

Abstract
Finding UDP-glucuronosyltransferases (UGT) require protein kinase C-mediated phosphorylation is important information that allows manipulation of this critical system. UGTs glucuronidate numerous aromatic-like chemicals derived from metabolites, diet, environment and, inadvertently, therapeutics to reduce toxicities. As UGTs are inactivated by downregulating PKCs with reversibly-acting dietary curcumin, we determined the impact of gastro-intestinal glucuronidation on free-drug uptake and efficacy using immunosuppressant, mycophenolic acid (MPA), in mice. Expressed in COS-1 cells, mouse GI-distributed Ugt1a1 glucuronidates curcumin and MPA and undergoes irreversibly and reversibly dephosphorylation by PKC-specific inhibitor calphostin-C and general-kinase inhibitor curcumin, respectively, with parallel effects on activity. Moreover, oral curcumin-administration to mice reversibly inhibited glucuronidation in GI-tissues. Finally, successive oral-administration of curcumin and MPA to antigen-treated mice increased serum free-MPA and immunosuppression up to 9-fold. Results indicate targeted inhibition of GI-glucuronidation in mice markedly improved free-chemical uptake and efficacy using MPA as a model.
Whereas the primary role of UDP-glucurononsyltransferases (UGT) is to detoxify numerous structurally diverse lipophilic chemicals, including metabolites, dietary constituents, environmental toxicants, carcinogens and, inadvertently, therapeutic chemicals, there is a limited capacity to manipulate glucuronidation to improve protection or reduce premature clearance of medications. As UGTs inactivate aromatic-like chemicals by attaching each to a glucuronic acid molecule(s) to promote water-solubility with high excretability, investigators have used recombinant UGTs under in-vitro conditions [1] to explore substrate suitability to gain information on the potential clearance of chemicals. While we know glucuronidation indiscriminately removes chemicals for protective purposes, glucuronidation can have a negative impact due to premature clearance of therapeutic drugs, which is inherently overcome by administering compensatorily higher levels, sometimes with side-effects. Despite the longstanding biochemical value associated with glucuronidation of chemicals, it has not been possible to establish or measure impact of this system, and it is not known whether biochemical manipulation of this system is warranted for protection against toxicants or to enhance drug efficacy. Unfortunately, it has not been possible to establish an effective, reversible biochemical inhibitor for all UGT activities. Because our recent studies demonstrated all UGTs tested require protein kinase C (PKC)-mediated phosphorylation [2,3] that is subject to reversible antioxidant down-regulation, we have developed a strategy to determine the impact of UGTs found in the mucosa layer of the gastrointestinal (GI) tract [1] on drug efficacy. We have downregulated glucuronidation by orally administering the kinase inhibitor and dietary condiment, curcumin, to block UGT phosphorylation on a transient basis [3], followed by administration of a test drug.
As we considered a UGT test substrate for in-vivo glucuronidation studies, immunosuppressant mycophenolic acid (MPA), administered to transplant patients as the mofetil-formulated prodrug MMF (4), was considered ideal for assessing the impact of GI glucuronidation on drug efficacy because it undergoes high levels of glucuronidation [5], it avoids the pharmacokinetics of in-vivo esterase-dependent cleavage of MMF and it is a potent immunosuppressant of cytotoxic T-lymphocyte (CTL) proliferation in renal transplant recipients [5] that is assessed in mice with a sensitive immunosuppression assay [6]. Potent MPA-inhibition of the rate-limiting enzyme, inosine monophosphate dehydrogenase (IMPDH) [7], is highly selective toward type II IMPDH isozyme over type I because of its specific role in de-novo synthesis of guanosine monophosphate in activated mononuclear cell proliferation. Selective inhibition of type II IMPDH by MPA prevents proliferation of activated T- and B-lymphocytes and allows the agent to exert a potent immunosuppressant effect, as well as it induces apoptosis of activated T-lymphocytes [8]. Immunosuppression is less toxic than anti-rejection therapy to alleviate acute allograft rejection of transplanted organs.
Whereas MPA readily forms ether-linked- (MPAG) or acyl-glucuronides (AcMPAG) [9–11], we studied MPA glucuronidation in vitro with human UGTs and found 4 isozymes are avid metabolizers [12] that reach saturation kinetics between 1.6 and 2.4 mM with Km values between 0.25 and 0.55 mM. These UGT isozymes, encoded at the UGT1 complex locus, were found strategically and differentially expressed in the gastrointestinal (GI) mucosa [1]. In this report, we established under both in-vitro and in-vivo conditions that mouse Ugt1a1 also requires phosphorylation, which can be transiently downregulated by nontoxic kinase inhibitor, curcumin [2,3,13], and irreversibly by calphostin-C, similar to that for human UGTs [2,3]. Moreover, effects of curcumin-pretreatment elicited a marked increase in free MPA levels in blood and a comparable improvement in immunosuppression in antigen-stimulated mice detected by the spleen CTL assay [6]. In short, our use of MPA demonstrates a ‘proof of principle’ that glucuronidation at the GI level can greatly compromise chemical uptake, including drug efficacy in mice.
