

Abstract
Gene conversion plays an important part in shaping genetic diversity in populations, yet estimating the rate at which it occurs is difficult because of the short lengths of DNA involved. We have developed a new statistical approach to estimating gene conversion rates from genetic variation, by extending an existing model for haplotype data in the presence of crossover events. We show, by simulation, that when the rate of gene conversion events is at least comparable to the rate of crossover events, the method provides a powerful approach to the detection of gene conversion and estimation of its rate. Application of the method to data from the telomeric X chromosome of Drosophila melanogaster, in which crossover activity is suppressed, indicates that gene conversion occurs ∼400 times more often than crossover events. We also extend the method to estimating variable crossover and gene conversion rates and estimate the rate of gene conversion to be ∼1.5 times higher than the crossover rate in a region of human chromosome 1 with known recombination hotspots.
AN important concept in the description of genetic variation is linkage disequilibrium (LD), the nonrandom association of alleles at different locations along the genome. Disease association studies rely heavily on knowledge of patterns of LD, both in pinpointing complex disease genes precisely and in performing genomewide studies (Pritchard and Przeworski 2001). Over long ranges, LD is mainly affected by crossover, which has been studied and modeled by many authors and is reviewed by Stumpf and Mcvean (2003). A less well-known form of recombination is homologous gene conversion, a nonreciprocal process acting on short lengths of DNA, where genetic material from one parental chromosome is incorporated into the alternate chromosome during meiotic exchange (Szostak et al. 1983). Crossover events in fact include a gene conversion tract but this cannot be detected using population-based methods, and we therefore use the term gene conversion only to refer to gene conversion events that are not accompanied by crossover.
In humans, gene conversion is thought to occur ∼4–15 times as frequently as crossover (Jeffreys and May 2004), but is more difficult to detect due to the short lengths of DNA transferred. Estimates of the tract length vary between studies and between organisms/regions but tend to lie between 50 and 2000 bp (e.g., Borts and Haber 1989; Hilliker et al. 1994; Jeffreys and May 2004). For a full description of the gene conversion process see Stahl (1994) and references therein.
Patterns of linkage disequilibrium in humans can be satisfactorily explained only by models including gene conversion (Frisse et al. 2001). Simulations show that in genomic regions that have been subject to gene conversion, estimates of the crossover rate are inflated when gene conversion is ignored (Smith and Fearnhead 2005). Przeworski and Wall (2001) showed, using human population genetic data, that gene conversion is likely to be an important factor in explaining a marked difference between estimates of the population recombination rate obtained through comparing genetic and physical maps and those found through analysis of nucleotide sequence polymorphism data. These factors have made gene conversion the subject of much investigation in recent years.
Although highly localized, the effects of gene conversion may also have a significant impact on association studies, which seek a genotyped marker that is in strong LD with an untyped allele responsible for the phenotype of interest. If gene conversion is ignored, the extent of LD over short distances is likely to be overestimated, while LD at longer distances will be underestimated due to the inflated rate of crossover needed to explain the short-range LD (Frisse et al. 2001). These two influences on LD may affect the choice of the number of markers to genotype for a study (Schork 2002).
Gene conversion also affects our ability to detect the effects of natural selection on a population (Andolfatto and Nordborg 1998). Tests for deviation from the null model typically rely on an estimate of the recombination rate in a region, and ignoring the effects of gene conversion will reduce the power of tests for selection and can also increase the false positive rate of such tests.
Finally, learning about gene conversion could help us to gain biological and mechanistic insights into recombination.
It is therefore desirable to be able to estimate the frequency at which gene conversion events occur, at a fine scale, over genomic regions many megabases in length and to detect variation in gene conversion rates within such a genomic region.
Rates of both crossover and gene conversion can be estimated directly using sperm-typing experiments such as those of Jeffreys and May (2004) that give highly accurate fine-scale rates, but cannot be performed on a genomic scale or on the X chromosome or in females.
Pedigree studies (e.g., Kong et al. 2002) can give further information such as sex-specific differences in crossover rates, but because of the infrequency of events, cannot give accurate fine-scale maps.
For genomewide fine-scale characterization of recombination rates a practical solution is statistical modeling of genetic data, based on simplified assumptions about the historical processes that resulted in the population genetic data seen today. Methods of inference can be performed in many different ways:
Summary statistics (e.g., Wiehe et al. 2000; Padhukasahasram et al. 2006) can sometimes be quick to calculate but make use of partial information only and are not able to detect fine-scale variation.
Composite likelihoods calculated using pairs or triplets of segregating sites (e.g., Frisse et al. 2001; Ptak et al. 2004) can provide a “reasonable” estimate of the gene conversion rate (i.e., within a factor of 2 of the truth) given sufficient data (Wall 2004). Fearnhead et al. (2005) applied one such method (Hudson 2001) to bacterial data sets and obtained some interesting results, including tract length estimates. However, for densely typed SNP data where there are likely to be high levels of LD, composite-likelihood methods may be unsuitable as they ignore the dependency between nearby pairs/triplets of SNPs.
Full-likelihood methods approximate the probability of the data under the assumed population genetic model (exact probabilities are not available due to the unknown history of the sample). Some use techniques such as importance sampling (Fearnhead and Donnelly 2001) to make the approximation, while others use a simplified model under which exact probabilities can be found (e.g., Li and Stephens 2003; Hellenthal 2006). The main benefit of the full-likelihood approach is to make use of as much of the information in the data as possible, and in the case of gene conversion we expect this to be important.
In this article we describe a statistical model of population genetic data that includes both crossover and gene conversion, where a gene conversion tract can include any number of markers. The model can be used to estimate the rates of crossover and/or gene conversion in a given region using maximum-likelihood techniques or could be implemented in a Bayesian framework. The model does not require that either rate be constant across the region of interest and could, for example, be used to obtain an estimate of the gene conversion rate in a region known to include a crossover hotspot. As well as performing tests on simulated data, we examine single-nucleotide polymorphism (SNP) data from a genomic region thought to be free from crossover hotspots and then consider a region of the human genome that contains several crossover hotspots (Jeffreys et al. 2005).
Our results on simulated data show that gene conversion rates can be estimated fairly accurately from population genetic data, and the inclusion of gene conversion in our model results in improved estimates of the crossover rate, particularly when gene conversion is present at high levels. In a region near the telomere of the X chromosome of Drosophila melanogaster we find that gene conversion events occur >400 times as frequently as crossovers, while in a region of human chromosome 1, there is only 1.5 times as much gene conversion activity as crossover.
MODEL
Our model is an extension of the coalescent-based model of Li and Stephens (2003) (henceforth abbreviated to LS model) to include gene conversion as well as crossover. Li and Stephens modeled the probability of seeing a particular chromosomal segment, given any other homologous segments already seen, and given the rates of mutation θ and crossover ρ = 4Nec, where Ne is the effective population size and c is the per-generation probability of crossover between adjacent base pairs. We use the terms haplotype and chromosome interchangeably to refer to a chromosomal segment and assume the method will be applied to resequenced or densely genotyped SNP data, although it could also be applied to microsatellite data with a suitably adjusted emission probability.
Our approach has the following properties, some of which are novel:
Gene conversion tract lengths may be arbitrarily long.
SNPs can be arbitrarily densely situated in the region of interest, allowing for multiple-SNP gene conversion tracts.
Crossover and gene conversion rates may vary across the region of interest.
Estimates can be obtained jointly for the crossover rate and the gene conversion rate (and in theory, also for the gene conversion tract length, but in settings where the tract length is short relative to the average SNP spacing there is little information in the data to pinpoint this).
It is model-based and calculates (an approximation to) the likelihood, so can provide estimates of uncertainty.
