

Summary
Drosophila expresses several muscle myosin isoforms from a single gene by alternatively splicing six of the nineteen exons. Here we investigate exon 7, which codes for a region in the upper 50k domain near the nucleotide binding pocket. This region is of interest because it is also the place where a large insert is found in myosin VI and where several cardiomyopathy mutations have been identified in human cardiac myosin. We expressed and purified chimeric muscle myosins from Drosophila, each varying at exon 7. Two chimerics exchanged the entire exon 7 domain between the indirect flight muscle (IFI, normally containing exon 7d) and embryonic (EMB, normally containing exon 7a) isoforms to create IFI-7a and EMB-7d. The second two chimerics replaced each half of the exon 7a domain in EMB with the corresponding portion of exon 7d to create EMB-7a/7d and EMB-7d/7a. Transient kinetic studies of the motor domain from these myosin isoforms revealed changes in several kinetic parameters between the IFI or EMB isoforms and the chimeras. Of significance were changes in nucleotide binding, which differed in the presence and absence of actin – consistent with a model in which the exon 7 domain is part of the communication pathway between the nucleotide and actin binding sites. Homology models of the structures suggest how the exon 7 domain might modulate this pathway.
Keywords: muscle, myosin, Drosophila, kinetics
Introduction
The conventional myosin II mechanoenzyme uses the energy from ATP hydrolysis to power muscle contraction. Myosin II, the main component of the muscle thick filament, is a hexameric protein formed by two myosin heavy chains (MHC), two essential light chains and two regulatory light chains. The catalytic myosin heads protrude from the surface of the thick filament and form strong transient interactions with the actin-containing thin filament. Summation of the interactions by many myosin heads with the thin filament generates force and motion that result in muscle fiber shortening.
Striated muscle fibers, which include skeletal and cardiac, exhibit a range of shortening velocities. A large body of evidence supports the hypothesis that variation in myosin heavy and light chain isoforms accounts for the diversity in contraction velocities of these fibers (see reviews: 1, 2). Early studies described a relationship between the steady-state ATPase activity of myosin isoforms and contraction velocity of muscle fibers expressing the same myosin isoforms suggesting that biochemical changes in MHC contribute to the observed diversity 3, 4. Numerous subsequent analyses established a correlation between myosin isoform kinetic properties and their actin filament in vitro motilities or in vivo muscle shortening velocities. Recent crystallographic and cryo-electron microscopy observations have revealed a conserved structural mechanism (see reviews: 5, 6) for these myosin isoforms further supporting the proposal that differences in the kinetic properties rather than mechanical properties of the myosin isoforms govern the contraction velocities of striated muscle fibers.
Recent investigations using an invertebrate model, Drosophila melanogaster, assessed whether a similar kinetic mechanism exists for muscle myosin isoforms in this organism. The studies focused on two of the fifteen known myosin isoforms 7-9, the embryonic body wall muscle isoform (EMB) and the indirect flight muscle isoform (IFI). Kinetic analyses of these isoforms using myosin or myosin subfragment 1 (S1) revealed less than a two-fold difference in nearly all measured steady-state and transient kinetic parameters 10, 11. These small kinetic differences are in clear contrast to the results from measuring in vitro actin filament velocities and the frequency of maximum power generation and the rate of tension redevelopment in fibers, which together show that IFI supports substantially faster actin filament and fiber contraction velocities than the EMB isoform 10-13. A recent study of the effects of ATP, ADP and Pi concentration on IFI and EMB fiber kinetics may shed some light on this lack of correlation. The results from this study show that the IFI and EMB-containing fibers respond very differently to ATP, ADP and Pi; the authors concluded that the rate-limiting step for the IFI isoform is Pi release while the EMB isoform rate-limiting step is the isomerization step prior to ADP release14.
To better understand the relationship between myosin kinetics and in vivo fiber shortening velocities in Drosophila, analyses have focused on the regions that vary between the IFI and EMB isoform. Myosin isoform diversity in Drosophila is generated by alternatively splicing six of the nineteen exons from a single Mhc gene 7, 9, 15; the IFI and EMB isoforms differ in all six regions. To address the function of each variable region, chimeric myosin isoforms were created by sequentially exchanging each variable domain between the IFI and EMB isoforms. Subsequent generation of transgenic flies expressing these chimeric isoforms allowed for assessment of the changes to muscle function in vivo and MHC function in vitro 10, 11, 13, 16-19.
The exon 7 domain (colored red in Fig. 1) forms the upper lip of the ATP-binding site (residues 319 – 335) and a portion of the adjacent external surface (residues 301 - 318). IFI and EMB spliced transcripts include exon 7d and 7a, respectively, which are two of four isovariants in this exon cassette. Previously, exons 7d and 7a were exchanged between the IFI and EMB transcripts to produce chimeric myosin isoforms IFI-7a and EMB-7d. Initial in vitro and in vivo results suggested the exon 7 domain modulates myosin isoform function by affecting the lifetime of a state where myosin is detached or weakly attached to actin 16. A second study of the indirect flight muscle (IFM) mechanical properties of IFI-7a and EMB-7d transgenic flies showed that the exon 7d domain also affects the duration of a strongly bound state that influences fiber speed and flight performance 17.
Figure 1.
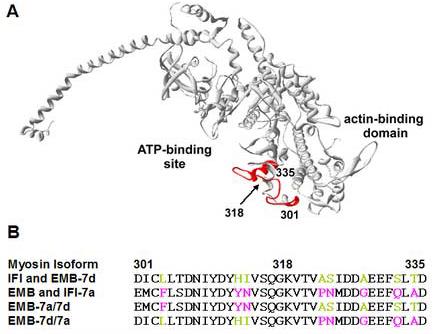
Location and amino acid sequence of the Drosophila exon 7 domain
A) The chicken skeletal S1 crystal structure minus the light chains (2MYS.pdb) is shown with residues 301-335, which correspond to the Drosophila exon 7 domain, colored red; the location of residues 301, 318 and 335 are indicated. To create EMB-7a/7d and EMB-7d/7a myosin isoforms, the exon 7 sequence was split in half: 1) 301-318 (surface domain) and 2) 319-335 (lip domain). In EMB-7d/7a, the 7a surface domain was replaced by 7d and in EMB-7a/7d the 7a lip domain was replaced by 7d. B) The sequences of exon 7 in the IFI, EMB and four chimeric isoforms are aligned; nonconserved residues are colored according to their origin (IFI/exon 7d are green and EMB/exon 7a are magenta).
Here we used transient kinetic analyses to perform a detailed investigation of the modulatory role of the exon 7 domain on Drosophila myosin isoform biochemical kinetics. Four chimeric isoforms were used; two were the IFI-7a and EMB-7d isoforms previously created 16. The other two isoforms were designed to independently test the influence of the residues that vary between the exon 7a and 7d domains on EMB isoform kinetic properties. We created these by replacing separately each sub-domain (lip or surface domain) of exon 7a in EMB by the same region of exon 7d, thus producing the EMB-7d/7a (includes 7d surface domain residues 301-318) and EMB-7a/7d (includes 7d lip domain residues 319-335) isoforms (Fig. 1B). Using these four isoforms, we measured several biochemical transitions in the myosin cross-bridge cycle.
We found that substitution of the entire exon 7a domain in the EMB isoform with exon 7d (EMB-7d) slightly reduced the ATP-induced dissociation rate of acto-S1 and significantly enhanced the rate of coumarin-ADP (eda-deac ADP) release but had no influence on the ADP affinity for acto-S1. Exchanging the entire exon 7d domain of IFI with exon 7a (IFI-7a) reduced the ATP-induced dissociation rate and the rate of eda-deac ADP release from IFI but enhanced the ADP affinity for acto-S1. When exchanging either the exon 7d lip or surface domain, we observed no change in the steady-state ATPase rate as seen with EMB-7d, but there were changes in both the eda-deac ADP release rate from S1 and in the affinity of ADP for acto-S1. Collectively, the kinetic differences measured in these four isoforms indicate that exchanging or splitting the exon 7 domain produced novel biochemical interactions governing Drosophila myosin kinetic transitions. Homology models derived for the IFI, EMB and four chimeric isoforms suggest a structural mechanism by which sequence changes in the exon 7 domain influence Drosophila myosin kinetics. Together the kinetic perturbations and modeling data reveal the exon 7 domain is part of the communication pathway between the nucleotide and actin-binding sites.