Materials and methods
Materials and methods are provided as supporting online materials.
Results
Metabolism of MPA and curcumin by mouse Ugt1a1
As shown in Fig. 1A, mouse Ugt1a1 metabolized MPA, generating 64 % MPA-7-0- and 36 % acylMPA-glucuronides, respectively, and reaching saturation kinetics at 2.0 mM driven by Km of 0.250 mM. Whereas the 9 mouse Ugt1a isozymes, except stomach-specific Ugt1a2, are distributed in all GI tissues [18], the level of MPA turnover by other mouse isozymes is unknown. Previously, we showed 4 out of 5 different human GI-distributed UGTs avidly metabolized MPA; human UGT1A1 [12,19] poorly metabolized MPA.
Fig. 1.
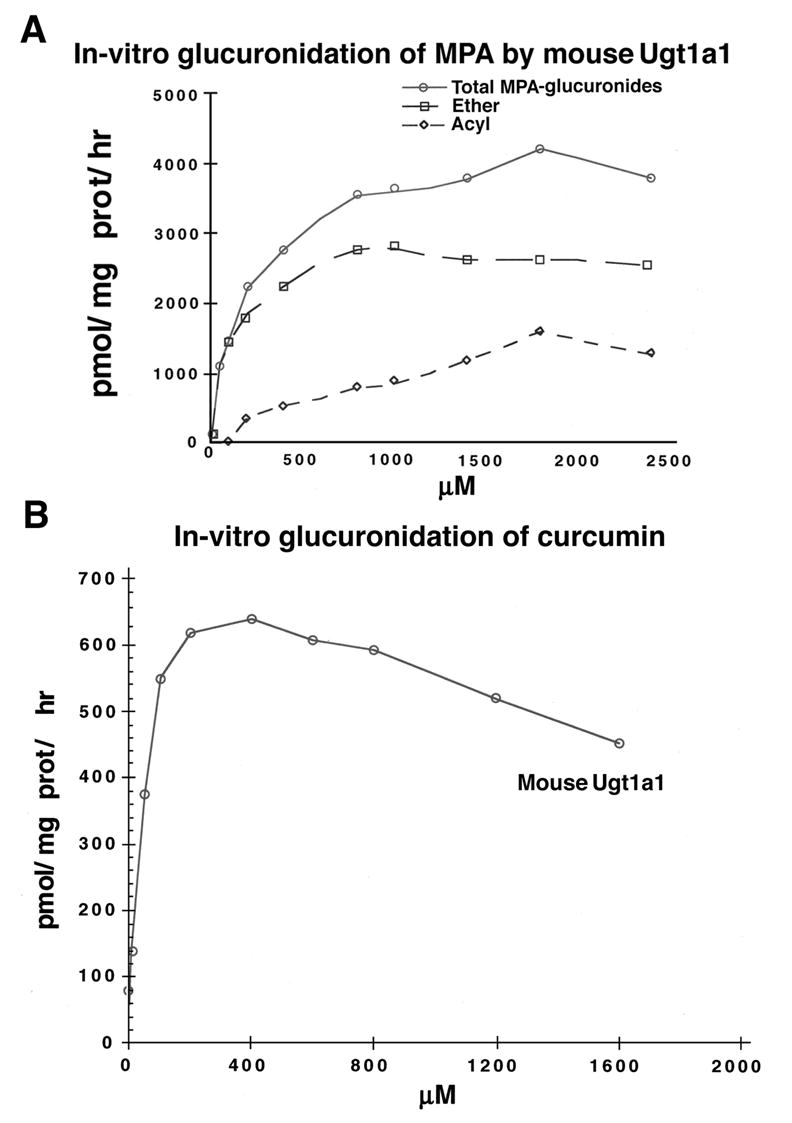
(A) In-vitro glucuronidation of mycophenolic acid by mouse Ugt1a1. Analysis of MPA glucuronidation was carried out with 150μg protein in 2 hr incubation with ether (MPAG) and acyl-glucuronides (AcMPAG) distinguished as described under Methods. Total MPA-glucuronides, MPAG and AcMPAG are pmol MPA-β-glucuronide/mg prot./hr. Experiments, repeated 3 times, have S.E. of ±1 to 5 %. (B) In-vitro glucuronidation of curcumin by mouse Ugt1a1. One hundred fifty μg of mouse Ugt1a1 (pH 6.4) expressed in COS-1 cells incubated 2 hr with increasing concentration of curcumin according to Methods. Activity is expressed as pmol/mg protein/hr. The experiment, repeated 3 times, has S.E. of ±1 to 4 %.
Since we propose to use curcumin, previously demonstrated [1] to be an in-vitro substrate for several human UGTs accounting for eventual recovery from inhibition in cell lines, we demonstrate it undergoes concentration-dependent glucuronidation by mouse recombinant Ugt1a1 (Fig. 1B). Moreover, we have shown that 98 %-inhibited LS180 cells by HPLC analysis [16] with 2 % remaining glucuronidation initiated curcumin removal [Basu, N.K., Kole, L., Garza, A., Mitra, P.S., and I. S. Owens, Manuscript submitted]; analysis revealed initially all free curcumin and no glucuronide, which progressed to nondetectable levels and 98.7 % curcumin glucuronides, respectively, concurrent with recovery of activity. Because curcumin is an effective general-kinase inhibitor (see Discussion) and an excellent substrate for certain UGTs, our results can account for its transient in-cellulo inhibition of UGT.