We chose the LS model because it does not rely on summary statistics, but attempts to use all the available information, albeit under an approximation to the likelihood, making it an ideal candidate for extension to the gene conversion model. We expect the trace of gene conversion to be difficult to detect and therefore wish to use the maximum information that can be extracted from the data.
We first introduce briefly the LS model for crossover alone and then describe the addition of gene conversion to this model. We validate our method using tests on data simulated with a range of parameter values and evaluate its robustness to deviations from our assumptions about population demographics. Finally we generalize the model to allow for variation in the rate of gene conversion.
Modeling crossover using a likelihood-based approach:
The objective of maximum-likelihood methods is to maximize the function L(Θ) = Pr(H | Θ), i.e., the likelihood of a set of model parameters Θ given the sampled data (haplotypes) H = h1, h2,…, hn.
If we knew the underlying genealogy of the sampled individuals, this could be calculated directly. However, this information, in a population genetic sample of unrelated individuals, is not available. In the presence of recombination, the individuals sampled may be related by a different (correlated) phylogenetic tree at each polymorphic site along the sequence [which, together, form the ancestral recombination graph (ARG) of Griffiths and Marjoram 1997], and phylogenetic methods are unreliable under these circumstances (Schierup and Hein 2000). It is therefore useful to develop an approximation to L that is not conditional on the ARG G relating the sampled individuals, using
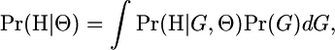 |
(1) |
where Pr(G) is the probability density function of the ARG relating the haplotypes H. One highly robust and flexible way to model Pr(G) is the coalescent with recombination (Kingman 1982; Hudson 1983; Griffiths and Marjoram 1997). This assumes a panmictic population of constant size, undergoing only neutral evolution. We base our model and the majority of our simulations on the standard coalescent, but we also investigate the accuracy of our method when it is applied to data that deviate from the assumed coalescent model.
Li and Stephens (2003) noted that
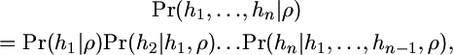 |
(2) |
where hi denotes the ith haplotype in the data set of n haplotypes, and ρ = 4Nec is the population crossover rate. By approximating each of the terms on the right-hand side in turn, they arrived at an approximation to the likelihood known as a product of approximate conditionals (PAC) model.
Their approximation 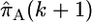 to the conditional probability Pr(hk+1 | h1,…, hk, ρ) is a modification of the imperfect mosaic model of Fearnhead and Donnelly (2001). Haplotype k + 1 is considered to be made up of segments copied from any or all of the preceding k haplotypes, and at marker l the haplotype being copied from is known as the “nearest neighbor.” The copying process can also be imperfect, giving rise to a difference between the new haplotype and its nearest neighbor; this is considered to be a mutation. When the nearest neighbor changes between marker i and marker i + 1 this is considered to be a crossover. The sequence of nearest neighbors taken when traversing the (k + 1)th haplotype from one end to the other can be modeled as a Markov chain where the nearest neighbor at a given marker is dependent only on that at the previous marker and on the crossover probability. The likelihood given a particular value of the parameter ρ (which may vary across the region) is then calculated by summing over all possible mosaic structures and a maximum-likelihood estimate
to the conditional probability Pr(hk+1 | h1,…, hk, ρ) is a modification of the imperfect mosaic model of Fearnhead and Donnelly (2001). Haplotype k + 1 is considered to be made up of segments copied from any or all of the preceding k haplotypes, and at marker l the haplotype being copied from is known as the “nearest neighbor.” The copying process can also be imperfect, giving rise to a difference between the new haplotype and its nearest neighbor; this is considered to be a mutation. When the nearest neighbor changes between marker i and marker i + 1 this is considered to be a crossover. The sequence of nearest neighbors taken when traversing the (k + 1)th haplotype from one end to the other can be modeled as a Markov chain where the nearest neighbor at a given marker is dependent only on that at the previous marker and on the crossover probability. The likelihood given a particular value of the parameter ρ (which may vary across the region) is then calculated by summing over all possible mosaic structures and a maximum-likelihood estimate 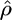 can therefore be found.
can therefore be found.
It is worth noting that this approximation to the likelihood is dependent on the order in which the haplotypes are observed. This unwelcome influence can be greatly reduced by averaging the likelihood over a number of different random orders. We find 20 orders sufficient to ensure that our estimates were consistent between different runs of the program and >20 to be cumbersome in terms of computational time. All results shown in this article are based on 20 orders chosen uniformly at random except where stated otherwise.
The LS model was previously extended by Hellenthal (2006) to include gene conversion, assuming each gene conversion tract includes only one SNP. In essence, the emission probability for the Markov chain is modified to mimic a gene conversion. This has the benefit of keeping the computational cost the same (O(N2)), but suffers from a difficulty in distinguishing gene conversion and genotyping error. Our adaptation of the LS model is much more computationally intensive but can be applied to more densely typed SNP data.
Modeling gene conversion:
We now consider our sample to have been affected both by crossover and by gene conversion events throughout its history. This scenario is well modeled by the coalescent model with gene conversion developed by Wiuf and Hein (2000). The imperfect mosaic model can be easily adapted to allow for gene conversion, by allowing a second process to alter our nearest neighbor from x to x′, with the proviso that we must eventually return to copying from x. See Figure 1 for an illustration of this. The distribution of lengths of gene conversion tracts can be approximated by a geometric distribution (Hilliker et al. 1994), giving a constant probability of ending the tract at any particular position and returning to copy from x, irrespective of the length of the tract so far. This lack of memory property allows us to use a Markov chain implementation of the model as above.
Figure 1.—

Illustration of the imperfect mosaic model with gene conversion. We construct the new haplotype h4 as a mosaic of pieces copied from existing haplotypes h1–h3. The haplotype copied at a particular point is known as the nearest neighbor at that point, and the nearest neighbor can change when we encounter a gene conversion event, such as the one between a and b, or a crossover event (c).
We make the following additional assumptions:
Crossover events occur independently of gene conversion events.
Gene conversion events cannot overlap or be nested.
The gene conversion rate may change instantaneously at each typed marker but cannot change within the interval between adjacent SNPs.
The first of these assumptions allows us to separate the gene conversion and crossover processes in our model, which simplifies the calculation of the transition probabilities in our Markov chain. It is also biologically reasonable in that we would not expect that the fact that a crossover had once occurred in a particular region to influence the probability that a gene conversion occurs in any given meiosis in that region, except in that a higher rate of crossover might point to a potentially higher rate of gene conversion. Our assumption does not disallow dependence between rates of gene conversion and crossover, only between events.
In our second assumption, we specify the conditions on entering and exiting a gene conversion event. We may begin a new gene conversion tract only when we are not already in a tract, and we may end a tract only by returning to copy from the haplotype we were copying from before the tract began. Allowing tracts to be nested and/or to overlap would violate the Markovian property of our model or necessitate the addition of one or more further dimensions to the model. There is no clear biological interpretation of this assumption. It is certainly reasonable to state that any gene conversion event taking place during a particular meiosis cannot overlap with or be nested within another tract occurring in the same meiosis. The trace left in population genetic data by many independent gene conversion events over a long period of time, perhaps occurring in hotspots and likely to overlap with previous events, is less obvious. When tract lengths are short compared to SNP spacing, we do not expect this assumption to have any effect (two or more SNPs must be in a gene conversion tract for overlapping or nesting to be detectable). When tract lengths are long, we might expect to miss some overlapping gene conversion tracts or see them as crossovers, thus giving a slight underestimate of the gene conversion rate and overestimate of the crossover rate.