Results
Generation of transgenic flies
To address which nonconserved residues between exon 7a and 7d contribute to the functional differences in myosin isoforms EMB and EMB-7d 16, we generated two constructs, EMB-7a/7d and EMB-7d/7a. Both chimeric myosin isoforms substituted exon 7d residues in place of the exon 7a residues normally present in the EMB isoform (Fig. 1). In EMB-7a/7d residues 319-335 (lip sub-domain) were exchanged and in EMB-7d/7a residues 301-318 (surface sub-domain) were exchanged. To mediate germline transformation of Drosophila, each P element construct was injected separately into yw embryos along with a helper plasmid, Δ2-3, containing a transposase source required for insertion of the P element into chromosomal DNA 20, 21. Transformed flies with appropriate insertion events were crossed into a Mhc10 background. Mhc10 is null for MHC in the indirect flight and jump muscles 22. This genetic background eliminates myosin isoform expression only in these muscle types and allows for assessment of in vivo function and purification of transgenic myosin without contamination from endogenous myosin. These techniques yielded three independent fly lines for the EMB-7a/7d isoform and five independent fly lines for the EMB-7d/7a isoform. A single fly line expressing the greatest amount of MHC for each mutant was used for myosin purification. Each myosin preparation yielded 300-400 μg from approximately 150-180 flies.
Steady-state Mg2+ATPase assays of chimeric myosin isoforms
A previous study showed an increase in the basal and actin-activated Mg2+ATPase activity when the exon 7d domain was exchanged for exon 7a in the EMB background (Table 1, EMB-7d isoform) 16. We determined the Mg2+ATPase activity for the chimeric myosin isoforms EMB-7a/7d and EMB-7d/7a. The basal and maximal (Vmax) actin-activated activities for both isoforms were similar to the EMB isoform and did not show any increase in activity as seen with the EMB-7d isoform (Fig. 2 and Table 1). The elevation of the ATPase activity therefore requires the whole exon 7 exchange, i.e., neither half of the exon can produce the effect on its own. However, the KM for actin with either isoform showed a 3-4 fold decrease relative to the EMB isoform and a 1.5-2 fold decrease relative to the EMB-7d isoform. The decrease of the KM is not simple to interpret but could reflect an increased affinity of these myosin isoforms for actin.
Table 1.
Steady-state kinetic parameters for the exon 7 domain chimeric myosin isoforms
| Myosin Isoform | EMBa | EMB-7da | EMB-7a/7d | EMB-7d/7a |
|---|---|---|---|---|
| Basal ATPase rate (s−1 head−1) |
0.07 ± 0.01 | 0.13 ± 0.02 | 0.06b ± 0.01 | 0.08b ± 0.02 |
| Vmax (s−1 head−1) | 0.7 ± 0.05 (5) | 0.91 ± 0.04 (6) | 0.6 ± 0.02 (5) | 0.6 ± 0.01 (5) |
| KM actin (μM) | 0.8 ± 0.2 | 0.5 ± 0.09 | 0.3 ± 0.04c | 0.2 ± 0.02c |
Values are the mean ± S.E. with parentheses indicating the number of preparations.
p < 0.05 determined by Student's t-test as compared to EMB-7d
p < 0.05 determined by Z-test on the parameter fits of Vmax and KM (EMB-7a/7d and EMB-7d/7a vs. EMB-7d or EMB)
Figure 2.
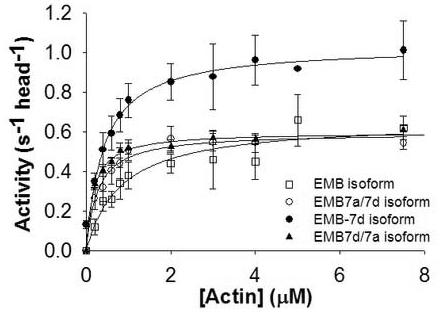
Actin-activated Mg2+ATPase activity of EMB-7a/7d and EMB-7d/7a isoforms
The maximal actin-activated ATPase activities for the exon 7 domain chimeric myosin isoforms (closed triangles: EMB-7d/7a, open circles: EMB-7a/7d) show no difference compared to the EMB isoform (open squares) but are decreased relative to the EMB-7d isoform (closed circles). Data were collected and fit with V = Vmax [actin] / (KM + [actin]) to determine Vmax and KM (Table 1). EMB data are from Littlefield et al. 10 and EMB-7d data are from Miller et al 16.
Production of Drosophila S1 isoforms
In order to measure the transient kinetic parameters, each myosin isoform was digested with α-chymotrypsin to produce S1, the catalytic globular domain. Chymotryptic digestion eliminates the α-helical rod domain and the regulatory light chain, but leaves the essential light chain intact. The S1 is soluble at low salt and produces minimal aggregation. The samples contain relatively pure S1 (∼100-200 μg) with minor contamination resulting from over-digestion of the myosin 11.
Dissociation of coumarin ADP (eda-deac ADP) from S1 (k−D)
To estimate the rate constant of ADP dissociation from S1 in the absence of actin, the change in fluorescence of a coumarin labelled ADP (eda-deac ADP42) was measured upon displacement of eda-deac ADP by ATP-binding to S1 23. Previously it was shown that this coumarin-labeled analog has very similar kinetic properties compared to ADP. When using edadeac ADP the ADP release rate (k−D) was unchanged for rabbit fast-skeletal myosin S1, the Dictyostelium myosin head fragment (M765-1R) and rat EDL myosin S1 23.
A single laser flash released 15-20 μM ATP from cATP (100 μM) and the fluorescence change resulting from eda-deac ADP release was well described by a single exponential function. Exchange of the full exon 7a or exon 7d domain into the IFI or EMB isoform, respectively, resulted in an alteration of the rate constant for eda-deac ADP release (k−D) in the direction of the donor domain; the eda-deac ADP release rate constant from IFI-7a (4.7 ± 0.8 s−1) and EMB-7d (4.3 ± 0.7 s−1) were very similar and the values lie between those of IFI (7.5 ± 1.3 s−1) and EMB S1 (1.8 ± 0.4 s−1; Table 2 and Fig. 3).
Table 2.
Transient kinetic parameters measured for the exon 7 chimeric S1 isoforms Values are mean ± S.D. based on a minimum of 3 preparations. K1k+2 is the second order rate constant for ATP-induced dissociation of acto-S1. KD and KAD are dissociation equilibrium constants determined by division of the dissociation rate constant by the association rate constant (e.g. K3 = k−3 / k+3). k−D and k−AD are the ADP dissociation rate constants in the absence and presence of actin, respectively. KAD / KD is the thermodynamic coupling constant describing the relationship between actin and ADP affinities 28.
| S1 Isoform |
IFI† | IFI-7a | EMB† | EMB-7d | EMB- 7a/7d |
EMB- 7d/7a |
|---|---|---|---|---|---|---|
|
K1k+2 (106 M−1 s−1) |
0.75 ± 0.08 | 0.33 ± 0.07 | 0.91 ± 0.13 | 0.64 ± 0.05 | 0.67 ± 0.18 | 0.66 ± 0.14 |
| k−D (s−1) | 7.5 ± 1.3d | 4.7 ± 0.8c,d | 1.8 ± 0.4c | 4.3 ± 0.7c,d | 2.6± 0.04c,d | 6.1 ± 0.08d |
| KD (μM)a | 7.5 | 4.7 | 1.8 | 4.3 | 2.6 | 6.1 |
| KAD (μM) | 409 ± 26d | 239 ± 52c,d | 587 ± 48c | 596 ± 39c | 191 ± 59c,d | 220 ± 33c,d |
| k−AD (s−1)b | 4090 | 2390 | 5870 | 5960 | 1910 | 2200 |
| KAD / KD | 55 | 51 | 326 | 139 | 73 | 36 |
Data are estimated from k−D assuming an association rate constant of 106 M−1 s−1.
Data are estimated from KAD assuming an association rate constant of 107 M−1 s−1.
Data are from Miller et al. 11
p < 0.05 determined by Student's t-test as compared to IFI
p < 0.05 determined by Student's t-test as compared to EMB
Figure 3.

Rate of eda-deac ADP dissociation (k−D) from Drosophila S1 isoforms
The rate constant for ADP dissociation (k−D) from S1 in the absence of actin was determined. Addition of ATP (15 μM) from cagedATP (100 μM) by a single laser flash displaced the eda-deac ADP bound to S1. The change in eda-deac ADP fluorescence upon release from S1 was measured and fit with a single exponential to determine k−D. Exchange of either the exon 7a or 7d domain resulted in a modulation of the eda-deac ADP release rate. The dissociation rate for EMB-7d (A: 4.3 s−1) is faster than EMB (A: 1.8 s−1) while that of IFI-7a (B: 4.7 s−1) is slower than IFI (7.5 s−1). The observed rate constant of eda-deac ADP dissociation (k−D) from EMB-7a/7d (C) and EMB-7d/7a (D) S1 gave mean values of 6.1 s−1 and 2.6 s−1, respectively. The conditions were: 20 mM MOPS, 30 mM KCl, 5 mM MgCl2, 10 mM DTT, pH 7.