Sensitivity of mouse Ugt1a1 to curcumin and calphostin-C
To assess whether mouse Ugt phosphorylation requirement is similar to that for human isozymes, we examined in-cellulo effects of curcumin and calphostin-C on Ugt1a1 with activity measured in-vitro. Curcumin treatment of Ugt1a1 caused concentration-dependent inhibition within 15-min that was reversible (Fig. 2, top) and did not affect specific protein kevel assessed with anti-UGT-1168 (Fig. 2, bottom). Calphostin-C also caused concentration-dependent inhibition of Ugt1a1 that was irreversible (Fig. 2, top), again without affecting specific protein level (Fig. 2, bottom). The parallel loss and recovery of both activity and phosphoserine content for Ugt1a1 following curcumin treatment indicates the mouse isozyme, like human UGTs [1–3], undergoes required phosphorylation without affecting Ugt1a1 protein (Fig. 2, bottom). The lack of curcumin and calphostin-C toxicity was confirmed by the MTT assay. Furthermore, we have shown that curcumin and calphostin-C inhibited [33P] orthophosphate incorporation into UGTs in LS180 colon cells in a time- and dose-dependent manner [Basu, N.K., Kole, L., Garza, A., Mitra, P.S., and I. S. Owens, Manuscript submitted]. In addition Ugt1a1 has 3 computer-detected serine-specific PKC phosphorylation sites at position 203, 437, and 529, similar to human isozymes [3].
Fig. 2.
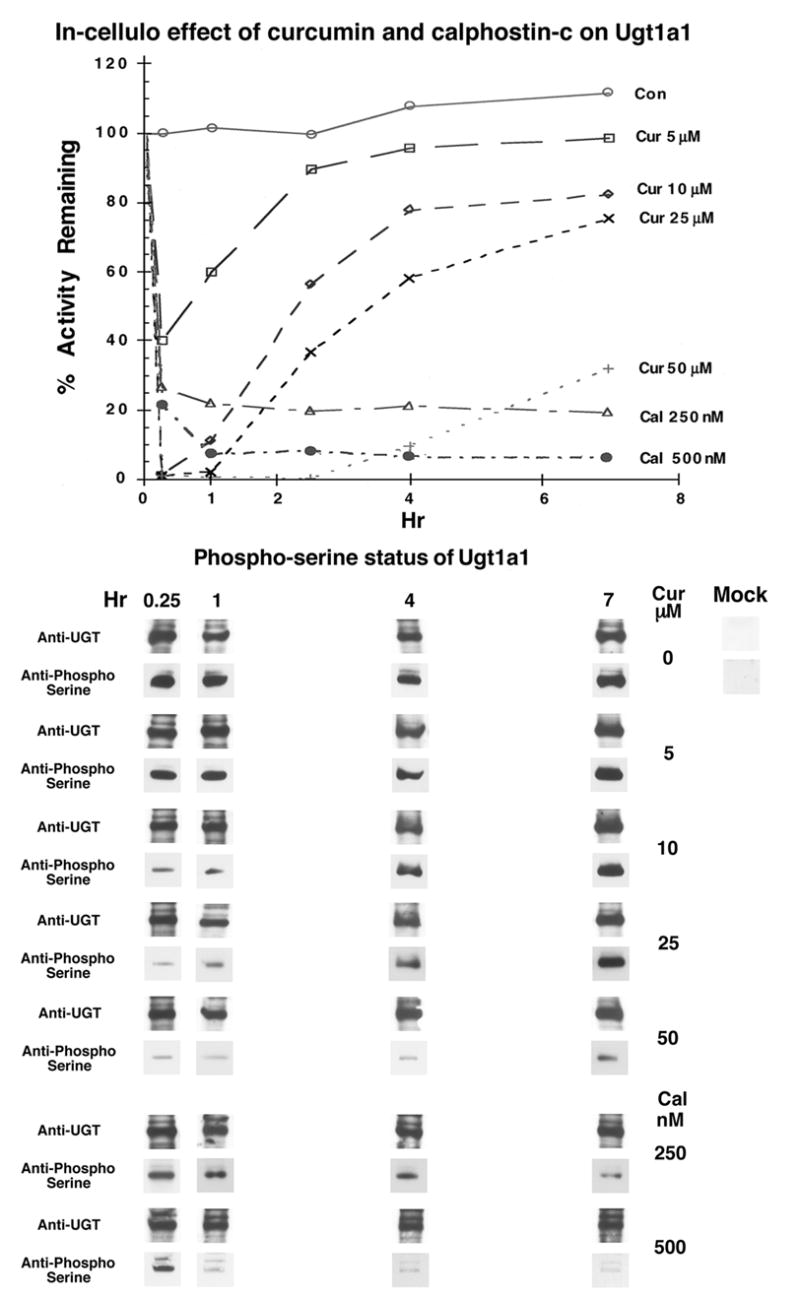
Effects of different concentrations of curcumin- or calphostin-C on Ugt1a1 activity (top), its expression level and phosphoserine status (bottom) in COS-1 cells. MPA glucuronidation was carried out in-vitro with cell homogenates containing 150 μg protein and 800 μM MPA, which incubated 2 hr before analysis as described under Methods. Western blot analysis with anti-UGT (1168) [2,3,12,15] and its phosphoserine status were carried out as described under Methods showing Mock transfected COS-1 cells as controls. Experiments were repeated 3 times and have S.E. of ±2 to 4 %.