Our final assumption is also one of convenience. We have no information about any variation in the rates of gene conversion and crossover in the gap between any pair of adjacent typed SNPs, and we therefore assume the rate is constant. In this article we are mainly considering regions where the rates of crossover and gene conversion are considered to be uniform, but the method also allows for rate variation. When rate variation exists, in this model the rate is permitted to change instantaneously only at a typed marker, and the rate in an interval will correspond to the average rate over the gap between the SNPs.
Details of our implementation of this model are in the appendix. In the next section we describe the results of applying this model to simulated data with the aim of jointly estimating γ = 4Neg and ρ = 4Nec, where g denotes the gene conversion rate per meiosis per unit distance.
RESULTS
Simulation study:
To test the performance of our method we undertook a simulation study. Data sets were simulated using the program ms (Hudson 2002). Each data set contains 50 haplotypes of length 20 kb, simulated with mutation rate θ = 0.5, 1, or 2.5; crossover rate ρ = 0, 0.5, 1 or 2.5; and gene conversion rate γ = 0, 1, or 10 (per kilobase). We focus on the data sets with θ = 1 as this corresponds to the human population-scaled mutation rate of ∼0.7–1.0/kb (Ptak et al. 2004). The mean gene conversion tract length, 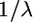 , was fixed at 500 bp (cf. Frisse et al. 2001) for the simulations and during estimation of parameters. The number of SNPs in each data set varied with the mutation rate and, when θ = 1, averaged 89 SNPs per simulated data set.
, was fixed at 500 bp (cf. Frisse et al. 2001) for the simulations and during estimation of parameters. The number of SNPs in each data set varied with the mutation rate and, when θ = 1, averaged 89 SNPs per simulated data set.
Our estimates 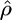 and
and 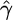 are shown in Figures 2a and 3 and summarized in Table 1. In each case we fixed the gene conversion tract length parameter at the value used to simulate data.
are shown in Figures 2a and 3 and summarized in Table 1. In each case we fixed the gene conversion tract length parameter at the value used to simulate data.
Figure 2.—

Comparison of maximum-likelihood estimates of ρ on data simulated with different values of f (all data simulated with ρ = θ = 1/kb) using our model (a) as compared to the LS model without bias correction (b). Our model gives good estimates of the crossover rate regardless of the amount of gene conversion present. It shows little bias, and when gene conversion is present (at least in simulated data), estimates of crossover rates can be inflated when gene conversion is not taken into account.
Figure 3.—

Maximum-likelihood estimates of γ on data simulated with ρ = θ = 1/kb using our model. Estimates obtained for data with high levels of gene conversion activity are very encouraging (999 of 1000 within a factor of 2 of the truth), but we tend to overestimate the level of gene conversion present when it is low or nonexistent. This is inevitable due to the true value being at the boundary of the range of possible values.
TABLE 1.
Summary of results of testing done on simulated data
| Simulated parameters | Deviation from model | Median 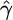
|
Median 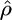
|
Median 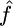
|
|---|---|---|---|---|
| γ = 0, ρ = 1 (f = 0) | None | 0.5332 (11.5) | 0.9706 (96.9) | 0.55 (11.5) |
| Growth | 0.5863 (16) | 0.9045 (89) | 0.586 (16) | |
| Bottleneck | 0.4777 (18) | 0.8678 (95) | 0.516 (18) | |
| Structure | 0.5472 (5) | 0.9415 (97) | 0.634 (5) | |
| Selection | 0.6031 (8) | 0.9464 (98) | 0.667 (8) | |
| γ = 1, ρ = 1 (f = 1) | None | 1.348 (77.3) | 0.9323 (95.8) | 1.45 (62.9) |
| Growth | 1.344 (71) | 0.8907 (94) | 1.36 (55) | |
| Bottleneck | 1.286 (75) | 0.8868 (97) | 1.43 (64) | |
| Structure | 1.401 (84) | 0.9077 (98) | 1.48 (67) | |
| γ = 10, ρ = 1 (f = 10) | None | 9.428 (99.9) | 0.9708 (94.0) | 9.54 (90.1) |
| Growth | 7.805 (97) | 0.8738 (92) | 9.08 (87) | |
| Bottleneck | 8.315 (97) | 0.8997 (94) | 8.91 (82) | |
| Structure | 8.455 (99) | 0.8927 (93) | 9.50 (89) | |
| Genotype | 7.49 (97) | 1.21 (93) | 6.24 (68) |
We first simulated data sets according to standard model assumptions (constant sized, panmictic population with neutral evolution), with θ = 1, and a variety of values of f. For each set of parameters 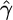 ,
, 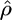 , and
, and 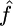 , the median estimate for 1000 independent simulations is given, with the proportion of data sets for which the estimate lies within the range (truth/2, truth × 2) in parentheses. In the case f = 0, we see that in 11.5% of cases we found
, the median estimate for 1000 independent simulations is given, with the proportion of data sets for which the estimate lies within the range (truth/2, truth × 2) in parentheses. In the case f = 0, we see that in 11.5% of cases we found 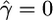 . However, for 82.8% of these data sets,
. However, for 82.8% of these data sets, 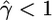 /kb. We also show the corresponding results for 100 data sets simulated with a range of deviations from the above assumptions (see text). These results are broadly similar to those with no deviation, except in the case of rephased data where the crossover rate is overestimated.
/kb. We also show the corresponding results for 100 data sets simulated with a range of deviations from the above assumptions (see text). These results are broadly similar to those with no deviation, except in the case of rephased data where the crossover rate is overestimated.
Estimation of ρ:
The distributions of our estimates 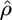 for data sets simulated with no gene conversion, equal rates of gene conversion and crossover (f = 1), and more gene conversion than crossover (f = 10) are shown in Figure 2a. The presence of gene conversion does not seem to have a detrimental effect on our ability to estimate ρ, and estimates of ρ are within a factor of 2 of the truth >90% of the time for each value of f. All the simulations shown used ρ = 1/kb, and simulations with ρ = 0.5 and ρ = 2.5/kb had similar results with slightly reduced accuracy. For comparison, estimates for ρ obtained using the LS model (without bias correction) on the same data sets are shown in Figure 2b. In the case where f = 10, these estimates are highly inflated, not surprisingly since this method is not intended for data sets where gene conversion is present. However, the fact that for all 1000 data sets with f = 10 this method gives an estimate for ρ that is more than twice the true value serves as a reminder of the effect that undetected gene conversion can have on estimates of the crossover rate.
for data sets simulated with no gene conversion, equal rates of gene conversion and crossover (f = 1), and more gene conversion than crossover (f = 10) are shown in Figure 2a. The presence of gene conversion does not seem to have a detrimental effect on our ability to estimate ρ, and estimates of ρ are within a factor of 2 of the truth >90% of the time for each value of f. All the simulations shown used ρ = 1/kb, and simulations with ρ = 0.5 and ρ = 2.5/kb had similar results with slightly reduced accuracy. For comparison, estimates for ρ obtained using the LS model (without bias correction) on the same data sets are shown in Figure 2b. In the case where f = 10, these estimates are highly inflated, not surprisingly since this method is not intended for data sets where gene conversion is present. However, the fact that for all 1000 data sets with f = 10 this method gives an estimate for ρ that is more than twice the true value serves as a reminder of the effect that undetected gene conversion can have on estimates of the crossover rate.
Estimation of γ:
The distributions of our estimates 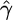 for the same data sets as those above are shown in Figure 3. In the case where f = 10, our estimated γ was within a factor of 2 of the value used to simulate data, for 999 of the 1000 simulated data sets. Results are summarized in Table 1.
for the same data sets as those above are shown in Figure 3. In the case where f = 10, our estimated γ was within a factor of 2 of the value used to simulate data, for 999 of the 1000 simulated data sets. Results are summarized in Table 1.