The eda-deac ADP release rate constants for EMB-7a/7d and EMB-7d/7a S1 were 2.6 ± 0.4 s−1 and 6.1 ± 0.8 s−1, respectively (Table 2). Including the IFI exon 7d lip domain (EMB-7a/7d) results in a rate close to the IFI S1 rate (7.5 ± 1.3 s−1). On the other hand, including the exon 7d surface domain (EMB-7d/7a) yields a rate closer to the EMB S1 rate 11. With EMB-7a/7d the eda-deac ADP fluorescence decreased on displacement, in the same direction as for the IFI, EMB and EMB-7d isoforms (Fig. 3A and C). There was no correlation between the direction of the fluorescence change and the affinity of the analogue for S1. Surprisingly, the results with EMB-7d/7a showed a fluorescence increase as did IFI-7a (Fig. 3B and D). Similar variation in the direction of fluorescence changes has been observed previously with vertebrate and Dictyostelium S1 isoforms 23 and reflects differences in the local environment of the protein bound fluorophore.
Determination of the ATP-induced dissociation rate (K1k+2) of acto-S1
To determine the second order rate constant governing the dissociation of acto-S1 by ATP (K1k+2: see Scheme 1), 1 μM actin and 1-3 μM S1 were preincubated and then dissociated by ATP released from cATP. A single sample was subjected to a series of laser flashes decreasing in intensity to release varying concentrations of ATP. The rate of change in light scattering (kobs) was recorded for each laser flash and could be well described by a single exponential at each ATP concentration (data not shown). The linear slope of a graph of kobs vs. ATP concentration determines the apparent second-order rate constant for the dissociation of acto-S1 by ATP (Fig. 4). Both the values for IFI-7a acto-S1 (0.33 ± 0.07 μM−1 s−1) and EMB-7d acto-S1 (0.64 ± 0.05 μM−1 s−1) showed decreases relative to the previously reported values for IFI (0.75 ± 0.08 μM−1 s−1) and EMB acto-S1 (0.91 ± 0.13 μM−1 s−1), respectively11 (Table 2). The kinetic rates for the ATP-induced dissociation of EMB-7a/7d (0.67 ± 0.18 μM−1 s−1) and EMB-7d/7a (0.66 ± 0.14 μM−1 s−1) acto-S1 (Fig. 4B) were similar to the dissociation rate observed for EMB-7d, but lower than the dissociation rate for EMB acto-S1 (Table 2).
Scheme 1.

The interaction of S1 with actin, ATP and ADP. M, A, T and D symbolize S1, actin, ATP and ADP, respectively.
Figure 4.
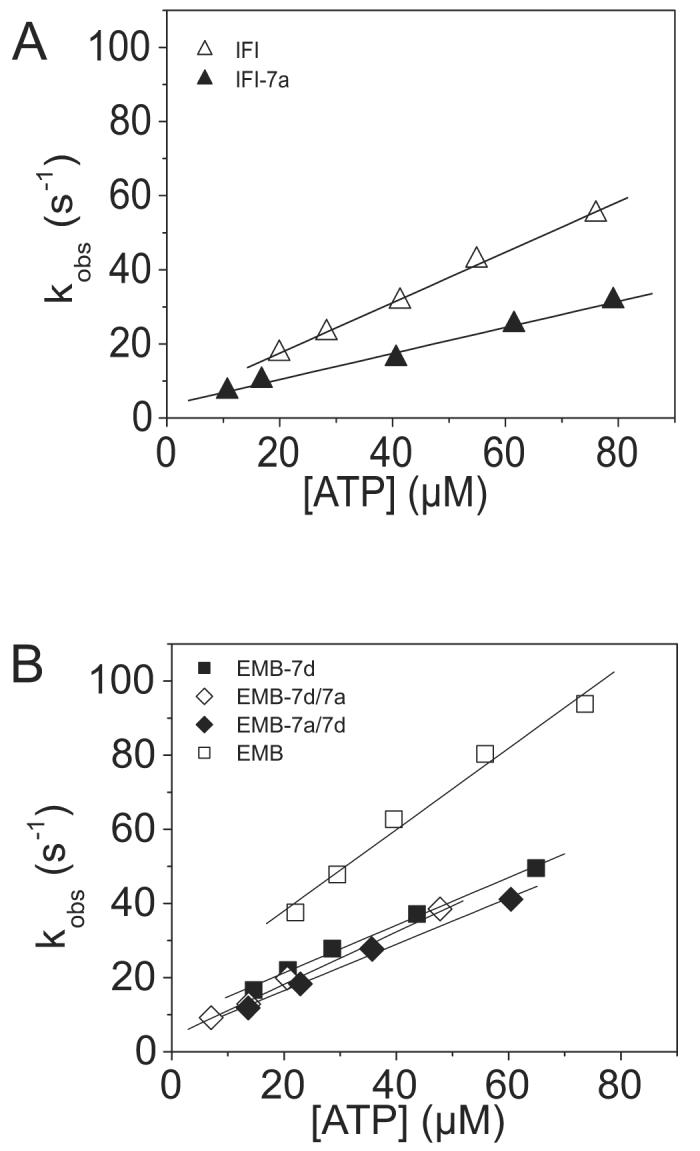
Kinetic determination of the ATP-induced dissociation rate (K1k+2) of the S1 isoforms from actin
The second order rate constant for the dissociation of S1 from actin is determined from a linear fit to the plot of the kobs vs. [ATP]. Light scattering transients monitoring dissociation of the acto-S1 complex (data not shown) were fitted with a single exponential at each ATP concentration in order to determine the kobs. A) The linear fits yielded values of 0.33 ± 0.07 106 M−1 s−1 for IFI-7a (filled triangles) as compared to as compared to 0.75 ± 0.08 106 M−1 s−1 for IFI (open triangles). B) The linear fits yielded mean values of 0.67 ± 0.18 106 M−1 s−1 for EMB-7a/7d S1 (filled triangles) and 0.66 ± 0.14 106 M−1 s−1 for EMB-7d/7a S1 (open triangles) as compared to 0.91 ± 0.13 106 M−1 s−1 for EMB S1 (empty squares) and 0.64 ± 0.05 106 M−1 s−1 for EMB-7d S1 (closed squares).
Determination of ADP affinity (KAD) for acto-S1
The equilibrium dissociation constant for ADP (KAD) from acto-S1 provides a measure of the ADP affinity for S1 in the presence of actin. Competition between ADP and ATP binding to acto-S1 allows the measurement of KAD; for details see Weiss et al. 24. To determine the ADP affinity in a single S1 sample, the ATP-induced rate of acto-S1 dissociation was measured repeatedly as the concentration of ADP increased. The change in the light scattering measured for each ADP concentration was fit with a single exponential to determine kobs. The KAD values for the isoforms (except for EMB-7a/7d S1, see below) determined from the applied hyperbolic fits (equation 1) to the graph of krel vs. ADP concentration (Fig. 5) are presented in Table 2. The KAD for EMB-7d is not different from EMB 11 but the IFI-7a KAD is 1.5-2 fold lower than that of IFI 11. Unexpectedly, the KAD for EMB-7d/7a S1 showed a 3-fold decrease (tighter ADP affinity) relative to EMB-7d and EMB S1 KAD values and a 2-fold decrease relative to the KAD for IFI S1 (409 ± 26 μM) 11. The significance of these KAD changes was confirmed by running a Student's t-test for all the mutants, as compared to IFI and EMB (see Table 2). Interestingly, the plot of the krel vs. ADP concentration for EMB-7a/7d exhibited a sigmoidal shape; this behavior was consistent in each of the preparations for this isoform. As a result, the curve could not be fit with equation 1; instead an apparent KAD was determined by calculating the ADP concentration at which 50% inhibition was observed. This yielded a value of 191 ± 59 μM for EMB-7a/7d actoS1 ADP affinity. Again the KAD for this isoform was 2-3 fold lower (tighter acto-S1 ADP affinity) than the values determined for the IFI, EMB and EMB-7d S1 isoforms.
Figure 5.

The affinity of ADP (KAD) for acto-S1
The dissociation of acto-S1 was induced by ATP in the presence of ADP ranging from 0-1500 μM. The light scattering traces were fitted with single exponentials to determine the kobs. Hyperbolic plots of the kobs vs. ADP concentration were fitted with an equation derived from Scheme 1 (kobs = K1k+2([ATP]/(1 + [ADP]/KAD)) to determine KAD. Shown are the relative kobs (krel) vs. ADP concentration plots for easier comparison. A) The fits yielded a value of 607 ± 44 μM for EMB-7d (empty squares) as compared to 587 ± 38 μM for EMB (filled squares). B) The fits yielded values of 409 ± 26 μM (open circles) for IFI and 239 ± 32 μM for IFI-7a (closed circles). The KAD for EMB-7a/7d (C) and EMB-7d/7a (D) S1 isoforms were measured with the following alterations. The dissociation of acto-S1 was calculated in the presence of 0 – 400 μM ADP and the KAD for EMB-7a/7d S1 was determined to be the ADP concentration at 50% inhibition. All preparations for EMB-7a/7d produced the sigmoidal-shaped curve observed here. The fits yielded values of 191 ± 59 μM for EMB-7a/7d and 220 ± 33 μM for EMB-7d/7a.
