arranged in a right-handed helix with sixteen and 1/3 subunits per turn. In the non-helical structure, seventeen subunits are arranged as a flat disk with the tops of two disks facing each other.
Figure 1.
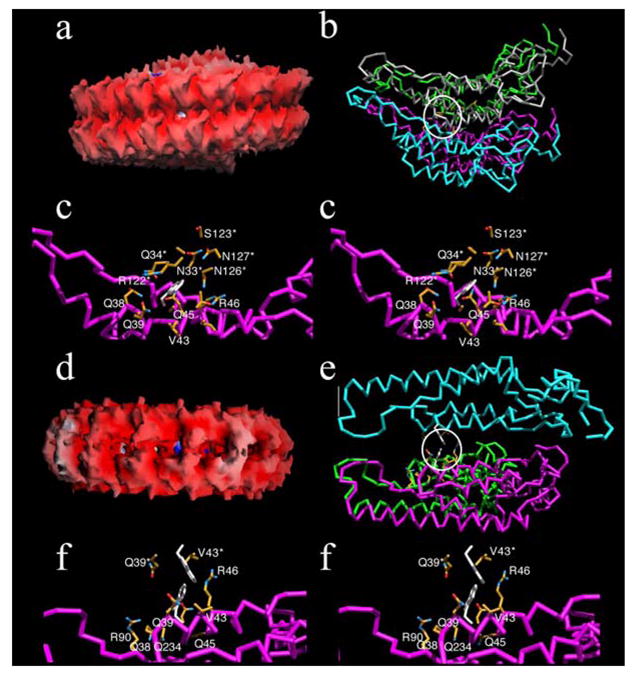
Structural models of helical (a–c) and non-helical (d–f) assemblies of TMV CPT42W. Panels a and d represent the molecular surfaces of two disks of helical and non-helical assembly of the protein, respectively. Images are colored according to electrostatic potential with positive and negative charges represented by blue and red, respectively. Panels b and e, show the general locations of the T42W mutation (highlighted by the white circle) in the helical and non-helical assemblies, respectively. Note that there is significantly more open space between the non-helical stacked disks than in the helical fibers. Each subunit of the aggregate is represented by a differently colored C-α backbone. Panels c and f are stereo images showing the environments of a.a. 42W in the helical and non-helical aggregates, respectively. The residues in contact with 42W are colored according to atom type with carbon, nitrogen, and oxygen colored yellow, blue, and red, respectively. The side chains for the 42W residue are represented by white stick models. Note that the 42W side chain environment is more crowded and hydrophilic than observed in the stacked disk conformation.
Table 1.
Residues in contact with the CP42W sidechains in the helical and nonhelical aggregates. The numbers shown in the table are the calculated contact surface areas in units of Å2. The residues in contact with W42 from subunits found within the same disks are denoted by single asterisks while those contacts made by subunits from adjacent disks are marked by multiple asterisks.
| Aggregate | Residue | Main Chain | Side Chain |
|---|---|---|---|
| Helical | |||
| Q38 | 6.3 | 4.1 | |
| Q39 | 10.3 | 8.9 | |
| V43 | 5.0 | 7.4 | |
| Q45 | 0.0 | 14.4 | |
| R46 | 0.0 | 17.7 | |
| G32* | 4.5 | 0.0 | |
| N33* | 2.1 | 2.1 | |
| Q34* | 5.2 | 21.0 | |
| P78** | 9.8 | 0.0 | |
| T81** | 0.0 | 11.0 | |
| A82** | 7.5 | 18.3 | |
| R122*** | 4.5 | 20.9 | |
| S123*** | 7.4 | 7.7 | |
| N126*** | 0.0 | 24.7 | |
| N127*** | 0.0 | 7.2 | |
| Non-helical | |||
| Q38 | 4.4 | 10.5 | |
| Q39 | 8.4 | 2.7 | |
| V43 | 10.1 | 8.5 | |
| Q45 | 0.0 | 9.0 | |
| R46 | 0.0 | 27.7 | |
| R90 | 0.0 | 3.8 | |
| G32* | 4.9 | 0.0 | |
| N33* | 4.3 | 3.8 | |
| Q34* | 4.2 | 15.7 | |
| Q139** | 0.0 | 9.5 | |
| W42** | 0.0 | 17.6 | |
| V43** | 0.0 | 17.8 | |
Table 2.