Estimation of f:
We used our estimates of ρ and γ for each data set to obtain an estimate 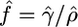 . These estimates were also generally close to the truth but suffered slightly from being a ratio of two other estimates with uncertainty in both. For data sets simulated with θ = 1 our median estimates of f were 0.55, 1.45, and 9.54 for data sets simulated with true f 0, 1, and 10, respectively.
. These estimates were also generally close to the truth but suffered slightly from being a ratio of two other estimates with uncertainty in both. For data sets simulated with θ = 1 our median estimates of f were 0.55, 1.45, and 9.54 for data sets simulated with true f 0, 1, and 10, respectively.
Robustness to deviation from assumed demography:
Here we use additional simulated data to evaluate the robustness of our model to deviations from the assumed neutral model. We consider our three major demographic assumptions: constant population size, panmixia (random mating), and neutral evolution. In each case, we simulated 100 data sets with 50 haplotypes, 20 kb in length, with mutation and crossover rates of 1/kb, and with various gene conversion rates.
Variation in population size:
To test our model in the presence of population size variation, we simulated 100 data sets with a bottleneck 0.15 Ne generations ago that reduced diversity to 85% of that expected without the bottleneck and a further 100 data sets with scaled exponential population growth parameter 1 (cf. McVean et al. 2004). Results under these demographic variations (summarized in Table 1) do not deviate far from those obtained on data simulated under the standard model, although we see a slight increase in our underestimation of the gene conversion rate when f is high.
Population structure:
To test our model in a nonrandom mating scenario, we simulated 100 data sets, where 25 of the 50 chromosomes were sampled from each of two subpopulations corresponding to a level of population differentiation of FST ≈ 0.2 (cf. Pritchard and Przeworski 2001). Properties of the resulting estimates for 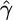 are shown in Table 1. Although in each simulation, the variance (not shown) of
are shown in Table 1. Although in each simulation, the variance (not shown) of 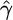 was higher than that for a single-population data set, this did not have a big effect on the median estimate or the proportion of results within a factor of 2 of the truth. The estimates for
was higher than that for a single-population data set, this did not have a big effect on the median estimate or the proportion of results within a factor of 2 of the truth. The estimates for 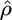 were similarly affected.
were similarly affected.
Selection:
As a final test we simulated 100 data sets where a positive selective sweep had just finished. These data were generated using the program SelSim (Spencer and Coop 2004). The strength of selection was chosen to be σ = 2Nes = 50, where s is the selective coefficient between homozygotes (cf. Smith and Fearnhead 2005), and was applied to a single site in the center of the 20-kb region. Again we saw no major difference in our results, implying that this method is robust to low to moderate levels of selection (see Table 1).
Genotype data:
To use our method on genotype data it is necessary to first phase the data. Currently available programs to phase genotype data do not take gene conversion into account, so we investigated the effect of performing this preprocessing of the data. For 100 of the above data sets simulated with f = 10, we randomly paired the 50 haplotypes into 25 individuals and then used the program PHASE (Stephens et al. 2001; Stephens and Scheet 2005) to rephase the data. We then obtained estimates for the gene conversion and crossover rates on these data sets, which are summarized in Table 1. We found that our method overestimates the crossover rate under these circumstances, as well as underestimating the gene conversion rate, which leads to an underestimate of f.
Comparison with other methods:
We now compare our results with two other methods: Hudson's pairwise composite-likelihood method (Hudson 2001) and a method based on the summary statistics method of Padhukasahasram et al. (2006). For the former, we used the program maxHap, freely available from the author's website. For the latter, we adapted a program (also available on the author's website). Our implementation differs from that described in Padhukasahasram et al. (2006) only in that we do not fix the positions of the segregating sites in our simulations. Results are shown in Table 2. For these calculations, maxHap took ∼1.7 sec per data set, summStat between 5 and 24 hr (depending on f), and GenCo just under 1 hr on a standard desktop computer.
TABLE 2.
Comparison with other methods
| Parameters | Method | γ | ρ | f |
|---|---|---|---|---|
| γ = 1, ρ = 1 (f = 1) | SummStat | 0.58 | 0.76 | 0.49 |
| MaxHap | 0.63 | 0.89 | 0.52 | |
| GenCo | 0.77 | 0.96 | 0.63 | |
| γ = 10, ρ = 1 (f = 10) | SummStat | 0.83 | 0.77 | 0.57 |
| MaxHap | 0.99 | 0.81 | 0.77 | |
| GenCo | 1 | 0.94 | 0.9 |
For 1000 data sets simulated with ρ = 1 and γ ∈ (1, 10), we compare our results (GenCo) with those from maxHap (Hudson 2001), and for 100 of the same data sets we also show results obtained using a third method (summStat) based on that of Padhukasahasram et al. (2006) (see text). MaxHap was run over a grid of 11 f values ranging from 0 to 2.5 or 25 (inclusive) for the simulated data sets with f = 1 and f = 10, respectively. SummStat was run on a coarse grid of ρ ∈ (8, 10, 20, 40, 45), and γ ∈ (8, 10, 20, 40, 45) for the first test and γ ∈ (80, 100, 200, 400, 450) for the second test. For each parameter γ, ρ, and f we present the proportion of data sets for which the estimated value was within a factor of 2 of the value used to simulate data.
D. melanogaster:
A particularly interesting organism in the study of LD is D. melanogaster, because of its unusual patterns of recombination. We applied our method to SNP data from two genes near the telomere of the X chromosome of African D. melanogaster (Langley et al. 2000). This data set consists of 87 SNPs within the su(s) and su(w) genes, which are involved in the regulation of gene expression (Fridell and Searles 1994). The genes are ∼4 and 2.5 kb long, respectively, and are separated by a region of 400 kb in which no SNPs were typed. Like chromosome 4 in Drosophila (Hochman 1976), the region near the X chromosome telomere is subject to a severely reduced level of crossover per physical length (Aguade and Langley 1994) compared to the genomewide average rate of 1.5 cM/Mb (Nachman 2002), perhaps due to regulation of double-strand-break repair mechanisms (McKim et al. 2002).
In addition to obtaining maximum-likelihood estimates for ρ and γ for this data set we also constructed a likelihood surface over a grid of values of ρ and γ, shown in Figure 4. We fixed the mean gene conversion tract length at 352 bp (Hilliker et al. 1994) and obtained 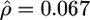 and
and 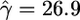 /kb (f = 432). Such a strong signal is unlikely to be explained by repeat mutation or genotyping error. These estimates support the conclusion of Langley et al. (2000) that while crossover is suppressed in the region, gene conversion is not. This could indicate that gene conversion and crossover are completely separate processes or, if both are initiated by the same process, that in this region of D. melanogaster there is a strong tendency for recombination events to be resolved as gene conversion rather than crossover. Whether this is the cause of or a consequence of the suppression of crossover is as yet unknown.
/kb (f = 432). Such a strong signal is unlikely to be explained by repeat mutation or genotyping error. These estimates support the conclusion of Langley et al. (2000) that while crossover is suppressed in the region, gene conversion is not. This could indicate that gene conversion and crossover are completely separate processes or, if both are initiated by the same process, that in this region of D. melanogaster there is a strong tendency for recombination events to be resolved as gene conversion rather than crossover. Whether this is the cause of or a consequence of the suppression of crossover is as yet unknown.
Figure 4.—

Likelihood surface for D. melanogaster data set. The maximum-likelihood point on the surface is γ = 26.8, ρ = 0.062/kb. The surface is fairly flat around this region but drops off steeply when ρ gets close to zero or γ drops below ∼15.
Variation in the rate of gene conversion:
To date there are few data regarding fine-scale variation in gene conversion rates. The clearest examples of such variation are the gene conversion hotspots experimentally identified in the center of two crossover hotspots by Jeffreys and May (2004). Padhukasahasram et al. (2006) estimated nonuniform gene conversion rates in simulated data and found that their method underestimates the gene conversion rate under these circumstances. Their method produces a single estimate of the total amount of gene conversion in a given region and does not attempt to pinpoint hotspots or to measure their intensity.