Curcumin effect on chemical uptake using mouse duodenal loop
As each in-cellulo study has confirmed UGT is reversibly inhibited by curcumin treatment, we initiated in-vivo effects of curcumin on glucuronidation by monitoring changes in free chemical uptake by sampling portal blood from an in-situ mouse duodenal loop using bilirubin as a marker. Although bilirubin-specific UGT1A1 was recently found at high levels in human duodenal mucosa as well as liver [1], the metabolite is primarily glucuronidated in liver (see discussion) and essentially by a single UGT in rat and human. Thus, bilirubin allowed us to assess glucuronidation by a single isozyme in duodenum. Recently, mouse Ugt1a1 was also shown to be highly abundant in duodenum [18]. Sampling at particular sites in the duodenal loop after 1 hr shows curcumin caused a 2-fold increase in free bilirubin and a 53 % reduction in bilirubin glucuronide in portal blood compared to an untreated loop (Fig. 3A, top), and it caused a 7- and 8-fold reduction in bilirubin glucuronide in duodenal tissue and its lumen, respectively. Evidently, high duodenal levels of UGT isozyme(s) [1] with broad substrate activities facilitate glucuronidation and removal of numerous dietary agents.
Fig. 3.
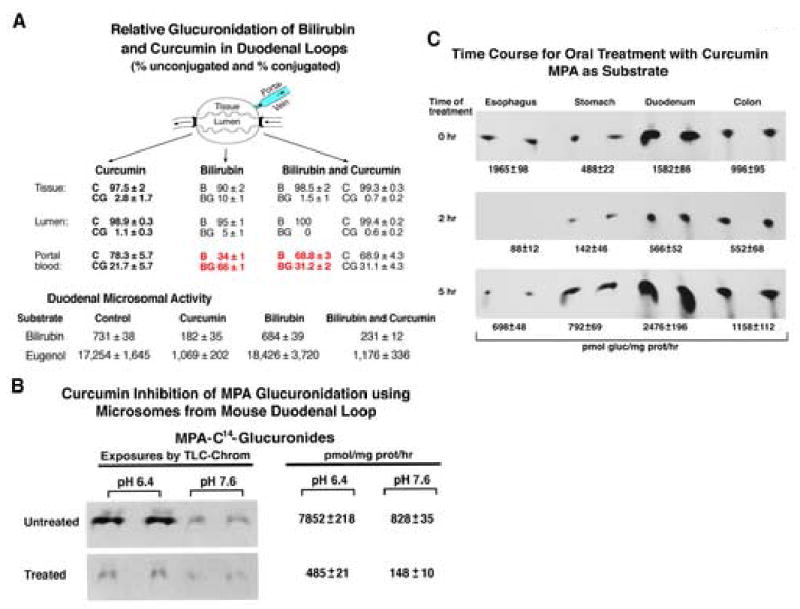
(A) Effect of curcumin treatment on uptake of bilirubin by mouse duodenal loops. Either curcumin (100 mg curcumin/kg), bilirubin (0.6 mg/animal), or curcumin (100 mg curcumin/kg) followed by bilirubin (0.6 mg) was injected into the lumen of in-situ duodenal loops with needle and syringe allowing 1 hr exposure; portal blood was collected and stored at 4°C before shipping for analysis. Duodena were collected, and microsomes were prepared [12]. Microsomal incubations were for 2-hr using 300 μg protein with bilirubin (100 μM) or eugenol (200 μM) or MPA (200 μM), and products were processed. B and C represent bilirubin and curcumin, respectively; BG and CG represent their corresponding glucuronides. Each group contained 3–5 animals. The study was repeated thrice with triplicate determinations. The unit for bilirubin and eugenol glucuronides is pmol/mg prot/hr. (B) MPA-glucuronidation by duodenal-loop microsomes. The left panel is an autoradiogram of a TLC plate that resolved MPA glucuronides generated in 2-hr incubations using 300 μg protein and 200 μM MPA; results represent the mean ± S.E for 3 determinations. (C) Time-course of curcumin treatment using mouse duodenal microsomes. Mice, treated with 100 mg curcumin/kg b. w. for 2 and 5 hr, were sacrificed; 0 hr represents untreated controls; duodena were removed and stored as described in (A) until microsomes were prepared. Microsomal MPA glucuronidation was determined using 300 μg protein and 200 μM MPA. MPA-[14C] glucuronides were separated by TLC and quantitated [12]; the amounts of [14C]glucuronide product are given below photographs. TLC plates, exposed to x-ray films, were developed for photographs, and product was reported as mean ± S.E for 3 determinations. Each group contained 3–5 animals.