Summary of the nature of the contacts between CPT42W and adjacent subunits in the two different kinds of aggregates. The numbers represent the contact surface area in units of Å2. Note that the total contact area and main chain contacts are significantly greater in the helical fiber, suggesting a tighter packing arrangement. Also note that there is significantly more hydrophobic contact in the non-helical aggregate.
| Contact Type | Helical | Non-helical | ||
|---|---|---|---|---|
| Mainchain | Sidechain | Mainchain | Sidechain | |
| Basic (H,K,R) | 4.5 | 38.6 | 0.0 | 31.6 |
| Polar (N,Q,S,T) | 31.4 | 101.1 | 21.3 | 51.4 |
| Small (A,G) | 12.0 | 18.3 | 4.9 | 0.0 |
| Nonpolar (C,I,L,M,P,V) | 14.8 | 7.4 | 10.1 | 26.3 |
| Aromatic Polar (W,Y) | 0.0 | 0.0 | 0.0 | 17.6 |
| Total Area | 62.7 | 165.4 | 36.3 | 126.9 |
As is apparent from these analyses, the region around residue 42 is more compact in the helical than in the non-helical configuration. In the helical arrangement, the side chain of residue 42W contacts three adjacent subunits whereas it contacts two subunits in the non-helical conformation. As summarized in Tables 1 and 2, the contact area for 42W is larger and involves more main chain interactions in the helical aggregate than in the non-helical configuration. The other striking difference between the two forms is that residue 42W in the helical configuration is entirely enclosed by hydrophilic residues whereas the contacts in the non-helical conformation are more evenly divided among the hydrophobic and hydrophilic side-chains. Furthermore, there is more space around the 42W contact residues in the non-helical aggregate, allowing it to more easily accommodate the bulky, hydrophobic tryptophan. Indeed, most of the contacts between 42W and neighboring hydrophilic residues are made via the aliphatic portions of the side-chains (compare Figs. 1C and 1F). Together, this suggests that the T42W mutation is more easily accommodated by the looser, and more hydrophobic, packing found in the non-helical fibers.
The most unexpected result was that, in the non-helical arrangement, 42W residues from adjacent disks can interact with each other. In the flat disk arrangement, the top of one disk contacts the top of the adjacent disk by virtue of 2-fold axes that lie perpendicular to the long axes of the non-helical fibers (Figs 1E and 1F). This places the 42W residue of one subunit in contact with the 42W residue from the adjacent subunit. When compared with the helical conformation in which 42W residue is tightly packed amongst hydrophilic residues, this 42W-42W interaction would be expected to significantly stabilize the non-helical structure. Therefore, the modeling studies suggest that the T42W mutation simultaneously causes unfavorable subunit contacts in the helical fiber while creating favorable interactions in the non-helical stacked disk conformation. It therefore follows that the T42W mutation should increase the propensity for the subunits to form aggregates composed of stacked disks comprised of AA dimers. This is in agreement with our earlier studies that showed virus-like particles recovered from plants infected with TMV-CPT42W lack RNA and that the virus like particles (VLP) contain stacked disks (Bendahmane et al., 1997).
Characterization of TMV-CPT42W VLPs
The working hypothesis for the modeling studies was that mutation T42W favors the formation of non-helical disks aggregates in vivo. We conducted tests to determine if the hypothesis was correct. TMV particles and VLPs containing TMV-CPT42W were purified from infected tobacco leaves as described in Methods. The identity and purity of the wt CP and CPT42W were confirmed by MALDI-TOF mass spectrometry (data not shown).
To confirm that the purification method did not affect the equilibrium of CP aggregates of CPT42W, we compared the length of purified VLP particles with those immuno-captured (on EM grids) immediately after lysing infected protoplasts. Particle lengths were somewhat longer in the immuno-capture sample than in the purified sample, and the frequency of certain particles were somewhat different between the two samples (Figure 2). Nevertheless, the overall conclusion of the study is that extraction and purification of particles did not significantly alter the range of sizes of VLPs formed by TMV-CPT42W.
Figure 2.
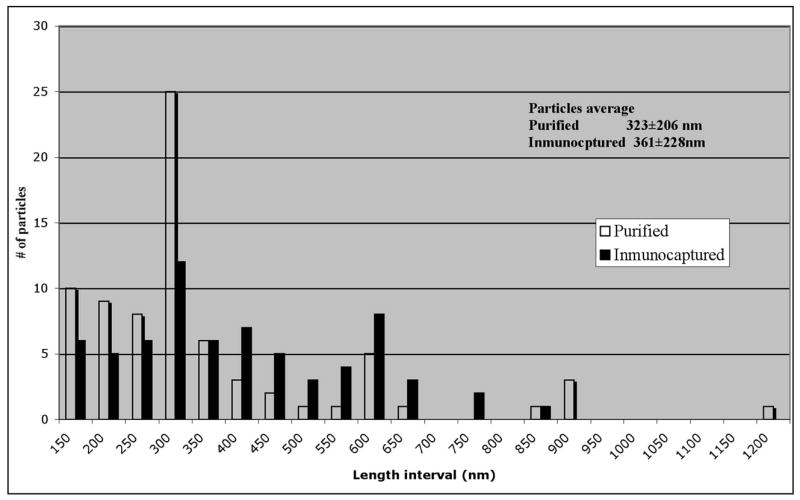
Histogram of the distribution of sizes of the particles. Comparison of the distribution of sizes of particles of TMV-CPT42W between of particles and immuno-captured particles.