Our model allows for a different rate of gene conversion between each pair of adjacent SNPs, so it was possible to implement an expectation-maximization algorithm to determine 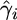 for each interval i. It should be noted that to reflect biological reality and maintain symmetry we would prefer to model the rate at which gene conversion initiation sites are encountered (i.e., somewhere around the middle of the tract), but due to the way the model is implemented we are in fact modeling the rate at which the left-hand sides of gene conversion tracts are encountered. It would be preferable to model a gene conversion tract extending in both directions from an initiation point (Hellenthal and Stephens 2007), but this would greatly increase the complexity and hence the computation time of our method.
for each interval i. It should be noted that to reflect biological reality and maintain symmetry we would prefer to model the rate at which gene conversion initiation sites are encountered (i.e., somewhere around the middle of the tract), but due to the way the model is implemented we are in fact modeling the rate at which the left-hand sides of gene conversion tracts are encountered. It would be preferable to model a gene conversion tract extending in both directions from an initiation point (Hellenthal and Stephens 2007), but this would greatly increase the complexity and hence the computation time of our method.
Instead, we map our estimates to the gene conversion rate by assuming the initiation is in the exact center of the gene conversion tract, according to the equation
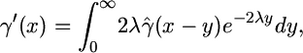 |
(3) |
where 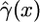 is our maximum-likelihood estimate (MLE) of the gene conversion rate at distance x from the beginning of the observed region, and a constant rate γ0 is assumed for all x < 0.
is our maximum-likelihood estimate (MLE) of the gene conversion rate at distance x from the beginning of the observed region, and a constant rate γ0 is assumed for all x < 0.
When the distances between markers are long compared to the length of a gene conversion tract, or when rates change only gradually between intervals, the difference between modeling the center and modeling the end of a gene conversion tract will be negligible. However, if very narrow hotspots of gene conversion are found, it may be necessary to convert the rate of encountering the left-hand side into the rate of gene conversion tract initiation to provide a useful gene conversion rate estimation.
To examine the power and reliability of our method when recombination rates vary, we used the program msHOT (Hellenthal and Stephens 2007) to simulate 100 data sets containing a hotspot for both gene conversion and crossover. Each data set consisted of 50 haplotypes, 20 kb in length, with θ = 1/kb, mean tract length 500 bp, and γ = 0.5 and ρ = 0.05/kb (f = 10), except in a “hotspot” 2 kb wide in the center of the region, where γ = 50 and ρ = 5/kb (f = 10).
Assuming that f was constant across the region, we obtained maximum-likelihood estimates for γ and f for each simulated data set. The estimates for γ and their median (sampled every 100 bp) are shown in Figure 5.
Figure 5.—

Variable rate simulations: we estimated gene conversion and crossover rates for 100 data sets, simulated with a hotspot in which the gene conversion and crossover rates are 100 times the background rate (simulated gene conversion rate shown in red). To estimate the gene conversion rate for each of these data sets, we assume the ratio f of gene conversion to crossover is fixed throughout the region and run our program 20 times with one independent random ordering each time. The results from each run are transformed using Equation 3 and we use the median as our best estimate for that data set. Here we show (in black) the median of these 100 estimates and the 5th and 95th percentiles (gray).
Individual estimates of γ show high levels of variance, but on average, the position, width, and heat of the estimated hotspot are close to the values used to simulate data, and there is little bias in our estimates of γ. However, estimates of ρ are downwardly biased, resulting in an overestimate of f (median 25.1). More work is needed to develop a method that can produce a less biased estimate of f in this variable-rate scenario, even under the restriction that f is constant. To obtain a more reliable gene conversion rate estimate on a single data set, a hotspot model would be needed, such as the reversible-jump MCMC crossover model of Auton and McVean (2007). However, accurate estimation of the strength of a crossover hotspot is problematic in general, because the regions on opposite sides of any hotspot above a certain size are in near-complete linkage equilibrium with each other (Auton and McVean 2007).
Despite some evidence that f may vary between regions in humans (Hellenthal and Stephens 2006; Padhukasahasram et al. 2006), we do not consider this scenario, mainly because population genetic data are unlikely to contain sufficient information to obtain an accurate fine-scale map of gene conversion rates independently of crossover rates, but also because the existence of gene conversion hotspots within crossover hotspots implies some correlation between the two rates, and finally because the processing time needed to maximize such a likelihood would be immense.
Human chromosome 1:
Many crossover hotspots have been identified in the human genome, but of particular interest is the MS32 hotspot on chromosome 1, the existence of which is supported by strong experimental evidence, but has not left a significant imprint on LD (Jeffreys et al. 2005). We applied our method to a 206-kb region, including this hotspot and several others. The SNP data (Jeffreys et al. 2005) consist of 214 SNPs on 80 genotypes; we used PHASE v2.1.1 (Stephens et al. 2001; Stephens and Scheet 2005) to infer the haplotypes and missing data and averaged our results over 20 independent random subsamples of 50 haplotypes, each taken in 10 random orders. For comparison, the crossover rate for this region, estimated using LDhat (Mcvean et al. 2002), is shown below. LDhat was run for 108 iterations with block penalty 5, results were sampled every 104 iterations, and the first 100 samples discarded. To reflect the idea that crossover and gene conversion hotspots tend to coincide (Jeffreys and May 2004), we allowed gene conversion rates to vary independently in each interval between SNPs while keeping f constant everywhere in the region. Our median estimated gene conversion rate is shown in Figure 6. Our median estimate of f for this region was 1.5. This estimate is strongly influenced by the gene conversion tract length parameter. In this study we assumed the mean tract length was 100 bp, but note that a longer mean tract length would lead to a lower 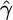 and a correspondingly lower
and a correspondingly lower 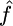 .
.
Figure 6.—

Median maximum-likelihood estimate for the gene conversion rate (blue) in the 206-kb region around the MS32 gene. For comparison, the crossover rate for the same region is shown in green. The gene conversion rate estimate assumes that f is constant throughout the region, and the mean tract length is 100 bp. Red triangles at the top and bottom show the centers of hotspots identified experimentally. Assuming no gene conversion, the hotspot found experimentally at MS32 (Jeffreys et al. 2005) shows little signal in population genetic data. However, under the gene conversion model this hotspot can clearly be seen.
For comparison, we also analyzed the TAP2 region of the human MHC and found f to be higher in this region, ∼9. This lies centrally within the range of 4–15 suggested by Jeffreys and May (2004) although as above, is dependent on the gene conversion tract length. The difference between this estimate and the one for the MS32 region could reflect variation in f between different regions of the human genome, also reported by Hellenthal and Stephens (2006).
DISCUSSION
We have developed a powerful and robust method for estimating gene conversion rates from population genetic data. Our accuracy is at its best when analyzing data that have been affected by fairly high levels of gene conversion and where the mean tract length is at least comparable to the mean SNP spacing. Our model also provides a reliable estimate of the rate of crossover in a region, regardless of the gene conversion rate. Our results are not seriously damaged by the most common deviations from standard model assumptions: nonrandom mating, changing population size, and nonneutral evolution.
Our model allows multiple SNPs to be included in a gene conversion tract. SNP density varies widely between data sets, but also within data sets: for example, in the MS32 data set analyzed above there are 214 SNPs in a 206-kb region, giving an average interval of 967 bp between adjacent markers. However, 45 intervals (21%) are <100 bp long and 133 (62%) are <500 bp long. Our simulations show that when the mean tract length is 100 bp, 9% of gene conversion tracts that initiate within this region will encompass the positions of two or more markers. With 500-bp tracts, this rises to 38%.