Homology Models
Both Drosophila IFI and EMB myosin head sequences showed significant sequence identity with various other myosin super family members. A sequence identity of 60 % (± 1%) was found when aligning IFI and EMB myosin head sequences with class II scallop myosin, thus enabling us to build well-resolved homology models. SWISS-MODEL generates reliable models for target-template pairs sharing > 40 % sequence identity. When the sequence identity is as high as 60% then the quality of target-template pairs is generally very good, with the majority of the models deviating between 0.5 – 1.0 Å from the template 25. Crystal structures are available of the various states of scallop myosin during the cross-bridge cycle and these were chosen to generate 3D homology models of the IFI and EMB myosin heads and also of the chimeras obtained after swapping exon 7 completely (IFI-7a and EMB-7d) or after swapping a part of exon 7 (EMB-7a/7d and EMB-7d/7a). Three of the scallop myosin structures used here contain a nucleotide in the binding pocket and they represent various conformational states of myosin during the cross-bridge cycle: the actin-detached state (ExPDB 1kk8) contains ADP-BeFx, the pre-powerstroke state (ExPDB 1qvi) contains ADP-VO4 and a novel conformation contains partially-bound ADPSO4 (ExPDB 1s5g). The other scallop myosin structure does not have a nucleotide in the binding pocket (ExPDB 1sr6) and this one represents the near-rigor state of myosin. The generated models have Ramachandran Z-scores of −0.475 (± 0.182) and average packing Z-scores of −1.069 (±0.064), which were within the expected range for well-refined structures. The overall topology of the models was similar to that of the scallop myosin II templates (average rmsd of the backbone is 0.45 ± 0.25 Å). Overlaying the two wild-type models for Drosophila IFI and EMB and their respective exon 7 chimeras revealed a high degree of similarity in backbone structure, apart from the region in the converter area (exon 11). The kinetic properties of this region have been described in detail elsewhere 11 and therefore will not be discussed here.
General description of the exon 7 area
In the exon 7 area, the rmsd of the myosin backbone between IFI and EMB was < 0.05 Å and followed the same topology for both isoforms. For descriptive purposes, we use the same nomenclature for secondary structure elements as described previously 26. A small helix (helix J) at the N-terminus of the exon 7 domain is followed by a longer loop and then another longer helix (helix K: 327-341). The N-terminus (327-335) of helix K is the C-terminal part of the exon 7 area. Figure 6 shows the structure of the exon 7 region (colored red) for both IFI and EMB using the scallop structure, ExPDB 1qvi, as a template. The residues that vary between the IFI and EMB isoform are indicated in Figure 1B. In the N-terminal part of exon 7, residue 304 is located before helix J and residues 313 and 314 immediately after this helix. The loop region between the two helices contains variable residues 323 and 324 while helix K in the C-terminal part of exon 7 has three variable sites: 328, 332 and 334.
Figure 6.

Interactions of the exon 7 domain with nearby structural elements
Homology model of IFI in the pre-power-stroke state (ExPDB 1qvi: bound nucleotide is ADP-VO4), showing the exon 7 area (residues 301-335 in red), the relay area (magenta), switch I (green) and helix L (yellow). The highly conserved salt-bridge between exon 7 residue D327 and relay area residue R283 is indicated. Note the proximity of switch I toward the relay area. Interactions from the non-conserved exon 7 residues Y313 and D327 toward helix L (yellow) are also shown.
Exon 7 area: highly conserved interactions.
Sequence alignments (Fig. 7) showed that only two exon 7 residues are highly conserved: G318 and D327. Inspection of the homology models revealed that D327, located at the N-terminus of helix K, is involved in a strong salt-bridge toward R283 (Fig. 6). This salt-bridge was present in all homology models and has also been found in crystal structures of other myosins, including skeletal and smooth chicken myosin II (ExPDB 2MYS and 1BR4) and Dictyostelium myosin II (1FMV), in addition to members of other myosin classes, such as myosin I (myosin-1E, 1LKX), myosin V (1OE9) and myosin VI (2BKH). Sequence alignments also showed that R283 and its neighboring residues are highly conserved. These residues interact with the well conserved switch 1 region, in particular K236, or with residues that are located around G318 in the exon 7 domain. Together, these residues around R283 form the ‘relay area’ and provide a mechanism by which the exon 7 domain is able to ‘sense’ the conformation of switch 1. The location of G318 in the middle of the loop between helix J and K may function to orient the interacting residues properly toward the relay area (Fig. 6). No variable residues in exon 7 are involved in direct contacts with either switch I or this ‘relay area’, indicating this switch I sensitivity is a conserved feature of both isoforms.
Figure 7.

Myosin sequence alignments
Sequence alignment of various myosins showing exon 7 (in the boxed area), together with loop 1, switch I and the relay area. “*” means that the residues are identical in all sequences in the alignment.“:” represents conserved substitutions, whereas “.” indicates semi-conserved substitutions. Only two residues in exon 7 are 100 % conserved (G318 and D327) of which D327 forms a salt-bridge towards the highly conserved R283 (scallop numbering). Note that myosin VI has an insert in the middle of the exon 7 domain. The corresponding cardiomyopathy sites in the exon 7 domain and helix L are indicated with a ▼.
Exon 7 area: Non-conserved interactions
Careful inspection of the exon 7 area in the homology models revealed close interactions between the ribose oxygen and the side-chain of the non-conserved residue 324 in all three states containing a nucleotide. Measurements of the hydrogen-bond distances between the H-donor (either asparagine in IFI or serine in EMB) and the H-acceptor of the nucleotide indicated that the asparagine (N324) side-chain is closer to the ribose moiety than the serine (S324) side-chain. This difference was most pronounced in the pre-powerstroke state, with the N324 hydrogen donor almost 1 Å closer to the ribose oxygen than the S324 hydrogen donor (Fig. 8). Comparison of the conformations with a partially-bound nucleotide showed the N324 H-donor is about 0.5 Å closer than the S324 side-chain (not shown). Lastly, in the actin-detached state only the N324 side-chain was able to form a hydrogen bond toward the nucleotide (not shown). All these observations indicate that an asparagine at position 324 allows a greater number of direct contacts with the bound nucleotide than a serine.
Figure 8.

Interactions of residue 324 with the nucleotide
Close-up of the interactions between exon 7 residue 324 and the bound nucleotide in the prepowerstroke state. Inspection of the homology models indicates that the H-donor of the asparagine side-chain found in the exon 7d region (A) is almost 1 Å closer to the nucleotide oxygen than the H-donor of the serine side-chain found in 7a (B).
Other non-conserved residues in the exon 7 area were found to make contacts with nearby regions, in particular helix L (yellow in Fig. 6: 345-361), which follows the exon 7 region (Fig. 6). The N-terminus of the exon 7 area makes contacts with the C-terminus of helix L whereas the C-terminus of the exon 7 region makes contacts with the N-terminus of helix L. It is worth noting that the variable residues at position 313 and 314 (H313I314 in EMB or Y313N314 in IFI) are interacting with helix L (Fig. 6) and that these contacts vary between myosin conformational states. In the near-rigor state, i.e. the myosin conformational state without a nucleotide in the binding pocket (ExPDB 1sr6), residues 313 and 314 make very weak contacts toward residues 355-360 of helix L and only the side-chain of N314 is involved in the formation of hydrogen bonds towards helix L. In the three states containing a nucleotide (actin-detached, pre-powerstroke and a novel conformation with the partially-bound ADP-SO4), however, the contacts between helix L and these residues become much stronger thereby allowing the side-chain hydroxyl of Y313 to form a hydrogen bond toward helix L (K365). These results indicate that the exon 7d domain, which contains both Y313 and N314 (Fig. 1B), is able to form more contacts toward helix L than does the exon 7a domain. In addition, the homology models that used the actin-detached state (ExPDB 1kk8) and the pre-powerstroke state (ExPDB 1qvi) showed that the variable residue at position 304 (L/F) interacts with residue 357 on helix L (not shown).
Discussion
Previously, exchange of the exon 7 domain between the IFI and EMB isoforms was found to increase steady-state Mg2+ATPase rates in the resulting IFI-7a and EMB-7d chimeric isoforms, but not to alter actin filament velocities generated by these isoforms compared to IFI and EMB, respectively. Expression of the IFI-7a and EMB-7d chimeric isoforms in the IFM (in place of the IFI) resulted in minimal effects on locomotion when compared to IFI and EMB, respectively. The EMB-7d transgenic flies showed improved stability of the muscle structure (uniformity of length and packing of actin and myosin filaments in the muscle fiber) compared to EMB transgenic flies, which show significant deterioration of muscle structure within a few days after eclosion 27. Collectively, these data led to the conclusion that the exon 7 domain modulates kinetic transitions that govern the attachment of myosin to actin 16. A follow up study investigated the influence of the exon 7 region on IFM fiber mechanical properties and found the IFI-7a exchange produced minimal alterations to IFM function 17. The only observed difference was a slight decrement in flight ability at 15°C. Exchanging EMB-7d, however, produced significant increases in the frequency of maximum power generation and the rate of tension redevelopment and higher active stiffness in these fibers compared to the EMB fibers. In order to produce these effects, myosin must be strongly bound to actin. Therefore the EMB-7d exchange must additionally influence the lifetime of a strongly bound state of the EMB cross-bridge cycle that does not limit actin filament velocity 17. In this study we used a detailed kinetic analysis to understand how sequence variation in the exon 7 domain influenced biochemical transitions in Drosophila myosin isoforms to clarify the modulatory role of this domain.