Cryo-electron microscopy of TMV and VLPs of TMV-CPT42W
Particles produced during infection by TMV and TMV-CPT42W were purified and subsequently visualized by cryo-TEM. At low magnification (10000X), it was apparent that TMV-CPT42W formed extremely long particles (Fig. 3a). At higher magnification (25000X) there were two apparent differences between w.t. virions and VLPs of TMV-CPT42W. First, while end-to-end alignment of particles formed long rods in both cases (Fig. 3b TMV and Fig. 3c TMV-CPT42W), the VLPs of CPT42W are often joined at an angle not observed in w.t. TMV. Second, the ends of some VLPs and the ‘angled joints’ were different compared with other regions of the VLP (Fig. 3c, see the arrow in the enlargement of the ‘angled joint’, Fig 3c′). Interestingly, this ‘angled joint’ portion of the TMV-CPT42W VLPs resembles the structure of a stacked disk.
Figure 3.

Cryoelectron microscopy of purified particles. a, c, c′ and e virus-like particles of TMV-CPT42W; b, d and f virions of TMV. a, 10000X magnification; b and c, 25000X magnification. c′, Shows an enlargement of c, the arrow points to the stacked-disk like structure in the ’angled joints’ described in the text. d and e, Fast Fourier Transformation images. The black squares on panels b and c indicate the areas used in the FFT.
The most direct method to determine if such particles have a helical or non-helical character is to perform a Fourier transform of the fiber image. Fast Fourier Transformation (FFT) (Fig. 3d, e) was carried out on selected regions of both types of structures (black squares in Fig. 3b, c, respectively). This analysis showed the expected repetition in the diffraction pattern along the length of each type of particle. The distance between the patterns for TMV and regions of TMV-CPT42W that did not have a stacked disk appearance was 23Å. In contrast, FFT analysis of regions of stacked disk-like structures in TMV-CPT42W VLPs exhibited repeat distances of approximately 25Å and 52Å. These data are in agreement with the values reported for FFT of stacked disks (Diaz-Avalos and Caspar, 1998; Diaz-Avalos and Caspar, 2000); we therefore concluded that CPT42W forms stacked discs as well as helical discs. Based on the model described in Fig 1 we suggest that the formation of rods containing non-helical stacked disk composed of the AA pairs observed in the crystal structure of the four layer stacked disk (Bhyravbhatla, Watowich, and Caspar, 1998) is due to the increased affinity between T42W residues.
EM Immuno-gold-labeling assays
While FFT analyses supports the hypothesis that VLPs of TMV-CPT42W contain portions of stacked disks, it is important to determine whether or not the stacked disks forms are similar to the AA pairs described in the crystal structure of the four layer stacked disk in which the tops of two simple layers are facing each other, exposing equivalent surfaces on each face of the stack. (Diaz-Avalos and Caspar, 1998; Diaz-Avalos and Caspar, 2000).
To determine if this is the case, a monoclonal antibody (Mab#16) that binds to one end of TMV was used (Dore et al., 1988). This antibody binds to the surface defined as the ‘bottom’ of the particle that contains the 5′ end of viral RNA (Dore et al., 1990). If the working model is correct, the TMV-CPT42W particles should include non-helical stacked disks similar to AA pair and Mab16 should react with both ends of the particles. Similar results are expected with helical particles that are joined end-to-end by a non-helical stacked disks.
Purified preparations of TMV and TMV-CPT42W were subjected to an immuno-gold labeling assay using antibody Mab#16. TMV exhibited two different labeling patterns, namely labeling at one end or in the middle of particles greater than unit length (Figure 4a). VLPs of TMV-CPT42W exhibited three different patterns of labeling; labeling at a single end, in the middle of particles (Figure 4b), and at both ends of particles (Figure 4c). Table 3 presents the data from these studies with statistical analyses. The fact that only CPT42W VLPs yielded particles that are labeled at both ends lends support to the hypothesis that such VLPs contain non-helical stacked disks. In w.t. TMV labeling within particles occurred only in particles greater than unit length, suggesting that labeling occurs at the junction of two particles. TMV is known to align end-to-end to form long particles (Beachy and Zaitlin, 1977).
Figure 4.

Inmuno-gold labeling of purified particles using Mab #16. a) TMV particles: left panel shows a particle with a single labeled end; the panel on the right shows label within the particle, see arrows. b) TMV-CPT42W particles: left panel shows a particle with a single end-label, and the panel on the right shows labeling within the particle. c) TMV-CPT42W particles with both ends labeled. The bar represents 100 nm.
Table 3.
Number of particles with reaction to Mab#16 labeled with colloidal gold, with emphasis on site of location on the particle.
| Type of Particle | Label at single end of particle (%) | Label within the particle (%) | Label at both ends of particle (%) |
|---|---|---|---|
| TMV | 75 of 103(73)* | 28 of 103(27)* | 0 of 103(0) |
| VLPs of TMVCPT42W | 67 of 123(54)* | 46 of 123(38)* | 10 of 123(8) |
Chi2 test= Out of normality
In the case of VLPs composed of CPT42W, we observed a range of particle sizes that were labeled by antibody within the particle; we propose that these particles represent the junction of VLPs. Statistical analysis of the data shows a significantly higher proportion of particles with internal labeling in samples that contain the VLPs of CPT42W than in samples that contain TMV (Table 3). The long rods in samples that contain VLPs of CPT42W include helical regions containing or joined together by non-helical stacked-disks. This is in agreement with the FFT data showed in Fig 3E.