When applied to data with little or no history of gene conversion, our model tends to overestimate the gene conversion rate. This is mainly due to the true value lying on or near the boundary of the parameter space. Simulation results (Figure 2, first column) demonstrate that including gene conversion in the model results in improved estimation of the recombination rate, suggesting that modeling errors are preferentially interpreted as gene conversion events. This implies that the use of our model could result in improvements to the estimation of the underlying crossover rate, even in the case where gene conversion is not occurring.
Estimates of uncertainty cannot be obtained directly from this method, due to the approximate likelihood used. To obtain confidence intervals it is necessary to perform a simulation study tailored to the specifics of a given data set (such as the number of haplotypes and rate of mutation).
We analyzed a region of the X chromosome in D. melanogaster and found that, under the assumption that the rates of crossover and gene conversion are constant across the region, gene conversion events occur >400 times as often as crossovers. Application of this model to additional regions of the Drosophila genome could enhance our understanding of the unusual patterns of recombination in this species.
We also analyzed a region of human chromosome 1, around the MS32 gene, a region containing several known crossover hotspots. Our analysis, allowing the rates of crossover and gene conversion to vary across the region while keeping their ratio f constant, shows that the MS32 hotspot, previously difficult to detect using population genetic methods, appears highly active under a gene conversion model. This could indicate that this hotspot is more active in gene conversion than in crossover, but it seems to contradict conclusions that the hotspot has only recently emerged (Jeffreys et al. 2005).
The maximum-likelihood estimate 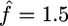 for this region should be treated with caution for two reasons: simulations show that this is a biased estimator (see above), but also
for this region should be treated with caution for two reasons: simulations show that this is a biased estimator (see above), but also 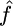 is highly dependent on the accuracy of our tract length estimate (here 100 bp). As with the method of Ptak et al. (2004), experiments with our method have shown that there is a strong correlation between the estimated values of λ and γ (data not shown).
is highly dependent on the accuracy of our tract length estimate (here 100 bp). As with the method of Ptak et al. (2004), experiments with our method have shown that there is a strong correlation between the estimated values of λ and γ (data not shown).
For this reason, we do not attempt to estimate the gene conversion tract length from the data. By misspecifying the mean tract length parameter for data sets simulated with known tract length t0, we find that using a mean tract length estimate t* that is double that simulated (t* = 2t0) results in a slightly lowered gene conversion rate estimate, while using an estimate 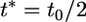 causes us to overestimate the gene conversion rate by approximately a factor of 2 (data not shown). These results will vary depending on the SNP spacing and the actual gene conversion rate. Fortunately, estimates of the gene conversion tract length are available for several organisms (e.g., Palmer et al. 2003; Jeffreys and May 2004; Nishant et al. 2004) although little is yet known about whether there is heterogeneity in this length between different genomic regions.
causes us to overestimate the gene conversion rate by approximately a factor of 2 (data not shown). These results will vary depending on the SNP spacing and the actual gene conversion rate. Fortunately, estimates of the gene conversion tract length are available for several organisms (e.g., Palmer et al. 2003; Jeffreys and May 2004; Nishant et al. 2004) although little is yet known about whether there is heterogeneity in this length between different genomic regions.
Our implementation of this model is of order ∼N3 and is linear in both the number of markers and the number of orders used. It takes ∼30 min of processing time on a standard desktop machine to jointly calculate the constant MLEs for ρ and γ when N = 50, L = 100, λ is fixed, and 10 random orderings are used. However, when γ and ρ are allowed to vary, with their ratio f constant, using the same data set it would take ∼9 hr to obtain 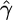 and
and 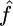 (and therefore
(and therefore 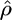 ). This time could be reduced by improvements to the implementation and by running on a faster computer. For very large data sets the method is impracticable with present computers, but results can still be obtained by taking several subsamples of the data and taking the median result. Subsamples should be as large as possible as smaller subsamples tend to produce underestimates of the gene conversion rate and have higher variance (see supplemental Table 1 at http://www.genetics.org/supplemental/), but when averaging over several subsamples, one order is sufficient. For reasonable-sized data sets we believe the accuracy obtained by this model's usage of all the information contained in the data makes its slow speed a worthwhile penalty.
). This time could be reduced by improvements to the implementation and by running on a faster computer. For very large data sets the method is impracticable with present computers, but results can still be obtained by taking several subsamples of the data and taking the median result. Subsamples should be as large as possible as smaller subsamples tend to produce underestimates of the gene conversion rate and have higher variance (see supplemental Table 1 at http://www.genetics.org/supplemental/), but when averaging over several subsamples, one order is sufficient. For reasonable-sized data sets we believe the accuracy obtained by this model's usage of all the information contained in the data makes its slow speed a worthwhile penalty.
In this article we do not consider the effects of genotyping error on our results. Genotyping error can have a similar effect on the patterns of genetic diversity to that of gene conversion (Ptak et al. 2004). In densely typed SNP data, allowing for multi-SNP gene conversion tracts should reduce the impact of genotyping error.
We anticipate that this model could be adapted to detect the signature of nonallelic gene conversion in population genetic data, which would be particularly useful when considering the evolution of multigene families such as the histones (see Nei and Rooney 2005).
As well as finding fine-scale variations in γ, it would be straightforward to adapt this model to compare estimates of f for different regions or genes within a given organism, allowing production of a broad-scale map of f. This could be used to discover whether large-scale genomic features such as proximity to centromeres affect this ratio.
Whether f is constant at a fine scale remains an open question.
Acknowledgments
The authors thank Daniel Falush, Chris Spencer, Matthew Stephens, and one anonymous reviewer for helpful comments on earlier versions of this work. Many of the simulations and data analyses were performed on the multinode computing cluster that was funded by a grant from the Wolfson Foundation to Peter Donnelly. J.G. is funded by the Engineering and Physical Sciences Research Council through the University of Oxford Life Sciences Interface Doctoral Training Centre.
APPENDIX
Hidden Markov model implementation:
Here we detail the implementation of the model described above, including the state transition probabilities and some algorithmic shortcuts used to reduce the computation time. The “hidden” data in our model are the “true” underlying mosaic structure of the current haplotype. Under the true coalescent genealogy, a mosaic consisting of haplotypes we have already seen may not exist, but under our approximation it always does. We do not try to infer this but sum over all mosaic structures using the hidden Markov model (HMM) formulation. Each of the terms Pr(hk+1 | h1, … , hk, ρ, γ) is approximated using its own HMM with k(k + 1) states and its own emission and transition probabilities that depend on k.
Our HMM has k(k + 1) distinct states (X = x, G = g), where 1 < x ≤ k denotes our nearest neighbor, unless g ≠ 0, in which case we are in a gene conversion, and the current nearest neighbor is g. Starting at the leftmost marker, we calculate the likelihood of the data for each possible state (Xj, Gj) at each marker j, on the basis of the k(k + 1) distinct state probabilities at the previous marker, and the transition (t) and emission (e) probabilites (see below).
Transition probabilities:
We model the process of crossover and that of gene conversion as separate processes, happening independently of each other. The variable Xj can be modified only by crossover, while Gj is affected only by gene conversion:
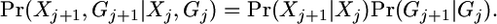 |
(A1) |
Initial state probabilities:
Since we have no data outside our region, 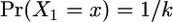 for all x. The probability that we start our Markov chain inside a gene conversion tract depends on how the rate of starting a gene conversion tract compares to the rate of ending one:
for all x. The probability that we start our Markov chain inside a gene conversion tract depends on how the rate of starting a gene conversion tract compares to the rate of ending one:
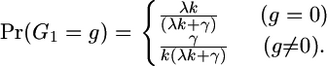 |
(A2) |
X transition probabilities:
The first term on the right-hand side of Equation A1 is given by Li and Stephens (2003) as
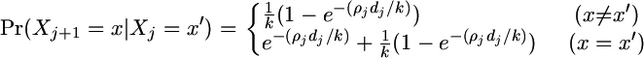 |
(A3) |
when we are considering the (k + 1)th haplotype and the distance between marker j − 1 and marker j is dj. Informally, in the case where x ≠ x′, we must have had at least one crossover between the two sites, and the probability that the new nearest neighbor was x′ is 1/k. When x = x′, we may have had no crossover event, or we may have crossed over one or more times but in the last event chose the same nearest neighbor.