Glucuronidation by microsomes isolated from the mouse duodenal loop
Bilirubin and eugenol glucuronidation, respectively, by microsomes isolated from treated duodenal-loop tissue versus untreated loop was: curcumin alone (25/6 %), bilirubin alone (94/107%) and curcumin/bilirubin (32/7 %), confirming Ugt inhibition (Fig. 3A, bottom). The fact that curcumin inhibition of mouse Ugt1a1 turnover of bilirubin (68 %) is less than that for eugenol (93 %) with duodenal microsomes is consistent with the fact that bilirubin is likely metabolized by a single enzyme, and eugenol is metabolized by many isozymes [1]. Curcumin inhibition of MPA glucuronidation using mouse duodenal loop microsomes is shown to be between 82 and 96 %, depending upon pH of the assay (Fig. 3B) (12). Glucuronidation of anticancer drug, SN-38, showed similar inhibition with microsomes from curcumin-treated loops, generating 86 % and 73 % inhibition at two different pH conditions (not shown). Results show curcumin inhibited in-vivo glucuronidation causing both a significant increase in free bilirubin in portal blood and a reduction in tissue bilirubin-glucuronide. This finding suggests normal glucuronidation necessarily significantly reduces free chemical uptake.
Time-course of curcumin-inhibition and recovery of glucuronidation in mouse duodenum
Previously, we determined dose-response, summarized under Method, for curcumin inhibition of MPA glucuronidation in mice following oral administration [12]. Preliminary results indicated oral administration of 10 mg curcumin/kg to mice caused 100 % inhibition of duodenal UGT activity with a 30-min delay that, no doubt, allowed transit time to reach the tissue. Activity returned to normal after 2 hr. Because the previous data [12] indicated 100 mg curcumin/kg b.w. maintained inhibition of MPA glucuronidation at 96 % for 1 hr, we carried out time-course experiments with 100 mg/kg.
To determine the time for reversal of inhibition in vivo, we tested MPA glucuronidation with microsomes islolated from all GI tissues between 2 and 5 hr following administration of 100 mg curcumin/kg. Overall, activity shows duodenum recovered: 36 and 157 % by 2 and 5 hr, respectively (Fig. 3C), whereas recovery for esophagus, stomach, and colon was 5/35, 24/135, and 55/116 % at 2 and 5 hr, respectively. After the 5-hr reversal, it is evident that MPA glucuronidation, except for esophagus, is between 16 and 57 % higher than control. The slower recovery by esophagus may reflect a more concentrated chemical application while traversing this organ compared to more distal tissues or esophagus-distributed Ugt(s) have lower curcumin glucuronidating activity, thereby, requiring longer recovery time.
Major improvement in serum free MPA uptake and its immunosuppression in antigen-treated mice
Since curcumin reversibly inhibited glucuronidation of MPA without detectable toxicity, which is consistent with its beneficial effects that include anti-oxidant [20,21], anti-inflammatory [22] and anti-carcinogenic [20,21] without detectable toxicity in mice or human, we tested its effect on free MPA levels in systemic blood [11] and immunosuppression [6] in antigen-treated mice. Oral administration of 100 mg curcumin/kg to SRBC-stimulated mice revealed a 6- and 7-fold increase in blood free MPA for 25 and 50 mg/kg, respectively; at the same time MPA glucuronide remaining in the blood was reduced by about 25 % (Fig. 4A). Pretreatment with 100 mg curcumin/kg before 50 mg MPA/kg compared to 150 mg MPA/kg, without curcumin pretreatment, also showed effectively a 6-fold improvement in blood free MPA and a 75 % decrease in MPA glucuronides. Using HPLC analysis [6], we were unable to detect AcMPAG in any mouse serum sample with pH values ranging from 8.0 to 9.0. Similarly, pretreatment with 100 mg/kg curcumin before administration of 25 and 50 mg/kg MPA versus no pretreatment caused a 2.5- and 6-fold improvement in immunosuppression of spleen CTL proliferation, respectively, in SRBC-stimulated mice (Fig. 4B). Again, pretreatment with 100 mg/kg curcumin before 50 mg/kg MPA versus no pretreatment with 150 mg/kg MPA caused nearly 9-fold more effective immunosuppression. In effect, MPA alone shows concentration dependent immunosuppression, and curcumin-inhibition of glucuronidation apparently caused a concentration-dependent improvement in MPA immunosuppression. Overall, the results show, at least, a 6-fold enhancement in both free MPA in blood and immunosuppression with curcumin-pretreatment at 100 mg/kg b.w.
Fig. 4.