Correlation between CP aggregation and CP-MR
To determine if other CP mutants that confer CP-MR activities similar to CPT42W have similar physical characteristics, we examined a subset of mutant CPs with single substitutions at a.a. Glu 50 and Asp 77 as described by Bendahmane et al (accompanying paper). Mutations were made to create carboxyl-carboxylate interactions between adjacent CP monomers in virions and VLPs. Modeling studies based on structural studies of TMV (Culver et al., 1995; Namba, Pattanayek, and Stubbs, 1989) suggested that these residues likely stabilize the quaternary structure of virions but not the tertiary structure of the CP monomer. Therefore, mutations at these sites are expected to alter the aggregation properties of mutant CPs. Bendhamane et al described the effects of mutations at positions 50 and 77 with regard to effects on assembly of virions and on systemic infection (indicative of encapsidation of vRNA). This work also described the effects of mutant CPs on TMV replication, and capacity to confer CP-MR [Summarized in Table I of the accompanying paper; Bendahmane et al.]. Mutants are grouped in three classes according to various characteristics. One group of mutants does not change virus replication and does not confer CP-MR. A second group enhances virus replication and confers CP-MR similar to w.t. CP. The third group reduces virus replication and virus replication complexes (VRCs) and confers elevated levels of CP-MR. The mutant CPT42W belongs to the latter group. Based on these and other characteristics we selected mutants representing the second group (CPE50R and CPD77K) and the third group (CPD77R and CPE50D) for analysis.
Infection caused by TMV-CPE50R and TMV-CPD77K produced virions with structures similar to wild type virus, i.e., with regular helical ultra-structure. Particles produced by TMV-CPE50R are longer than w.t. TMV while particles of TMV-CPD77K tend to be somewhat shorter (on average) than w.t. TMV. In contrast, virions/VLPs produced by TMV-CPE50D and TMV-CPD77R reveal ultra-structures with regions reminiscent of the stacked disks observed in VLPs produced by TMV-CPT42W (see Fig 2 of accompanying paper; Bendahmane et al).
Sucrose density gradients of virus mutants
To determine the nature of aggregates produced by CP mutants, a sucrose gradient sedimentation assay was performed. Extracts of infected leaves were loaded on linear 5% to 40 % sucrose gradients and following centrifugation, eight fractions and the pellet were collected. Figure 5 shows the results of western blot analyses of equal volume aliquots of each fraction. In agreement with previous data, most of the CP produced by w.t. TMV assembles to virions and is found in the pellet; a smaller but significant amount of CP sediments throughout the gradient. This pattern represents different levels of aggregation of CP, and sub-viral particles that contain segments of TMV-RNA (Beachy and Zaitlin, 1977). In contrast the mutant CPT42W produces VLPs (lacking vRNA; Bendahmane et al., 1997) and other aggregates that sediment in the upper part of the gradient (fraction 1 to 4); small amounts of CP are found in the pellet.
Figure 5.
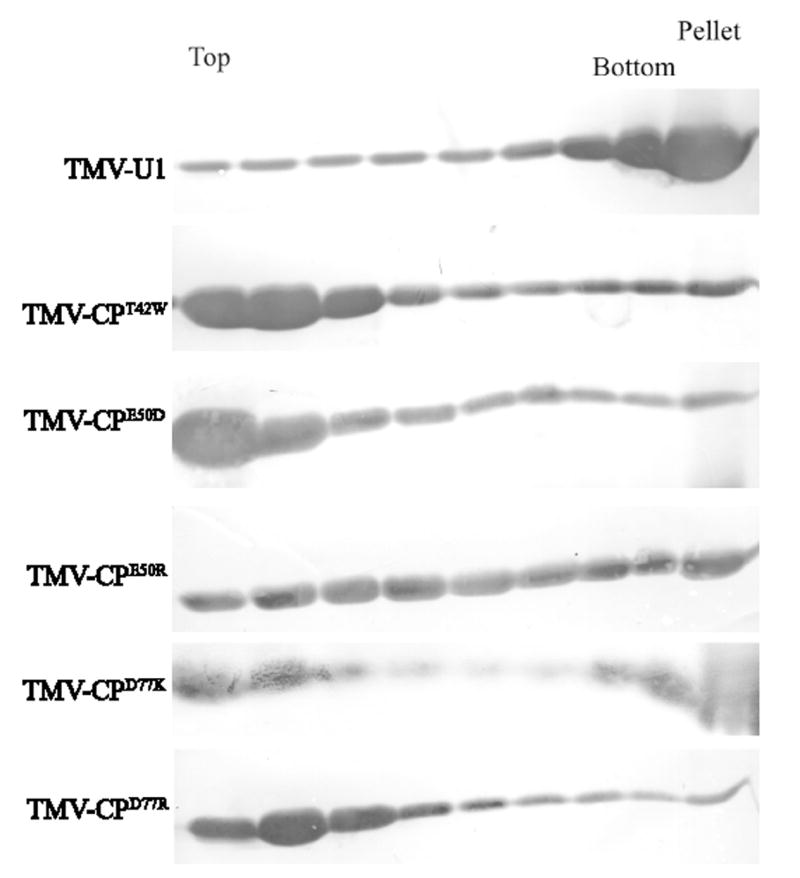
Sucrose gradient analysis. Leaf homogenates were prepared from infected plants and analyzed on linear 5 to 40% sucrose gradients. Gradients were collected in 8 fractions plus pellet, and aliquots were subjected to PAGE in SDA and western blot analysis using anti-TMV serum. T= top of the gradient, B= bottom and P = pellet.