Steady-state kinetics of Drosophila exon 7 chimeric isoforms
We sought to determine if either of two distinct areas of the IFI exon 7d domain might be responsible for driving the kinetic properties of myosin isoforms. Interestingly, exchanging only a part of the exon 7d domain in creating the EMB-7a/7d and EMB-7d/7a isoforms had little effect on the maximal actin-activated Mg2+ATPase activity but decreased the KM 2-3 fold as compared to the EMB isoform. This was in contrast to the EMB-7d isoform, which showed a 30% increase in maximal actin-activated ATPase activity but no effect on the KM as compared to the EMB isoform 10, 16. Overall, these data suggest that variable residues in either half of the exon 7d domain mediate an increase in actin affinity but both halves of the exon 7d domain coordinate to produce the increase in ATPase activity observed with the EMB-7d isoform.
Transient kinetics of Drosophila exon 7 chimeric isoforms
In general, the differences observed in the transient kinetic experiments of EMB-7d, EMB-7a/7d and EMB-7d/7a as compared to the IFI and EMB isoforms indicate that the interactions with the nucleotide made by the exon 7d domain are different from those for the exon 7a domain. Previously, a 4-fold difference between the rate constants of eda-deac ADP release from IFI and EMB in the absence of actin was observed 11. Presently, a three-fold change for the EMB-7d and EMB-7d/7a isoforms resulted from altering only exon 7. EMB-7d is more like IFI than EMB in this parameter and EMB-7d/7a is even more similar to IFI. These results suggest that the other variable domains do not play a major role in defining this parameter and that the surface domain of exon 7 has the most influence on ADP release in the absence of actin. The exon 7 domain might influence the interaction of nucleotide with the motor domain directly or indirectly. Homology modeling indicated that N324 (present in the lip of exon 7d in the IFI isoform) can make a shorter hydrogen bond to the oxygen of the ADP than can S324 in exon 7a of the EMB isoform. A simple argument therefore predicts a more tightly bound ADP, and a lower release rate constant (k−D) in IFI than in EMB. However, the result was opposite to what was predicted; the IFI ADP release rate is faster than the EMB isoform. Thus, the role of the exon 7 domain in ADP binding is more indirect than via a single hydrogen bond between the exon 7 domain and the nucleotide.
The effect of the exon 7 domain on eda-deac ADP affinity suggested that this domain could also affect ATP and ADP binding in the presence of actin. In the present work, we observed that the apparent second order rate constant of ATP binding (K1k+2) decreased by 30% when all or part of exon 7a was replaced by 7d in the EMB isoform and when 7a replaced 7d in the IFI isoform. In addition, the affinity of acto-S1 for ADP (KAD) was reduced more than 2-fold for either the lip or surface domain exchanges and the IFI-7a isoform, but was unchanged for EMB-7d. Together these results confirm the ability of the exon 7 domain to modulate nucleotide binding parameters in the presence of actin and may help to explain the differences in nucleotide affinity recently observed between the IFI and EMB-containing fibers 14.
Additional observations support the conclusion that the interactions between the exon 7 domain and the nucleotide differ depending on the presence of exon 7a or 7d. The plot of krel vs. ADP concentration for the EMB-7a/7d S1 isoform exhibited an unusual sigmoidal shape (Fig. 5C), rather than the characteristic hyperbolic shape observed with all other S1 isoforms. For IFI-7a and EMB-7d/7a S1 (Fig. 3B and D), we also observed a difference in the direction of the coumarin fluorescence signal relative to all other S1 isoforms; these results probably reflect local structural differences in the probe environment.
Myosin is capable of binding both actin and ADP simultaneously; the ratio KAD/KD (Table 3) or KDA/KA describes the effect of actin on ADP affinity or the effect of ADP on actin affinity, respectively, and is termed the thermodynamic coupling constant 28. Fast skeletal myosins exhibit a high coupling constant (∼50) indicating that the presence of actin significantly weakens ADP affinity and vice versa. The coupling for IFI-7a (51), EMB-7d (139), EMB-7a/7d (73) and EMB-7d/7a (36) S1 suggests that the exon 7 domain interactions with the nucleotide in these isoforms are more similar to IFI (55) than to EMB (326). For EMB-7d S1 the alteration in KD but not KAD reveals the exon 7d domain has more influence on interactions in the nucleotide-binding pocket in the absence of actin than in the presence of actin. On the other hand, the changes in both KAD and KD for the IFI-7a, EMB-7a/7d and EMB-7d/7a S1 indicate that these exon 7 regions interact differently with the nucleotide, thus modulating the response of myosin to the presence or absence of actin.
Molecular mechanism by which the exon 7 region modulates Drosophila myosin isoform function
The location of the exon 7 domain places certain residues in favorable positions for potential direct interactions with the nucleotide (Fig. 8). Several studies have established that the upper 50 kD domain, which includes the exon 7 area, moves as myosin transits through the biochemical cycle 29-32. At a minimum, these results show that the domain is shifting position in response to actin and/or the nucleotide state. Yet, only a single study of pre-powerstroke and near rigor scallop myosin crystal structures implicates direct interaction of an exon 7 domain residue with the nucleotide 33. This study observed an interaction between N321 (S324 in Drosophila exon 7a and N324 in exon 7d regions: Fig. 8) and the diphosphate moiety of ADP that strengthened as myosin transitioned from the pre-powerstroke to near rigor state. None of the other surrounding residues appeared to form any direct interactions. In Drosophila myosin isoforms, the variation at this position in exon 7a (a serine) versus exon 7d (an asparagine) suggests a possible direct mechanism for modulation of the kinetics observed here and in previous studies 10-13, 16-19. Examination of the remaining nonconserved residues (Fig. 1B) between exon 7a and exon 7d uncovers no obvious changes in charge (unless the histidine at 313 is protonated), a few changes in polarity (for instance, I/N314 and T/A334), a change in flexibility (A/P323) and a change in size (A/G328). If the direct interaction between the nucleotide and residue 324 exists in Drosophila myosin isoforms, variation in the other nonconserved residues may modulate the strength of this interaction and affect kinetic transitions.
The idea discussed above is supported by the homology models, which indicate that the exon 7 domain could act as part of the signal relay pathway between the nucleotide binding site and the actin binding site through the following mechanism. Residue 324 (‘lip domain’), which varies between IFI and EMB, makes direct contact with the nucleotide and is able to ‘sense’ the state of switch I via a highly conserved salt-bridge between D327 and R283 (and contacts around R283, the ‘relay’ area) toward switch I. Although one should interpret the measured distances with care, all the homology models which contain N324 show that the side-chain of this residue is consistently closer to the nucleotide than the models which have a serine at that position. Additional variable residues at the N-terminus of the exon 7 domain (304, 313 and 314) are subsequently involved in relaying the signal toward helix L. The exon 7d domain makes more contacts toward both the nucleotide and helix L than exon 7a, which might result in a more efficient signal relay and faster ADP release in the absence of actin. We observed that introduction of 7a into IFI reduces ADP release rates significantly (Table 3), whereas introducing 7d into EMB enhances ADP release rates. Comparing EMB-7d/7a, which has an increased ADP release rate, to EMB-7a/7d, which has a k−D close to EMB, suggests the N-terminus of the exon 7 domain may have more effect on the efficiency of signal relay than the C-terminus. This result is confirmed by the homology models, which indicate that the variable residues at the N-terminus of exon 7 are involved in relaying the signal towards helix L. In the presence of actin, the picture is more complex because other variable domains are involved, in particular those encoded by exons 3 and 9. One should also keep in mind that these homology models are based on myosin crystal structures without actin present, since actomyosin structures are not yet available. Apart from EMB-7d, all the other chimeric S1 isoforms show much higher ADP affinity in the presence of actin as compared to EMB (Table 2), indicating that the communication pathway between the actin-binding site and the nucleotide-binding site is altered in these isoforms.
The observation that myosin VI has two extra inserts 34, compared to other myosins, is also of interest because insert 1 is in the middle of the exon 7 area (Fig. 7). Kinetic measurements suggested that this insert has a crucial function in slowing ADP release and ATP-induced actomyosin dissociation 34. The myosin VI structure reveals that the presence of insert 1 does not alter the conformation of switch I relative to the upper 50 kDa domain. However, the presence of insert 1 does allow for a repositioning of helix K, which is now able to interact directly with switch 1 and also protrudes within the nucleotide-binding pocket, resulting in a decrease in nucleotide accessibility. Additional support for the hypothesis that the exon 7 domain is part of the signal relay pathway between the nucleotide and actin-binding sites comes from the observation that both the exon 7 domain and helix L in β-myosin heavy chain contain several sites that, when mutated, cause hypertrophic cardiomyopathy. Mutations have been reported for F312/C (Van Driest et al. 35), V320/M (Havndrup et al. 36), A326/P (Van Driest et al. 35), and E328/G (Yu et al. 37). The equivalent residues in Drosophila are I/N314, V322, A/G328 and E330 respectively (see Figure 7). We propose that these mutations alter myosin function by disrupting the relay pathway between the nucleotide and actin-binding sites.