G transition probabilities:
To calculate Pr(Gj+1 | Gj) we must consider not only the probability of beginning a gene conversion event within the current interval but also that of ending one. The rate of terminating a gene conversion tract is fixed at λ, regardless of the length the tract has so far covered. This geometric model allows us to consider the ending of a gene conversion event as a process in its own right, which goes on all the time, independently of our current state, and resets the state of the system to the base (non-gene-conversion) state. This is reasonable because although in biological terms there is no event corresponding to the end of a gene conversion that can occur outside of a gene conversion tract, any such event occurring when we are already in the base state has no effect and thus has no effect on our model.
For each type of transition between gene conversion states, we describe the sequence of events that could cause that transition to occur and give the probability of undergoing this transition. In each case, any events occurring before the last reset event within the interval will have no effect on the state at the right-hand side of the interval. We therefore integrate back from the right-hand side of the interval, over possible positions of the last reset event, so that we do not have to explicitly consider how many gene conversion events may have taken place prior to the final reset event.
There are five distinct types of transition between gene conversion states:
- We are currently not in a gene conversion, and we were not in one at the last marker. We break this down into two scenarios: there was no reset event in the interval [which happens with probability exp(−λdj)] and also no gene conversion event [probability
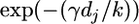 ] or there was a reset event and no gene conversion event has taken place since then. This last term can be written as the integral (over all possible places at which the last reset event might have happened) of the probability that no further reset event occurred multiplied by the probability that no gene conversion occurred:
] or there was a reset event and no gene conversion event has taken place since then. This last term can be written as the integral (over all possible places at which the last reset event might have happened) of the probability that no further reset event occurred multiplied by the probability that no gene conversion occurred: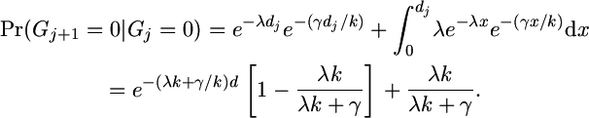
(A4) - We have moved from a non-gene-conversion state to a gene conversion state:
(either there was no reset event in the interval but there was a gene conversion event that made g our new nearest neighbor or there was a reset event and there was a gene conversion event after it).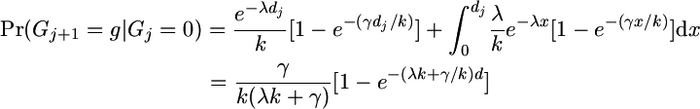
(A5) - Previously we were in a gene conversion event but now we are not:
(no gene conversion event has taken place since the last reset event).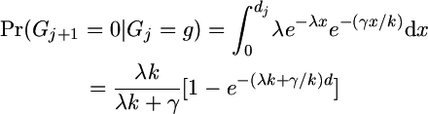
(A6) - We were previously in a gene conversion state where we were copying from haplotype g, and we are currently in a similar state:
(either no reset event has occurred or there has been a gene conversion event choosing the same value of g since the last reset event).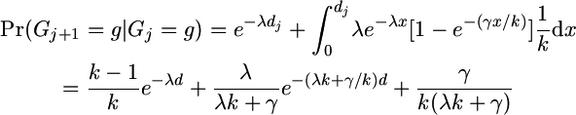
(A7) - We were previously in a gene conversion state copying from haplotype g and we have moved into a gene conversion state where we are copying from haplotype g′, where g ≠ g′:
(as above but without the option for no event occurring).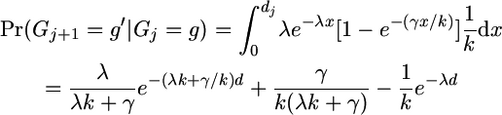
(A8)
These gene conversion state transition probabilities, together with the crossover state transition probabilities above, make up the state transition probabilities for our Markov chain. We write tG(g′ | g, j) = Pr(Gj+1 = g′ | Gj = g) and tX(x′ | x, j) = Pr(Xj+1 = g′ | Xj = x).
Emission probabilities:
When it is not known, we (as do Li and Stephens 2003) use Watterson's estimator (Watterson 1975) to approximate the per-site rate of mutation, θ/L:
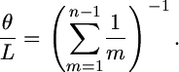 |
(A9) |
Conditional on the hidden state (Xj, Gj) at marker j, we could calculate the emission probability on the basis of whether or not a mutation had occurred, compared to the chromosome c from which we are copying (c = Xj if Gj = 0, otherwise c = Gj). This is simply
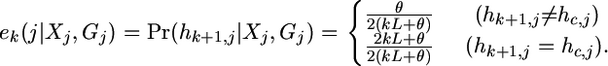 |
(A10) |
Likelihood:
Let px,g(j) be the relative probability of being in the state (x, g) at the marker j, given the data up to that marker. Then
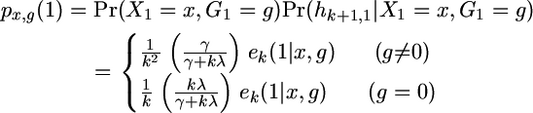 |
(A11) |
and
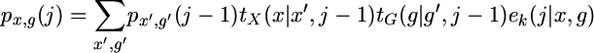 |
(A12) |
and the approximate likelihood of the data, for the (k + 1)th chromosome, for this chromosomal order, is given by
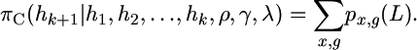 |
(A13) |
To overcome the issue of order dependency, we repeat this calculation n ≥ 10 times and take the average likelihood.
Optimization:
The basic algorithm described above is of approximate order N4, but we were able to reduce this to N3 by calculating sums of several subgroups of the px,g(j) and using these sums to facilitate the calculation of px′,g′(j + 1). First note that tX(x | x′, j) can take only two possible values, depending on whether x = x′. Similarly, tG can take five values as described above. So, for example, tX(x | x′, j) is the same for all values of x′ except x′ = x, and the sum 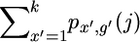 gives the total probability of all the states where Gj = g′. To calculate px,g′(j + 1), subtracting px,g′(j) from this sum leaves a group of states at marker j for which the transition probability is the same.
gives the total probability of all the states where Gj = g′. To calculate px,g′(j + 1), subtracting px,g′(j) from this sum leaves a group of states at marker j for which the transition probability is the same.
These sums, calculated once for each marker, can be used many times in different combinations.
Estimating parameters:
To find maximum-likelihood estimates we use a direct search algorithm (Hooke and Jeeves 1961) to find the 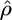 and
and 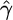 that maximize the average likelihood. When variable rate estimates are required, we use expectation maximization to find these rates independently in each interval.
that maximize the average likelihood. When variable rate estimates are required, we use expectation maximization to find these rates independently in each interval.
The C++ code and windows/linux executables for our implementation of this model are available from http://www.stats.ox.ac.uk/∼gay/.
References
- Aguade, M., and C. H. Langley, 1994. Non-Neutral Evolution: Theories and Molecular Data, pp. 67–76. Chapman & Hall, London/New York.