CPE50R and CPD77K exhibited a pattern of sedimentation in sucrose gradients similar but not identical to w.t. virus. In contrast, CPD77R and CPE50D showed a pattern of sedimentation similar to CPT42W. Mutants CPT42W, CPE50D and CPD77R produced a larger proportion of mid-sized aggregates in comparison with w.t. TMV, CPE50R, and CPD77K. The finding that mutants in these two groups confer similar biological functions with regard to virus replication and CP-MR (above) leads us to propose that the equilibrium of CP aggregates plays a role in CP functions other than in virus assembly per se.
Discussion
Modeling studies suggest that mutation of residue 42 of TMV CP from T to W has two effects on the formation of helical aggregates. Firstly, the mutation is likely to interfere with the formation of helical structures since the area of the mutation is tightly packed with amino acids that contain hydrophilic sidechains. Secondly, the mutation appears to promote the formation of AA dimers (Fig. 1E). In this conformation, the environment around residue 42 is less compressed and less hydrophilic than in w.t. virions. Furthermore, this conformation places residue 42W in adjacent disks in direct contact with each other. We propose that these two effects act together to drive the equilibrium of CP aggregates towards the non-helical formation. This hypothesis is supported by data that shows the presence of particles containing non-helical disks. Earlier studies showed that, in solutions that contain TMV CP, an equilibrium of different states of aggregation is established, and the proportion of each is correlated with pH and ionic strength of the solution (Butler, 1999; Klug, 1999). The data obtained in the present work are consistent with the hypothesis that, in the case of TMV-CPT42W, the relative abundance of the non-helical stacked disk form is increased.
CP mutants with biological properties similar to those of CPT42W revealed correlations between sedimentation of CP aggregates in sucrose gradients and biological features. For example, CPD77R is similar to CPT42W in reducing formation of VRCs during infection, in conferring high levels of CP-MR, in forming stacked disks, and in patterns of sedimentation in sucrose gradients. Mutant CPE50D reduced formation of VRCs and virus replication in protoplasts, and exhibited similar patterns of sedimentation in sucrose gradients as CPD77R and CPT42W. This mutant yielded high levels of CP-MR in the local lesion host but not in systemic hosts (Bendahmane et al, accompanying paper).
CPE50R and CPD77K have sedimentation patterns that are different than CPT42W, and more similar to patterns of w.t. TMV; like w.t CP, CPE50R provides CP-MR. The fact that CPD77K does not confer CP-MR may be due to the observation that VLPs produced by this mutant are associated with cellular debris and are therefore unavailable for CP-MR (Benhamane et al., accompanying paper).
It was previously demonstrated that CPT42W produces greater amounts of 20S aggregates compared with w.t. CP, and that the level of CP-MR produced by CPT42W was significantly greater than CP-MR produced by w.t. CP (Bendahmane et al., 1997). Similar results were obtained with the CPD77R (Bendahmane et al, accompanying paper). We propose, based on the results of previous and present studies, that the increased level of CP-MR in plants that express these mutants is a result of the shift in intracellular equilibrium of the CP aggregates.
Our data suggest that the multiple functions of TMV CP are mediated by the physical nature of aggregates of CP, in particular, that some of the functions are mediated by helical structures while others are mediated by stacked discs. CPs that produce small amounts of midsized aggregates (relative to total CP) have a positive effect on virus replication, while mutants that yield stacked discs and high amounts of smaller aggregates reduce replication. We proposed earlier that a function of the CP is to regulate the formation of VRCs by regulating production of MP; this likely regulates the production of other viral proteins and virus replication (Asurmendi et al., 2004; Bendahmane et al., 2002). It is known that production of MP decreases during late stages of the infection (Lehto, Bubrick, and Dawson, 1990; Watanabe et al., 1984). We propose that increased concentrations of CP during replication result in changes in the states of aggregation of CP which in turn modulates MP expression. In the case of CPT42W and other CP mutants the equilibrium is altered by the nature of the mutation, and regulates MP production and formation of VRCs.