Conclusions
This study was designed to investigate in detail the modulatory effects of the exon 7 domain on Drosophila myosin isoform kinetics. Our results indicate that the sequence differences created in the IFI-7a, EMB-7d, EMB-7a/7d and EMB-7d/7a isoforms induced biochemical and structural changes in the exon 7 domain, thereby influencing attached and detached myosin kinetic transitions. Homology models suggest a mechanism by which the exon 7 domain transmits these changes between the nucleotide and actin-binding sites. Interestingly, all Drosophila myosin isoform kinetic parameters that have been measured in vitro (Table 3 and Miller et al. 11) show small differences (4-fold or less) between isoforms. Thus, small kinetic modifications, controlled by sequence differences in the variable domains, are sufficient to tune Drosophila myosin isoforms for their physiological function.
Materials and Methods
Preparation of exon 7 chimeric subdomain transgenes
To create P element based constructs EMB-7a/7d and EMB-7d/7a, we exchanged small parts of exon 7d into the EMB-encoding transgene (P [w+Mhcemb] w3 or Mhcemb) 27 to replace portions of the endogenous exon 7a. The following primers to exon 4 (containing a Stu I site), to exon 8 (containing a Bgl II site), and to exon 7a and exon 7d were generated in order to amplify the replacement DNA by PCR: Primer W (exon 4): 5' GAAGGCCTTCGGTAACGCCA 3', Primer X (exon 7a reverse): 5' GGTGTAACAGAGGGTCCCATTTCATTGAC 3', Primer Y (exon 7d forward): 5' CCACATTGTCTCCCAGGGTAAAGTAACTG 3', Primer M (exon 7a forward): 5' ACAGTGACCTTGCCCTGGGATACGTTATAGT 3', Primer N (exon 7d reverse): 5' TACTATAACGTATCCCAGGGCAAGGTCACTGTA, Primer Z (exon 8): 5' GAAGATCTCAAACCAGCAATA 3'.
To produce the exon 4 to exon 8 fragment with either exon 7a/7d or 7d/7a, the final DNA segment was generated using three rounds of PCR followed by ligation of the final PCR fragment into a vector. To create EMB-7a/7d, the Emb DNA (Mhcemb) 27 was used as a template in the first PCR reaction with Primers W and X, yielding an ∼270 bp band. A second PCR reaction with EMB-7d DNA 16 as a template for Primers Y and Z generated a band of ∼460 bp. An overlapping region between the products of these two PCR reactions allowed them to be mixed together and serve as a template for the final PCR reaction. Prior to amplification of the full-length product (730 bp) using Primers W and Z, the 3' ends were extended during an incubation period to create the full length template. The final PCR product was gel purified and digested with Stu I (exon 4 – Primer W) and Bgl II (exon 8 – Primer Z) and ligated into the Litmus 29 vector (New England BioLabs, Beverly, MA). To generate the final EMB-7a/7d construct, Mhcemb DNA was digested with Stu I (exon 4) and Sph I (exon 17) to isolate a 12 kb fragment containing a portion of Emb DNA and the vector. A second digest of the Mhcemb DNA with Bgl II (exon 8) and Sph I (exon 13) isolated a fragment of ∼1.2 kb including exons 8 - 13. A triple ligation between these two fragments (12 kb and 1.2 kb) and the Stu I (exon 4) to Bgl II (exon 8) fragment digested from the Litmus 29 construct produced a plasmid containing exon 7a/7d but missing exons 13-17. To create the final EMB-7a/7d DNA, this plasmid was digested with Sph I, dephosphorylated and then ligated together with a fragment containing exons 13-17 that was isolated from a Sph I digest of Mhcemb DNA. All enzyme sites and the exon 7 region were sequenced to verify proper ligation and orientation of all fragments prior to large scale DNA purification.
To create EMB-7d/7a, a similar procedure was used. The differences include: the first PCR reaction contained EMB-7d DNA 16 as a template for Primers W and N while the second PCR utilized MhcEmb 27 as a template for Primers M and Z. Details for the final round of PCR and ligation of exon 4 to exon 8 into the Litmus 29 vector were the same as described above for EMB-7a/7d. In addition, the steps to produce the final EMB-7d/7a construct were analogous to that described for EMB-7a/7d. Again, all ligation sites and the exon 7 region were sequenced to verify that no errors were introduced during creation of the P element constructs. An Eco RI site present at the end of exon 7d was used to distinguish between the EMB-7a/7d and EMB-7d/7a DNA. When digested with Eco RI, the former construct produced 4 bands and the latter yielded 3 bands.
Transformation of Drosophila with chimeric exon 7 transgenes
To create the transgenic organisms, 600 - 800 yw embryos were injected with EMB-7a/7d or EMB-7d/7a DNA (0.4 mg ml−1) along with Δ 2-3 helper plasmid DNA (0.06 mg ml−1), serving as the source of transposase necessary for insertion of the P element vector into the germline DNA 20, 21. Viable flies were crossed to the yw fly line; progeny resulting from these matings were screened for pigmented eyes indicating germline transformation by insertion of the P element vector. Standard genetic crosses with the balancer line w1118; CyO/Bl; TM2/TM6b were used to map each chromosome insertion event. Individual fly lines with inserts mapping to chromosome 1 (X), 3 or 4 were crossed into a Mhc10 background, which is null for MHC production in the indirect flight and jump muscles 22, in order to eliminate endogenous MHC expression in these muscle types.
Transgene and protein expression
To determine proper transgenic expression of the altered Mhc gene, mRNA was extracted (RNeasy, Qiagen, Valencia, CA) from 60 – 80 transgenic fly dorsolongitudinal IFM's expressing either EMB-7a/7d or EMB-7d/7a transcripts in a Mhc10 background 22. In separate PCR reactions, the isolated mRNA from each transgenic fly line was used as a template for primers flanking the exon 7 domain. The sequence of the primers are as follows: exon 5 (+) 5' GGCTGGTGCTGATATTGAGA 3' and exon 10 (−) 5' TGTTGGTCAGCTTCTCCGAGA 3'. Each PCR reaction amplified a 900 bp band from exon 5 to exon 10 that was subsequently ligated into the pGEM – T easy vector (Promega, Madison, WI) and sequenced to confirm the change (exon 7a/7d or 7d/7a in place of exon 7a).
Protein expression studies determined the level of myosin isoform accumulation in the IFM's of transformed flies in the Mhc10 null background 22. The results are presented as mean ± S.E. This method was previously described 16. For each myosin isoform, a single fly line expressing the highest level of MHC was selected for characterization. To produce EMB-7a/7d line #2 was used, which expressed at 97 ± 3% of wild-type MHC. For EMB-7d/7a line #200A was used, which expressed 105 ± 3% of wild-type MHC.
Drosophila myosin isolation and S1 preparation
Myosin was purified, according to previously described methods 12, from the IFM tissue (dorsolongitudinal fibers) of 150-180 transgenic flies expressing the MHC isoform of interest in a Mhc10 background. For myosin steady-state Mg2+ATPase assays the resulting myosin pellets (300-400 μg) were resuspended in 30-50 μl of myosin storage buffer (pH 7.0: 0.5 M KCl, 20 mM MOPS, 2 mM MgCl2 and 10 mM DTT). The final myosin concentration was determined at 280 nm absorbance with an extinction coefficient of E1% = 0.53 cm−1 38 and a molecular mass of 200 kDa. To produce S1 by α-chymotrypsin (Worthington Biochemical Corp., Lakewood, NJ) digestion, the final myosin pellets were resuspended in 60-100 μl of digestion buffer (120 mM NaCl, 20 mM Na2PO4, 1 mM EDTA, pH 7.0, 4 mM DTT) and prepared as described 11. S1 concentration was quantified at 280 nm absorbance using an extinction coefficient of E1% = 0.75 cm−1 and a molecular mass of 115 kDa. A typical S1 preparation yielded 100-200 μg; all purified S1 isoforms were stored on ice at 4°C and used within two weeks.
Actin isolation
F-actin was extracted from chicken skeletal muscle acetone powder and quantified using previously described methods 39. Final actin pellets were resuspended in actin storage buffer (pH 7.0: 0.1 M KCl, 4 mM imidazole, 2 mM MgCl2, 0.5 mM ATP, 1 mM sodium azide and 1 mM DTT), stored on ice at 4°C, and used within one month of preparation for actin-activated Mg2+ATPase assays.