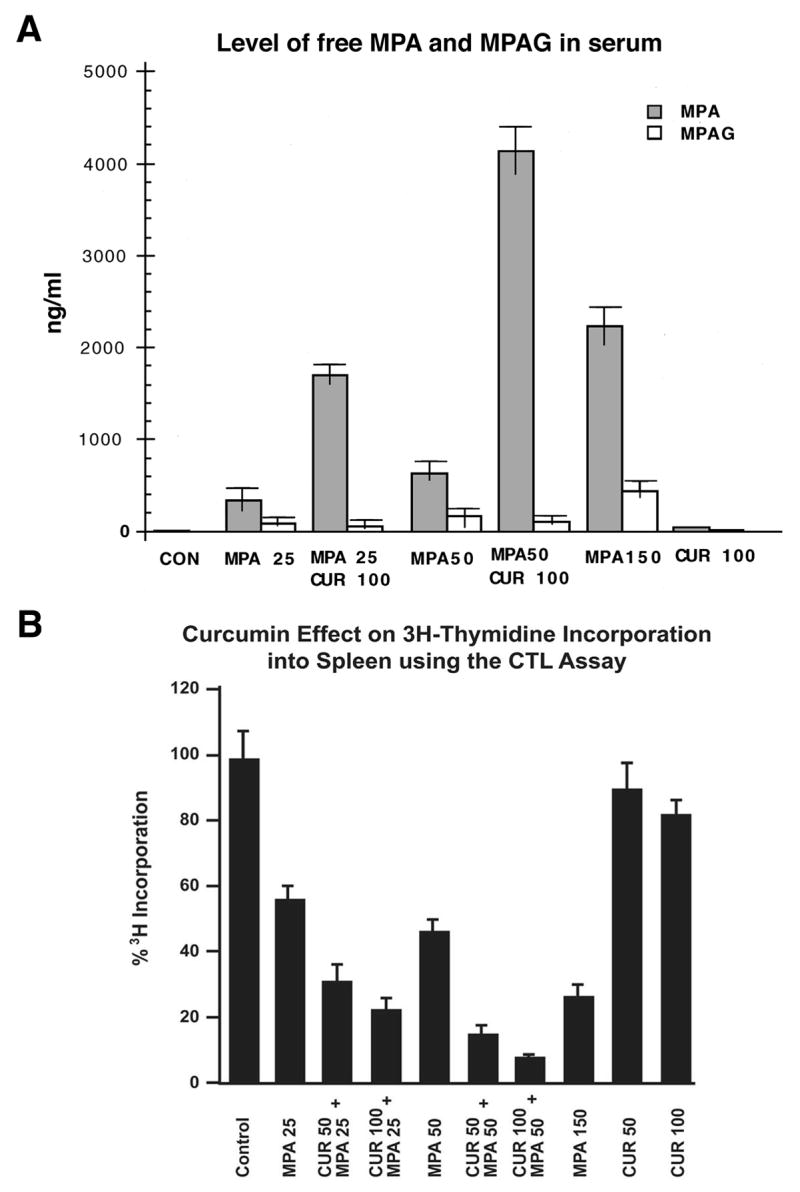
(A) Effect of oral pretreatment with curcumin on free MPA in blood and (B) MPA immunosuppression of CTL proliferation in mouse spleen. Mice were treated with SRBC to stimulate spleen CTL proliferation as described under Methods [6]. Immediately after SRBC administration, mice received curcumin (50 or 100 mg/kg) orally, which was followed by oral administration of MPA (25-, 50-, or 150- mg/kg b.w.) 20 min later or DMSO, as defined in the graph. [3H]Thymidine was injected intraperitoneally. Mice were sacrificed, and spleens were removed and processed for radioactivity as described under Methods. Simultaneously, blood was collected to ship for HPLC analysis [11] in order to distinguish free and MPA glucuronides. CPM/mg protein were determined and expressed as a percentage (treated/control) shown in (B). [3H]-Thymidine incorporation into SRBC-stimulated control spleen cells was 633,319 ± 43,364 CPM/mg protein after subtracting non-SRBC-stimulated control.
Discussion
Despite identification of UGTs in GI mucosa using molecular biology techniques [1–3,15], it has not been possible to determine the impact of these isozymes [1] on chemical uptake due to the lack of a strategy to reversibly remove all GI activities. As liver UGTs are critical to systemically-distributed chemicals, it is likely that GI glucuronidation in mucosal cells has a significant impact on removing ingested substrates. The observation that all UGTs tested require phosphorylation (see ref 2, page 6287, paragraph 1; N.K.Basu, L. Kole, A. Garza, P.S.Mitra, and I.S. Owens, Manuscript submitted), which is downregulated by dietary curcumin, provided an important strategy to analyze the impact of GI glucuronidation. Hence, we utilized curcumin to downregulate UGT phosphorylation reversibly and, thus, glucuronidation to demonstrate between a 6- and 9-fold improvement in free drug uptake and therapeutic efficacy, using the potent immunosuppressant MPA and the sensitive CTL proliferation assay in mice as a model. Kinetically, MPA glucuronidation by mouse recombinant Ugt1a1, like human UGT isozymes [12], required millimolar concentrations to reach saturation kinetics driven by Km value of at least 0.25 mM. Moreover, we were able to confirm with endogenous metabolite, bilirubin, that curcumin inhibition of GI glucuronidation caused a 2-fold increase in portal-blood free bilirubin in mouse duodenal loop with a corresponding 53 % decrease in its glucuronide. Because many orally-administered drugs have been shown to undergo glucuronidation under in-vitro conditions with recombinant UGTs, it is possible that GI-glucuronidation has an impact on a significant number of medications leading to premature removal. It remains to be seen, however, whether the significant improvement in both serum free-MPA level and immunosuppression in response to downregulation of GI-distributed glucuronidating activities in mice has human relevance.