Other viral CPs are known to possess activities in addition to encapsidating vRNA. For example, Alfalfa Mosaic Virus (AlMV; an Ilar virus) CP activates the viral genome and is a positive regulator of synthesis of plus-strand vRNA (Bol, 2005). Similar results were reported for tobacco streak ilarvirus (van Dun et al., 1988). Neverthless, CPs of AlMV and TSV confer CP-MR. Likewise, the CP of potato virus X confers CP-MR by reducing infection of single cells as well as spread of infection (Bazzini et al., 2006). The mechanisms of ‘negative regulation’ by the CPs of ilarviruses and PVX during CP-mediated resistance are not known. In contrast, accumulation of CP of rice yellow mottle virus (RYMV) in transgenic rice plants increased the rate of virus replication and disease in these plants (Kouassi et al., 2006), and leads to the suggestion that CP of RYMV is a positive, rather than a negative, regulator of virus replication. These observations, in combination with data reported here for mutants of TMV CP suggest that functions of CP can be altered by specific mutations, some of which can be to confer or enhance levels of CP-MR that will increase virus resistance in transgenic crops.
Material and Methods
In vitro transcription, protoplast isolation and transfection
To obtain infectious viral RNA from the full-length cDNA clone of TMV RNA, pU3/4-12(Holt et al., 1990) or pTMV-CPT42W (Bendahmane et al., 1997) the MEGAscript™ Transcription kit (Ambion, USA) was used according to the manufacturer’s recommendations. Reactions were supplemented with m7G(5′)ppp(5′)G Cap Analog (Ambion, USA). Tobacco BY-2 protoplasts were prepared from suspension cell cultures essentially as previously described (Watanabe, Ohno, and Okada, 1982) and 1.5 × 106 cells were inoculated by electroporation and incubated at 28°C in the dark.
Virus purification
The standard purification procedure was shown to be not effective in purifying VLPs of TMV-CPT42W (Asselin and Zaitlin, 1978). Therefore, we used a modified protocol in which ultracentrifugation steps were omitted (Fenczik et al., 1995). Large amounts of TMV and TMV-CPT42W were obtained by this procedure. The identity and purity of the CP was confirmed by MALDI mass spectrometry (data not shown).
In brief, TMV and VLPs were isolated from infected tobacco leaf tissues that were triturated in 0.5 M phosphate buffer pH 7.0, 143 mM β-mercaptoethanol (1 ml/g of fresh tissue). Two volumes of water-saturated chloroform/butanol (50:50) were added and the samples were centrifuged for 15 min at 3000 g. To precipitate the virions from the aqueous phase, 4% PEG-8000 (w/v final concentration) was added to the samples for 10 min, on ice. Virus was collected by centrifugation in a microfuge, resuspended in 10 mM phosphate buffer pH 7.0, and centrifuged again to remove insoluble material. Finally, virus was precipitated again with 4% PEG-8000 and 1% NaCl (w/v) and collected by centrifugation.
Sucrose density gradient analyses
Homogenates were prepared from three grams of fresh leaves harvested from transgenic plants and were analyzed on linear 5% to 40% sucrose gradients as previously described (Bendahmane et al., 1997). Fractions of 0.8 or 1 ml were collected from the top and aliquots of 10 μl were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot assays.
Electronic microscopy and immuno-capture of virus-like particles
Infected BY-2 protoplasts were harvested 20 h. after electroporation and homogenized in PBS. Formvar-coated nickel grids (200 mesh) were incubated twenty minutes with a 1/50 dilution of rabbit anti-TMV CP antibody, washed, and then blocked with a solution of 0.2 % BSA in PBS for fifteen minutes. Grids were floated on drops of the samples for 30 minutes, and then washed five times with PBS. Samples were negatively stained with 1% phosphotungstic acid solution (pH 6.8) for two minutes, dried, and examined on a Leo 912AB TEM. The images were recorded using a CCD camera (2Kb × 2Kb). In the case of the purified particles from infected leaves, the grids were deposited on top of the samples for 20 min and then negatively stained as described above.
Cryo-Electronmicroscopy
The samples were prepared by placing a 7 μl drop of virus suspension on a holey grid, blotted with filter paper, and plunged into ethane slush. The grids were stored in liquid N2.
The grids were examined using a LEO 912AB TEM equipped with a Gatan anticontaminator; a Gatan 626 cryoholder was used to mount the grids. Low-dose images were taken at 10 000 and 25 000X using an accelerating voltage of 80KeV. The Fast Fourier Transformation (FFT) was performed on selected zones of the images using the Software AnalySIS (Soft Imaging System, Gnbh, Munster, Germany).
Immuno-gold Labeling
EM grids (Formvar coated, 200 mesh) were floated on drops of the virus suspension samples for 20 min. The grids were treated with PBS BSA (1% w/v) for 20 min, and the first antibody [Mab #16, ATCC 931 (Dore et al., 1988)] was applied and incubated for 1 hour (1/50 in PBS 0.2% BSA). After washing four times with PBS/BSA (0.2% w/v), the second, gold conjugated antibody was incubated with the grid for 1 hour (1/200, goat anti-mouse, 12 nm colloidal gold, Jackson Immunoresearch, USA). The grids were washed five times with PBS/BSA (0.2% w/v) and rinsed twice with deionized water. Finally, the grids were negatively stained and observed as described above.