Rabbit skeletal muscle actin was used for transient kinetic experiments and prepared according to the method of Spudich and Watt 40. Quantification of the purified actin was based on a molecular mass of 42 kDa and an extinction coefficient of E1% = 1.104 cm−1 at 280 nm absorbance 41. Prior to use, rabbit F-actin was mixed with an equimolar concentration of phalloidin and incubated at 4°C for at least four hours in order to stabilize the filaments.
Steady-state Mg2+ATPase assays
Myosin steady-state basal and actin-activated Mg2+ATPase assays were performed immediately following purification using malachite green / ammonium molybdate to detect phosphate production according to methods described 10. Data collected from multiple myosin preparations (n ≥ 5) activated with 0-7.5 μM actin in a low salt buffer (20 mM KCl, 20 mM imidazole, 0.1 mM CaCl2, 5 mM MgCl2, pH 6.0, and 10 mM DTT) were averaged, fit with a two - parameter regression to determine the Vmax and KM and reported as mean ± S.E (SigmaPlot, v7.101, SPSS Inc).
Chemicals
Caged ATP (cATP) was purchased from Molecular Probes and coumarin labelled ATP (3'-O-{N-[2-(7-diethylaminocoumarin-3-carboxamido)ethyl]carbamoyl}ATP abbreviated to eda-deac ATP or ADP) was a gift from Martin Webb (National Institute for Medical Research, Mill Hill, London, UK). The cmATP (eda-deac ATP analog) was prepared for use in the transient kinetic studies according to methods previously described 42.
Flash photolysis system
Due to the small amount of protein available and the relatively poor fluorescence signal changes observed with pyrene labeled actin, flash photolysis was the method of choice to investigate the kinetic properties of the Drosophila myosin S1 isoforms. In particular, the ATP-induced dissociation of the acto-S1 complex and the ADP affinity, either in the presence (KAD) or absence of actin (KD), were investigated. The flash photolysis experimental system, described previously 11, 24, was used to monitor the ATP-induced dissociation of the acto-S1 complex by following changes in the light scattering signal and to monitor the dissociation of nucleotide from S1 by fluorescence changes. In brief, the 20 μl sample was held in a quartz cuvette and ATP was liberated by a single 5 ns flash at 353 nm from a neodymium-yttrium-aluminum-garnet laser (Surelite I-10, 70 mJ maximum power) along the vertical axis of the cell at a rate of 90 s−1 to start the reaction. Both absorbance (to determine the ATP concentration) and light scattering (to monitor the acto-S1 complex) or fluorescence (to determine eda-deac ADP release) were measured from the cuvette simultaneously following the laser flash. White light > 389 nm was introduced to the sample from a 100 watt halogen lamp and the change in the amount of light scattered at 90° was monitored after each flash. The absorbance at 405 nm was measured with a monochromator to determine the amount of ATP liberated from caged ATP during each laser flash. Coumarin fluorescence changes were detected by monitoring the emission through a 455 nm cut-off filter after excitation at 434 nm (75-watt xenon/mercury lamp).
All light scattering experiments were conducted in a low salt buffer (pH 7.0: 30 mM KCl, 5 mM MgCl2, 20 mM MOPS and 4 mM DTT) with 1 μM actin, 1-3 μM S1, 500 μM cATP, 10 mM DTT and either apyrase (2 units/ml: ATP-induced dissociation of acto-S1) or ADP (various concentrations) and a glucose-hexokinase system (0.03 units/ml hexokinase, 1 mM glucose and 100 μM Ap5A: KAD determination). Each sample was subjected to multiple laser flashes. During KAD determination ADP and cATP were added after each flash. eda-deac ADP dissociation experiments were also performed in this low salt buffer and contained 4 μM S1, 10 μM eda-deac ATP (source of eda-deac ADP) and 100 μM cATP.
Analysis of the transient kinetic data
The following equation was derived from the interaction of actin and S1 with ATP and ADP shown in Scheme 1 and was used to determine KAD.
| (equation 1) |
where kobs is the observed rate constant for the ATP-induced dissociation of acto-S1; K1k+2 is the second-order rate constant for ATP binding to acto-S1; KAD is the equilibrium dissociation constant for the binding of ADP to acto-S1. The equation krel = kobs/ko was used to determine the relative rate constant (krel) shown in Figure 6, where ko is the value when the [ADP] = 0.
Homology Modeling:
Three-dimensional homology models were generated for the Drosophila IFI and EMB myosin motor domain and the exon 7 mutants using the SWISS-MODEL automatic comparative protein modeling server, as described previously 43. Briefly, SWISS-MODEL uses four steps: template superposition, target-template alignment, model building and energy minimization. The templates are weighted by their sequence similarity to the target and significantly deviating atom positions are excluded. Since the template coordinates cannot be used to model regions with insertions or deletions in the target-template alignment, an ensemble of fragments compatible with the neighboring stems is generated using constraint space programming (CSP). The best loop is selected using a scoring scheme which includes force field energy, steric hindrance and hydrogen bond formation. If no suitable loop can be identified, the flanking residues are included in the rebuilt fragment to allow for more flexibility. Reconstruction of the model side-chains is based on weighted positions of corresponding residues in the template structures. Starting with conserved residues, the model side-chains are built by iso-sterically replacing template structure side-chains. Possible side-chain formations are selected from a backbone dependent rotamer library and the most likely conformation is selected using a scoring function that assesses favorable interactions (e.g. hydrogen-bonds, disulphide bridges) and unfavorable close contacts. Finally, the generated model is subjected to steepest descent energy minimization using the GROMOS96 force field. The primary sequences of the Drosophila IFI and EMB isoform were pairwise aligned with the sequence of four scallop myosin structures as templates (ExPDB 1kk8, 1qvi, 1s5g and 1sr6) using the CLUSTALW alignment protocol. The alignments were then submitted to the alignment interface of SWISS-MODEL and the generated models were validated using WHAT CHECK 44.
Acknowledgements
This work was supported by NIH research grant GM32443 (to SIB), Wellcome Trust Grant 070021 (to MAG and MJB), Hungarian National Research Foundation grants K60186 and K60968 (to MN), and a predoctoral fellowship (to BMM) from the Western States Affiliate of the American Heart Association. M.N. holds a Wellcome Trust International Senior Research Fellowship in Biomedical Sciences. We thank Martin Webb for the coumarin ATP/ADP used in this work and Corey Dambacher for excellent technical assistance. In addition, we thank Dr. Douglas Swank for helpful comments on the manuscript.