While UGT location in GI mucosa cells [1] suggests it is a strategic site for manipulating glucuronidation by either negative or positive regulators of activities, it is important that evidence for each UGT tested with curcumin [2,3] requires on-going phosphorylation. At appropriate concentrations, the literature indicates chemopreventive curcumin acts to scavenge free radicals and interrupt PKC signaling triggered by oxidants [23]. Specifically, we have acquired evidence [N.K. Basu, L. Kole, A. Garza, P.S. Mitra, and I. S. Owens, Manuscript submitted] that UGTs undergo phosphorylation-dephosphorylation cycling that is regulated via signaling. Moreover, our demonstration that mouse Ugt requires phosphorylation in this study showing curcumin or calphostin-C disrupts Ugt1a1 phosphoserine content in parallel with loss of activity provides additional evidence that UGTs in each species require phosphorylation.
An important observation is that control mouse microsomes isolated from surgically established duodenal loops, but otherwise untreated, showed MPA glucuronidation was 5-fold higher than that by non-surgical control microsomes: compare 7,852 units for MPA (Fig. 3B) and 1,582 units (Fig. 3C) for normal duodenal microsomes. This observation suggests trauma in GI-tissue alone can have a significant effect on UGT activity.
While chemopreventive curcumin [24] at appropriate concentrations downregulates phosphorylation, it also undergoes glucuronidation by both human and mouse GI-distributed UGT(s). Thus, low remaining activity conjugates the chemical leading to progressive reversal of inhibition making it, at least, a model for global inhibition of UGTs in GI. As endogenous toxin, bilirubin, is glucuronidated by hepato-distributed human UGT1A1 and mouse Ugt1a1 following directed transport to the liver from its site of synthesis in spleen, it is unlikely that orally administered curcumin inhibits liver glucuronidation. Since available human studies showed 8 gm daily, the highest bulk-volume acceptable to patients [21], provided the highest attainable serum level of 1.77 μM peaking between 1 and 2 hr, it is unlikely that curcumin reached sufficient levels to inhibit bilirubin glucuronidation in liver. Our in-cellulo data requiring 50 μM or more for inhibition (1,12) suggest 1.77 μM is more than an order of magnitude below inhibitory levels. Importantly, inhibition with 100 mg/kg administered orally in this study suggests GI mucosa cells accumulated much higher levels of curcumin than the maximum achievable serum level of 1.77μM. These are questions that require further study.
Whereas the supra level of activity in the mice after 5 hr of recovery (Fig. 3C) suggests curcumin was not toxic, this natural product and active component of turmeric is known to have highly beneficial medicinal effects, including anti-inflammatory, anti-tumorigenic [21,25] and anti-oxidant actions already cited [20,21,26, (N.K. Basu, L. Kole, A. Garza, P. S. Mitra, and I. S. Owens, Manuscript submitted)]. More importantly, literature searches show curcumin has been widely studied in human and animal models without a report of toxicity even when consumed in extraordinarily large quantities. Hence, use of ‘nontoxic’ curcumin in this model of immunosuppression by MPA provides strong evidence only for ‘proof of principle’ that glucuronidation can adversely affect drug efficacy.
In summary, the magnitude of the effect of inhibiting GI glucuronidation on chemical uptake and drug efficacy provides the first evidence that this metabolic process can have a substantial impact on uptake of orally-administered glucuronidatable drugs and/or dietary agents. Our findings also raise the possibility that consumption of certain agents may downregulate UGTs when maximum activity is necessary to remove toxins, an issue that also warrants further investigation.
Supplementary Material
Acknowledgments
We thank Dr. Abhijit Ghosh for his help with animal experimentations. We also acknowledge the funding from the NICHD Intramural Research Program that made this research possible.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Nikhil K. Basu, Heritable Disorders Branch, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892
Labanyamoy Kole, Heritable Disorders Branch, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892.
Mousumi Basu, Heritable Disorders Branch, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892.