Structural Modeling
The model for the helical assembly of TMV (Namba, Pattanayek, and Stubbs, 1989) and the ‘AA ring pair’ of the four layer disk (Bhyravbhatla, Watowich, and Caspar, 1998) structures were used as templates to model the CPT42W protein. Program O (Jones, Zou, and Cowan, 1991) was then used for ‘mutating’ the residue and selecting the rotomeric position that exhibited the fewest steric clashes. It should be noted that in the case of the non-helical structure, residue R46, which is structurally adjacent to 42W, could have been moved to an alternative rotomeric position, but was left unmodified to prevent bias in the subsequent contact calculations. From each of these aggregates, a cluster of four subunits was selected to insure that the entire environment of the T42W mutation was represented. These clusters of four subunits were then subjected to Powell energy minimization using the program X-Plor (Brünger, 1992). The contact environment of the 42W sidechain was then analyzed using the program MS (Connolly, 1981; Connolly, 1983a; Connolly, 1983b) and a 1.7Å probe radius.
Acknowledgments
This work was supported by grant AI27161 from the National Institutes of Health. We are grateful for the support of the NSF Major Research Instrumentation program in the purchase of the LEO TEM used in this study (NSF/MRI award number DBI-0116650).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
S. Asurmendi, Email: sasurmendi@cnia.inta.gov.ar.
R.H. Berg, Email: rhberg@danforthcenter.org.
T.J. Smith, Email: tsmith@danforthcenter.org.
M. Bendhamane, Email: mbendahm@ens-lyon.fr.
References
- Asselin A, Zaitlin M. Characterization of a second protein associated with virions of tobacco mosaic virus. Virology. 1978;91(1):173–81. doi: 10.1016/0042-6822(78)90365-3. [DOI] [PubMed] [Google Scholar]
- Asurmendi S, Berg RH, Koo JC, Beachy RN. Coat protein regulates formation of replication complexes during tobacco mosaic virus infection. Proc Natl Acad Sci U S A. 2004;101(5):1415–20. doi: 10.1073/pnas.0307778101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini AA, Asurmendi S, Hopp HE, Beachy RN. Tobacco mosaic virus (TMV) and potato virus X (PVX) coat proteins confer heterologous interference to PVX and TMV infection, respectively. J Gen Virol. 2006;87(Pt 4):1005–12. doi: 10.1099/vir.0.81396-0. [DOI] [PubMed] [Google Scholar]
- Beachy RN. Coat-protein-mediated resistance to tobacco mosaic virus: discovery mechanisms and exploitation. Philos Trans R Soc Lond B Biol Sci. 1999;354(1383):659–64. doi: 10.1098/rstb.1999.0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beachy RN, Zaitlin M. Characterization and in vitro translation of the RNAs from less-than-full-length, virus-related, nucleoprotein rods present in tobacco mosaic virus preparations. Virology. 1977;81(1):160–9. doi: 10.1016/0042-6822(77)90068-x. [DOI] [PubMed] [Google Scholar]
- Bendahmane M, Fitchen JH, Zhang G, Beachy RN. Studies of coat protein-mediated resistance to tobacco mosaic tobamovirus: correlation between assembly of mutant coat proteins and resistance. J Virol. 1997;71(10):7942–50. doi: 10.1128/jvi.71.10.7942-7950.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendahmane M, Beachy RN. Control of tobamovirus infections via pathogen derived resistance. Adv Virus Res. 1999;53:369–386. doi: 10.1016/s0065-3527(08)60357-7. [DOI] [PubMed] [Google Scholar]
- Bendahmane M, Szecsi J, Chen I, Berg RH, Beachy RN. Characterization of mutant tobacco mosaic virus coat protein that interferes with virus cell-to-cell movement. Proc Natl Acad Sci U S A. 2002;99(6):3645–50. doi: 10.1073/pnas.062041499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhyravbhatla B, Watowich SJ, Caspar DL. Refined atomic model of the four-layer aggregate of the tobacco mosaic virus coat protein at 2.4-A resolution. Biophys J. 1998;74(1):604–15. doi: 10.1016/S0006-3495(98)77819-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bol JF. Replication of alfamo- and ilarviruses: role of the coat protein. Annu Rev Phytopathol. 2005;43:39–62. doi: 10.1146/annurev.phyto.43.101804.120505. [DOI] [PubMed] [Google Scholar]
- Brünger AT. X-plor (Version 3.1) User’s Guide. Yale University; New Haven, CT: 1992. [Google Scholar]
- Butler PJ. Self-assembly of tobacco mosaic virus: the role of an intermediate aggregate in generating both specificity and speed. Philos Trans R Soc Lond B Biol Sci. 1999;354(1383):537–50. doi: 10.1098/rstb.1999.0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler PJ, Klug A. The assembly of a virus. Sci Am. 1978;239(5):62–9. doi: 10.1038/scientificamerican1178-62. [DOI] [PubMed] [Google Scholar]