- Andolfatto, P., and M. Nordborg, 1998. The effect of gene conversion on intralocus associations. Genetics 148: 1397–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton, A., and G. McVean, 2007. Recombination rate estimation in the presence of hotspots. Genome Res. 17: 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borts, R. H., and J. E. Haber, 1989. Length and distribution of meiotic gene conversion tracts and crossovers in Saccharomyces cerevisiae. Genetics 123: 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnhead, P., and P. Donnelly, 2001. Estimating recombination rates from population genetic data. Genetics 159: 1299–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnhead, P., N. G. C. Smith, M. Barrigas, A. Fox and N. French, 2005. Analysis of recombination in campylobacter jejuni from MLST populaton data. J. Mol. Evol 61: 333–340. [DOI] [PubMed] [Google Scholar]
- Fridell, R. A., and L. L. Searles, 1994. Evidence for a role of the Drosophila melanogaster suppressor of sable gene in the pre-mRNA splicing pathway. Mol. Cell. Biol. 14: 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisse, L., R. R. Hudson, A. Bartoszewicz, J. D. Wall, J. Donfack et al., 2001. Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium. Am. J. Hum. Genet. 69: 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, R., and P. Marjoram, 1997. An Ancestral Recombination Graph (IMA Vol. 87: Progress in Population Genetics and Human Evolution), pp. 257–270. Springer-Verlag, Berlin/Heidelberg, Germany/New York.
- Hellenthal, G., 2006. Exploring rates and patterns of variability in gene conversion and crossover in the human genome. Ph.D. Thesis, University of Washington, Seattle.
- Hellenthal, G., and M. Stephens, 2006. Insights into recombination from population genetic variation. Curr. Opin. Genet. Dev. 16: 565–572. [DOI] [PubMed] [Google Scholar]
- Hellenthal, G., and M. Stephens, 2007. msHOT: modifying Hudson's ms simulator to incorporate crossover and gene conversion hotspots. Bioinformatics 23: 520–521. [DOI] [PubMed] [Google Scholar]
- Hilliker, A. J., G. Harauz, A. G. Reaume, M. Gray, S. H. Clark et al., 1994. Meiotic gene conversion tract length distribution within the rosy locus of Drosophila melanogaster. Genetics 137: 1019–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochman, B., 1976. The fourth chromosome of Drosophila melanogaster, pp. 903–928 in The Genetics and Biology of Drosophila, Vol. 1b, edited by M. Ashburner and E. Novitski. Academic Press, New York.
- Hooke, R., and T. A. Jeeves, 1961. Direct search solution of numerical and statistical problems. J. Appl. Comp. Meth. 8: 212–229. [Google Scholar]
- Hudson, R., 2002. Generating samples under a Wright-Fisher neutral model. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 1983. Properties of a neutral allele model with intragenic recombination. Theor. Popul. Biol. 2: 183–201. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 2001. Two-locus sampling distributions and their application. Genetics 159: 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys, A. J., and C. A. May, 2004. Intense and highly localized gene conversion activity in human meiotic crossover hot spots. Nat. Genet. 36: 151–156. [DOI] [PubMed] [Google Scholar]
- Jeffreys, A. J., R. Neumann, M. Panayi, S. Myers and P. Donnelly, 2005. Human recombination hot spots hidden in regions of strong marker association. Nat. Genet. 37: 601–606. [DOI] [PubMed] [Google Scholar]
- Kingman, J., 1982. On the genealogy of large populations. J. Appl. Probab. 19A: 27–43. [Google Scholar]
- Kong, A., D. F. Gudbjartsson, J. Sainz, G. M. Jonsdottir, S. A. Gudjonsson et al., 2002. A high-resolution recombination map of the human genome. Nat. Genet. 31: 241–247. [DOI] [PubMed] [Google Scholar]
- Langley, C. H., B. P. Lazzaro, W. Phillips, E. Heikkinen and J. M. Braverman, 2000. Linkage disequilibria and the site frequency spectra in the su(s) and su(wa) regions of the Drosophila melanogaster X chromosome. Genetics 156: 1837–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, N., and M. Stephens, 2003. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics 165: 2213–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKim, K. S., J. K. Jang and E. A. Manheim, 2002. Meiotic recombination and chromosome segregation in Drosophila females. Annu. Rev. Genet. 36: 205–232. [DOI] [PubMed] [Google Scholar]
- McVean, G., P. Awadalla and P. Fearnhead, 2002. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160: 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean, G. A. T., S. R. Myers, S. Hunt, P. Deloukas, D. R. Bentley et al., 2004. The fine-scale structure of recombination rate variation in the human genome. Science 304: 581–584. [DOI] [PubMed] [Google Scholar]
- Nachman, M. W., 2002. Variation in recombination rate across the genome: evidence and implications. Curr. Opin. Genet. Dev. 12: 657–663. [DOI] [PubMed] [Google Scholar]
- Nei, M., and A. Rooney, 2005. Concerted and birth-and-death evolution of multigene families. Annu. Rev. Genet. 39: 121–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishant, K. T., H. Ravishankar and M. R. S. Rao, 2004. Characterization of a mouse recombination hot spot locus encoding a novel non-protein-coding RNA. Mol. Cell. Biol. 24: 5620–5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhukasahasram, B., J. D. Wall, P. Marjoram and M. Nordborg, 2006. Estimating recombination rates from single-nucleotide polymorphisms using summary statistics. Genetics 174: 1517–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, S., E. Schildkraut, R. Lazarin, J. Nguyen and J. A. Nickoloff, 2003. Gene conversion tract lengths in Saccharomyces cerevisiae can be extremely short and highly directional. Nucleic Acids Res. 31: 1164–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., and M. Przeworski, 2001. Linkage disequilibrium in humans: models and data. Am. J. Hum. Genet. 69: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., and J. D. Wall, 2001. Why is there so little intragenic linkage disequilibrium in humans. Genet. Res. 77: 143–151. [DOI] [PubMed] [Google Scholar]
- Ptak, S. E., K. Voelpel and M. Przeworski, 2004. Insights into recombination from patterns of linkage disequilibrium in humans. Genetics 167: 387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierup, M. H., and J. Hein, 2000. Consequences of recombination on traditional phylogenetic analysis. Genetics 156: 876–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schork, N. J., 2002. Power calculations for genetic association studies using estimated probability distributions. Am. J. Hum. Genet. 70: 1480–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, N. G. C., and P. Fearnhead, 2005. A comparison of three estimators of the population-scaled recombination rate: accuracy and robustness. Genetics 171: 2051–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer, C. C. A., and G. Coop, 2004. SelSim: a program to simulate population genetic data with natural selection and recombination. Bioinformatics 20: 3673–3675. [DOI] [PubMed] [Google Scholar]
- Stahl, F. W., 1994. The holliday junction on its thirtieth anniversary. Genetics 138: 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, M., and P. Scheet, 2005. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am. J. Hum. Genet. 76: 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, M., N. Smith and P. Donnelly, 2001. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68: 978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf, M. P. H., and G. A. T. McVean, 2003. Estimating recombination rates from population-genetic data. Nat. Rev. Genet. 4: 959–968. [DOI] [PubMed] [Google Scholar]
- Szostak, J. W., T. L. Orr-Weaver, R. J. Rothstein and F. W. Stahl, 1983. The double-strand-break repair model for recombination. Cell 33: 25–35. [DOI] [PubMed] [Google Scholar]
- Wall, J. D., 2004. Estimating recombination rates using three-site likelihoods. Genetics 167: 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G., 1975. On the number of segregation sites. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Wiehe, T., J. Mountain, P. Parham and M. Slatkin, 2000. Distinguishing recombination and intragenic gene conversion by linkage disequilibrium patterns. Genet. Res. 75: 61–73. [DOI] [PubMed] [Google Scholar]
- Wiuf, C., and J. Hein, 2000. The coalescent with gene conversion. Genetics 155: 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]