Antony F. McDonagh, Division of Gastroenterology and the Liver Center, Department of Medicine, University of California at San Francisco, San Francisco, CA 94143
Ida S. Owens, Heritable Disorders Branch, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892
References
- 1.Basu NK, Ciotti M, Hwang MS, Kole L, Mitra PS, Cho JW, Owens IS. J Biol Chem. 2004;279:1429–1441. doi: 10.1074/jbc.M306439200. [DOI] [PubMed] [Google Scholar]
- 2.Basu NK, Kovarova M, Garza A, Kubota S, Saha T, Mitra PS, Banerjee R, Rivera J, Owens IS. Proc Natl Acad Sci USA. 2005;102:6285–6290. doi: 10.1073/pnas.0407872102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basu NK, Kole L, Owens IS. Biochem Biophys Res Commun. 2003;303:98–104. doi: 10.1016/s0006-291x(03)00241-9. [DOI] [PubMed] [Google Scholar]
- 4.Allison AC, Eugui EM. Transplantation. 2005;80:S181–S190. doi: 10.1097/01.tp.0000186390.10150.66. [DOI] [PubMed] [Google Scholar]
- 5.MacPhee IAM, Spreafico S, Bewick M, Davis C, Eastwood JB, Johnston A, Lee T, Holt DW. Kidney Internat. 2000;57:1164–1168. doi: 10.1046/j.1523-1755.2000.00943.x. [DOI] [PubMed] [Google Scholar]
- 6.Eugui EM, Mirkovich A, Allison AC. Scand J Immunol. 1991;33:175–183. doi: 10.1111/j.1365-3083.1991.tb03747.x. [DOI] [PubMed] [Google Scholar]
- 7.Sintchak MD, Fleming MA, Futer O, Raybuck SA, Chambers SP, Caron PR, Murcko MA, Wilson KP. Cell. 1996;85:921–930. doi: 10.1016/s0092-8674(00)81275-1. [DOI] [PubMed] [Google Scholar]
- 8.Cohn RG, Mirkovich A, Dunlap B, Burton P, Chiu SH, Eugui E, Caulfield JP. Transplantation. 1999;68:411–418. doi: 10.1097/00007890-199908150-00014. [DOI] [PubMed] [Google Scholar]
- 9.Wieland E, Shipkova M, Schellhaas U, Shütz E, Niedmann PD, Armstrong VW, Oellerich M. Clin Biochem. 2000;33:107–113. doi: 10.1016/s0009-9120(99)00101-0. [DOI] [PubMed] [Google Scholar]
- 10.Shipkova M, Beck H, Voland A, Armstrong VW, Hermann-Josef G, Oellerich M, Wieland E. Proteomics. 2004;4:2728–2738. doi: 10.1002/pmic.200300836. [DOI] [PubMed] [Google Scholar]
- 11.Shipkova M, Armstrong VM, Wieland E, Niedmann PD, Schütz E, Brenner-Weiss G, Voihsel M, Braun F, Oellerich M. Br J Pharmacol. 1999;126:1075–1082. doi: 10.1038/sj.bjp.0702399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basu NK, Kole L, Kubota S, Owens IS. Drug Metab Dispos. 2004;32:768–773. doi: 10.1124/dmd.32.7.768. [DOI] [PubMed] [Google Scholar]
- 13.Lin JK. Arch Pharm Res. 2004;27:683–692. doi: 10.1007/BF02980135. [DOI] [PubMed] [Google Scholar]
- References 14–17 are in the Supplement under supporting material.
- 18.Zhang T, Haws P, Wu Q. Genome Res. 2004;14:79–89. doi: 10.1101/gr.1225204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mackenzie PI. Ther Drug Monit. 2000;22:10–13. doi: 10.1097/00007691-200002000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Garcea G, Berry DP, Jones DJ, Singh R, Dennison AR, Farmer PB, Sharma RA, Steward WP, Gescher AJ. Cancer Epidemiol Biomarkers Prev. 2000;14:120–125. [PubMed] [Google Scholar]
- 21.Cheng AL, Hsu CH, Lin JK, Hsu MM, Ho YF, Shen TS, Ko JY, Lin JT, Lin BR, Ming-Shiang W, Yu HS, Jee SH, Chen GS, Chen TM, Chen CA, Lai MK, Pu YS, Pan MH, Wang YJ, Tsai CC, Hsieh CY. Anticancer Res. 2001;21:2895–2900. [PubMed] [Google Scholar]
- 22.Lang S, Picu A, Hofmann T, Andratschke M, Mack B, Moosmann A, Gires O, Tiwari S, Zeidler R. Int J Immunopathol Pharmacol. 2006;19:409–419. doi: 10.1177/039463200601900217. [DOI] [PubMed] [Google Scholar]
- 23.Gopalakrishna R, Jaken S. Free Radic Biol Med. 2000;28:1349–1361. doi: 10.1016/s0891-5849(00)00221-5. [DOI] [PubMed] [Google Scholar]
- 24.Dorai T, Aggarwal BB. Cancer Lett. 2004;215:129–140. doi: 10.1016/j.canlet.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 25.Churchill M, Chadburn A, Bilinski RT, Bertagnolli MM. J Surg Res. 2000;89:169–175. doi: 10.1006/jsre.2000.5826. [DOI] [PubMed] [Google Scholar]
- 26.Pereira MA, Grubbs CJ, Barnes LH, Li H, Olson GR, Eto I, Juliana M, Whitaker LM, Kelloff GJ, Steele VE, Lubet RA. Carcinogenesis. 1996;17:1305–1311. doi: 10.1093/carcin/17.6.1305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.