The following abbreviations were used in the manuscript
- ATP
adenosine triphosphate
- MHC
myosin heavy chain
- EMB
embryonic body wall muscle
- IFI
indirect flight muscle isoform
- IFM
indirect flight muscle
- eda-deac ADP
coumarin ADP
- cATP
caged ATP
- ADP
adenosine diphosphate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lutz GJ, Lieber RL. Myosin isoforms in anuran skeletal muscle: their influence on contractile properties and in vivo muscle function. Microsc. Res. Tech. 2000;50:443–457. doi: 10.1002/1097-0029(20000915)50:6<443::AID-JEMT3>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 2.Tyska MJ, Warshaw DM. The myosin power stroke. Cell Motil. Cytoskeleton. 2002;51:1–15. doi: 10.1002/cm.10014. [DOI] [PubMed] [Google Scholar]
- 3.Barany M. ATPase activity of myosin correlated with speed of muscle shortening. J. Gen. Physiol. 1967;50(Suppl):197–218. doi: 10.1085/jgp.50.6.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamrell BB, Low RB. The relationship of mechanical Vmax to myosin ATPase activity in rabbit and marmot ventricular muscle. Pflugers Arch. 1978;377:119–124. doi: 10.1007/BF00582841. [DOI] [PubMed] [Google Scholar]
- 5.Geeves MA, Holmes KC. Structural mechanism of muscle contraction. Annu. Rev. Biochem. 1999;68:687–728. doi: 10.1146/annurev.biochem.68.1.687. [DOI] [PubMed] [Google Scholar]
- 6.Geeves MA, Fedorov R, Manstein DJ. Molecular mechanism of actomyosin-based motility. Cell Mol. Life Sci. 2005;62:1462–1477. doi: 10.1007/s00018-005-5015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.George EL, Ober MB, Emerson CP., Jr. Functional domains of the Drosophila melanogaster muscle myosin heavy-chain gene are encoded by alternatively spliced exons. Mol. Cell Biol. 1989;9:2957–2974. doi: 10.1128/mcb.9.7.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hastings GA, Emerson CP., Jr. Myosin functional domains encoded by alternative exons are expressed in specific thoracic muscles of Drosophila. J. Cell Biol. 1991;114:263–276. doi: 10.1083/jcb.114.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang S, Bernstein SI. Spatially and temporally regulated expression of myosin heavy chain alternative exons during Drosophila embryogenesis. Mech. Dev. 2001;101:35–45. doi: 10.1016/s0925-4773(00)00549-9. [DOI] [PubMed] [Google Scholar]
- 10.Littlefield KP, Swank DM, Sanchez BM, Knowles AF, Warshaw DM, Bernstein SI. The converter domain modulates kinetic properties of Drosophila myosin. Am. J. Physiol. Cell Physiol. 2003;284:C1031–C1038. doi: 10.1152/ajpcell.00474.2002. [DOI] [PubMed] [Google Scholar]
- 11.Miller BM, Nyitrai M, Bernstein SI, Geeves MA. Kinetic analysis of Drosophila muscle myosin isoforms suggests a novel mode of mechanochemical coupling. J. Biol. Chem. 2003;278:50293–50300. doi: 10.1074/jbc.M308318200. [DOI] [PubMed] [Google Scholar]
- 12.Swank DM, Bartoo ML, Knowles AF, Iliffe C, Bernstein SI, Molloy JE, Sparrow JC. Alternative exon-encoded regions of Drosophila myosin heavy chain modulate ATPase rates and actin sliding velocity. J. Biol. Chem. 2001;276:15117–15124. doi: 10.1074/jbc.M008379200. [DOI] [PubMed] [Google Scholar]
- 13.Swank DM, Knowles AF, Suggs JA, Sarsoza F, Lee A, Maughan DW, Bernstein SI. The myosin converter domain modulates muscle performance. Nat. Cell Biol. 2002;4:312–317. doi: 10.1038/ncb776. [DOI] [PubMed] [Google Scholar]
- 14.Swank DM, Vishnudas VK, Maughan DW. An exceptionally fast actomyosin reaction powers insect flight muscle. Proc. Natl. Acad. Sci. U S A. 2006;103:17543–17547. doi: 10.1073/pnas.0604972103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernstein SI, Mogami K, Donady JJ, Emerson CP., Jr. Drosophila muscle myosin heavy chain encoded by a single gene in a cluster of muscle mutations. Nature. 1983;302:393–397. doi: 10.1038/302393a0. [DOI] [PubMed] [Google Scholar]
- 16.Miller BM, Zhang S, Suggs JA, Swank DM, Littlefield KP, Knowles AF, Bernstein SI. An alternative domain near the nucleotide-binding site of Drosophila muscle myosin affects ATPase kinetics. J. Mol. Biol. 2005;353:14–25. doi: 10.1016/j.jmb.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 17.Swank DM, Braddock J, Brown W, Lesage H, Bernstein SI, Maughan DW. An alternative domain near the ATP binding pocket of Drosophila myosin affects muscle fiber kinetics. Biophys. J. 2006;90:2427–2435. doi: 10.1529/biophysj.105.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swank DM, Knowles AF, Kronert WA, Suggs JA, Morrill GE, Nikkhoy M, Manipon GG, Bernstein SI. Variable N-terminal regions of muscle myosin heavy chain modulate ATPase rate and actin sliding velocity. J. Biol. Chem. 2003;278:17475–17482. doi: 10.1074/jbc.M212727200. [DOI] [PubMed] [Google Scholar]
- 19.Swank DM, Kronert WA, Bernstein SI, Maughan DW. Alternative N-terminal regions of Drosophila myosin heavy chain tune muscle kinetics for optimal power output. Biophys. J. 2004;87:1805–1814. doi: 10.1529/biophysj.103.032078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cripps RM, Becker KD, Mardahl M, Kronert WA, Hodges D, Bernstein SI. Transformation of Drosophila melanogaster with the wild-type myosin heavy-chain gene: rescue of mutant phenotypes and analysis of defects caused by overexpression. J. Cell Biol. 1994;126:689–699. doi: 10.1083/jcb.126.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rubin GM, Spradling AC. Genetic transformation of Drosophila with transposable element vectors. Science. 1982;218:348–353. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- 22.Collier VL, Kronert WA, O'Donnell PT, Edwards KA, Bernstein SI. Alternative myosin hinge regions are utilized in a tissue-specific fashion that correlates with muscle contraction speed. Genes Dev. 1990;4:885–895. doi: 10.1101/gad.4.6.885. [DOI] [PubMed] [Google Scholar]
- 23.Clark RJ, Nyitrai M, Webb MR, Geeves MA. Probing nucleotide dissociation from myosin in vitro using microgram quantities of myosin. J. Muscle Res. Cell Motil. 2003;24:315–321. [PubMed] [Google Scholar]
- 24.Weiss S, Chizhov I, Geeves MA. A flash photolysis fluorescence/light scattering apparatus for use with sub microgram quantities of muscle proteins. J. Muscle Res. Cell Motil. 2000;21:423–432. doi: 10.1023/a:1005690106951. [DOI] [PubMed] [Google Scholar]
- 25.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 26.Cope MJ, Whisstock J, Rayment I, Kendrick-Jones J. Conservation within the myosin motor domain: implications for structure and function. Structure. 1996;4:969–987. doi: 10.1016/s0969-2126(96)00103-7. [DOI] [PubMed] [Google Scholar]
- 27.Wells L, Edwards KA, Bernstein SI. Myosin heavy chain isoforms regulate muscle function but not myofibril assembly. EMBO J. 1996;15:4454–4459. [PMC free article] [PubMed] [Google Scholar]
- 28.Cremo CR, Geeves MA. Interaction of actin and ADP with the head domain of smooth muscle myosin: implications for strain-dependent ADP release in smooth muscle. Biochemistry. 1998;37:1969–1978. doi: 10.1021/bi9722406. [DOI] [PubMed] [Google Scholar]
- 29.Conibear PB, Bagshaw CR, Fajer PG, Kovacs M, Malnasi-Csizmadia A. Myosin cleft movement and its coupling to actomyosin dissociation. Nat. Struct. Biol. 2003;10:831–835. doi: 10.1038/nsb986. [DOI] [PubMed] [Google Scholar]
- 30.Holmes KC, Angert I, Kull FJ, Jahn W, Schroder RR. Electron cryomicroscopy shows how strong binding of myosin to actin releases nucleotide. Nature. 2003;425:423–427. doi: 10.1038/nature02005. [DOI] [PubMed] [Google Scholar]
- 31.Volkmann N, Ouyang G, Trybus KM, DeRosier DJ, Lowey S, Hanein D. Myosin isoforms show unique conformations in the actin-bound state. Proc. Natl. Acad. Sci. U S A. 2003;100:3227–3232. doi: 10.1073/pnas.0536510100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Volkmann N, Hanein D, Ouyang G, Trybus KM, DeRosier DJ, Lowey S. Evidence for cleft closure in actomyosin upon ADP release. Nat. Struct. Biol. 2000;7:1147–1155. doi: 10.1038/82008. [DOI] [PubMed] [Google Scholar]
- 33.Risal D, Gourinath S, Himmel DM, Szent-Gyorgyi AG, Cohen C. Myosin subfragment 1 structures reveal a partially bound nucleotide and a complex salt bridge that helps couple nucleotide and actin binding. Proc. Natl. Acad. Sci. U S A. 2004;101:8930–8935. doi: 10.1073/pnas.0403002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menetrey J, Bahloul A, Wells AL, Yengo CM, Morris CA, Sweeney HL, Houdusse A. The structure of the myosin VI motor reveals the mechanism of directionality reversal. Nature. 2005;435:779–785. doi: 10.1038/nature03592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Driest SL, Jaeger MA, Ommen SR, Will ML, Gersh BJ, Tajik AJ, Ackerman MJ. Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2004;44:602–610. doi: 10.1016/j.jacc.2004.04.039. [DOI] [PubMed] [Google Scholar]
- 36.Havndrup O, Bundgaard H, Andersen PS, Allan Larsen L, Vuust J, Kjeldsen K, Christiansen M. Outcome of clinical versus genetic family screening in hypertrophic cardiomyopathy with focus on cardiac beta-myosin gene mutations. Cardiovasc. Res. 2003;57:347–357. doi: 10.1016/s0008-6363(02)00711-3. [DOI] [PubMed] [Google Scholar]
- 37.Yu B, Sawyer NA, Caramins M, Yuan ZG, Saunderson RB, Pamphlett R, Richmond DR, Jeremy RW, Trent RJ. Denaturing high performance liquid chromatography: high throughput mutation screening in familial hypertrophic cardiomyopathy and SNP genotyping in motor neurone disease. J. Clin. Pathol. 2005;58:479–485. doi: 10.1136/jcp.2004.021642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Margossian SS, Lowey S. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 1982;85(Pt B):55–71. doi: 10.1016/0076-6879(82)85009-x. [DOI] [PubMed] [Google Scholar]
- 39.Pardee JD, Spudich JA. Purification of muscle actin. Methods Enzymol. 1982;85:164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- 40.Spudich JA, Watt S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- 41.West JJ, Nagy B, Gergely J. Free adenosine diphosphate as an intermediary in the phosphorylation by creatine phosphate of adenosine diphosphate bound to actin. J. Biol. Chem. 1967;242:1140–1145. [PubMed] [Google Scholar]
- 42.Webb MR, Corrie JE. Fluorescent coumarin-labeled nucleotides to measure ADP release from actomyosin. Biophys. J. 2001;81:1562–1569. doi: 10.1016/S0006-3495(01)75810-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]