- Caspar DL, Namba K. Switching in the self-assembly of tobacco mosaic virus. Adv Biophys. 1990;26:157–85. doi: 10.1016/0065-227x(90)90011-h. [DOI] [PubMed] [Google Scholar]
- Connolly ML. PhD. University of California; Berkeley, Berkeley: 1981. [Google Scholar]
- Connolly ML. Analytical molecular surface calculation. J Appl Crystallogr. 1983a;16:548. [Google Scholar]
- Connolly ML. Solvent-accessible surfaces of proteins and nucleaic acids. Science. 1983b;221:709. doi: 10.1126/science.6879170. [DOI] [PubMed] [Google Scholar]
- Culver JN, Dawson WO, Plonk K, Stubbs G. Site-directed mutagenesis confirms the involvement of carboxylate groups in the disassembly of tobacco mosaic virus. Virology. 1995;206(1):724–30. doi: 10.1016/s0042-6822(95)80096-4. [DOI] [PubMed] [Google Scholar]
- Diaz-Avalos R, Caspar DL. Structure of the stacked disk aggregate of tobacco mosaic virus protein. Biophys J. 1998;74(1):595–603. doi: 10.1016/S0006-3495(98)77818-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Avalos R, Caspar DL. Hyperstable stacked-disk structure of tobacco mosaic virus protein: electron cryomicroscopy image reconstruction related to atomic models. J Mol Biol. 2000;297(1):67–72. doi: 10.1006/jmbi.1999.3481. [DOI] [PubMed] [Google Scholar]
- Dore I, Ruhlmann C, Oudet P, Cahoon M, Caspar DL, Van Regenmortel MH. Polarity of binding of monoclonal antibodies to tobacco mosaic virus rods and stacked disks. Virology. 1990;176(1):25–9. doi: 10.1016/0042-6822(90)90226-h. [DOI] [PubMed] [Google Scholar]
- Dore I, Weiss E, Altschuh D, Van Regenmortel MH. Visualization by electron microscopy of the location of tobacco mosaic virus epitopes reacting with monoclonal antibodies in enzyme immunoassay. Virology. 1988;162(2):279–89. doi: 10.1016/0042-6822(88)90467-9. [DOI] [PubMed] [Google Scholar]
- Fenczik CA, Padgett HS, Holt CA, Casper SJ, Beachy RN. Mutational analysis of the movement protein of odontoglossum ringspot virus to identify a host-range determinant. Mol Plant Microbe Interact. 1995;8(5):666–73. doi: 10.1094/mpmi-8-0666. [DOI] [PubMed] [Google Scholar]
- Holt CA, Hodgson RA, Coker FA, Beachy RN, Nelson RS. Characterization of the masked strain of tobacco mosaic virus: identification of the region responsible for symptom attenuation by analysis of an infectious cDNA clone. Mol Plant Microbe Interact. 1990;3(6):417–23. doi: 10.1094/mpmi-3-417. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Klug A. The tobacco mosaic virus particle: structure and assembly. Philos Trans R Soc Lond B Biol Sci. 1999;354(1383):531–5. doi: 10.1098/rstb.1999.0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouassi NK, Chen L, Sire C, Bangratz-Reyser M, Beachy RN, Fauquet CM, Brugidou C. Expression of rice yellow mottle virus coat protein enhances virus infection in transgenic plants. Arch Virol. 2006;151(11):2111–2122. doi: 10.1007/s00705-006-0802-3. [DOI] [PubMed] [Google Scholar]
- Lehto K, Bubrick P, Dawson WO. Time course of TMV 30K protein accumulation in intact leaves. Virology. 1990;174(1):290–3. doi: 10.1016/0042-6822(90)90077-5. [DOI] [PubMed] [Google Scholar]
- Namba K, Pattanayek R, Stubbs G. Visualization of protein-nucleic acid interactions in a virus. Refined structure of intact tobacco mosaic virus at 2.9 A resolution by X-ray fiber diffraction. J Mol Biol. 1989;208(2):307–25. doi: 10.1016/0022-2836(89)90391-4. [DOI] [PubMed] [Google Scholar]
- Raghavendra K, Kelly JA, Khairallah L, Schuster TM. Structure and function of disk aggregates of the coat protein of tobacco mosaic virus. Biochemistry. 1988;27(20):7583–8. doi: 10.1021/bi00420a002. [DOI] [PubMed] [Google Scholar]
- van Dun CM, Overduin B, van Vloten-Doting L, Bol JF. Transgenic tobacco expressing tobacco streak virus or mutated alfalfa mosaic virus coat protein does not cross-protect against alfalfa mosaic virus infection. Virology. 1988;164(2):383–9. doi: 10.1016/0042-6822(88)90551-x. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Emori Y, Ooshika I, Meshi T, Ohno T, Okada Y. Synthesis of TMV-specific RNAs and proteins at the early stage of infection in tobacco protoplasts: transient expression of the 30K protein and its mRNA. Virology. 1984;133(1):18–24. doi: 10.1016/0042-6822(84)90421-5. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Ohno T, Okada Y. Virus multiplication in tobacco protoplasts inoculated with tobacco mosaic virus RNA encapsulated in large unilamellar vesicle liposomes. Virology. 1982;120(2):478–480. doi: 10.1016/0042-6822(82)90048-4. [DOI] [PubMed] [Google Scholar]