

Abstract
Coactivator-associated arginine methyltransferase (CARM1) is a transcriptional coactivator that methylates Arg17 and Arg26 in histone H3. CARM1 contains a conserved protein arginine methyltransferase (PRMT) catalytic core flanked by unique pre- and post-core regions. The crystal structures of the CARM1 catalytic core in the apo and holo states reveal cofactor-dependent formation of a substrate-binding groove providing a specific access channel for arginine to the active site. The groove is supported by the first eight residues of the post-core region (C-extension), not present in other PRMTs. In vitro methylation assays show that the C-extension is essential for all histone H3 methylation activity, whereas the pre-core region is required for methylation of Arg26, but not Arg17. Kinetic analysis shows Arg17 methylation is potentiated by pre-acetylation of Lys18, and this is reflected in kcat rather than Km. Together with the absence of specificity subsites in the structure, this suggests an electrostatic sensing mechanism for communicating the modification status of vicinal residues as part of the syntax of the ‘histone code.'
Keywords: arginine, CARM1, histone, methylation, PRMT4
Introduction
The N-terminal tails of the core histones are subjected to a diverse array of post-translational modifications, such as acetylation, methylation, ubiquitination and phosphorylation. These covalent modifications play a role in regulating chromatin-dependent functions such as transcription, replication and repair (Kouzarides, 2007). Recent studies have implicated the arginine methylation of histones H3 and H4 in transcriptional regulation. In particular, methylation of histone H3 Arg17 by coactivator-associated arginine methyltransferase 1 (CARM1) has been linked with the transcriptional activation of hormone-responsive promoters in vivo (Ma et al, 2001; Bauer et al, 2002; Miao et al, 2006), and has a key role in defining cell fate in the early embryo (Torres-Padilla et al, 2007).
CARM1 was originally identified in a yeast two-hybrid screen as an interacting protein for the p160-family coactivator, glucocorticoid receptor-interacting protein-1 (GRIP1, also known as SRC2/TIF2) (Chen et al, 1999). CARM1 activates transcription mediated by nuclear receptors, by functioning synergistically with the primary coactivator GRIP1 and the histone acetyltransferase p300/CBP (Chen et al, 2000; Lee et al, 2002). CARM1 functions as a secondary coactivator: its recruitment to the promoters of hormone-responsive genes by GRIP1 coincides with gene activation and the methylation of H3 Arg17 and Arg26. In addition, CARM1 functions as a coactivator in non-nuclear receptor systems, associating and cooperating with NF-κB (Covic et al, 2005; Miao et al, 2006), p53 (An et al, 2004), IFN-γ (Zika et al, 2005), MEF2C (Chen et al, 2002) and β-catenin (Koh et al, 2002). Consistent with its pleiotropic roles, CARM1-null mice are small and die perinatally (Yadav et al, 2003).
CARM1 belongs to the family of protein arginine methyltransferases (PRMTs) of which there are nine mammalian members. They catalyse transfer of the methyl group from S-adenosyl-L-methionine (SAM) to the guanidino group of arginine residues with the concomitant production of S-adenosyl-L-homocysteine (SAH) (for review, see Bedford and Richard, 2005). This produces monomethyl arginine, which can be further methylated to form asymmetric dimethyl arginine (by type I PRMTs) or symmetric dimethyl arginine (by type II PRMTs). PRMTs contain a conserved catalytic core preceded by a variable N-terminal region (pre-core). CARM1 (also known as PRMT4) uniquely contains a substantial C-terminal region (post-core) (Figure 1A), which has been shown to have autonomous activation activity and to interact with the transcription coactivator TIF1α (Teyssier et al, 2002, 2006).
Figure 1.

CARM1 is a member of the PRMT family. (A) Top: domain architecture of PRMT family members. The catalytic core is coloured blue for N-domain, red for C-domain and yellow for dimerisation arm. While all PRMTs have different pre-core regions, CARM1 also has a unique post-core region (green). PRMT9, which has limited sequence homology with other PRMTs (Cook et al, 2006), is not shown. Bottom: the CARM1 constructs used in this study. (B) In vitro methylation of H3-H4 tetramer by CARM1-full, CARM1-ΔN or CARM1-ΔNC, detected by anti-H3 dimethyl-Arg2, Arg17 or Arg26 antibodies (top 3 panels). Loading controls for CARM1 and H3-H4 proteins (bottom 2 panels) are shown. (C) CARM1 interacts with the AD2 domain of GRIP1. Top: domain architecture of mouse GRIP1. Bottom: GST-tagged proteins were incubated with equivalent amounts of CARM1-full, CARM1-ΔN, CARM1-ΔNC or CARM1-N. Loading controls for GST-tagged proteins are shown. (D) CARM1-ΔNC interacts with the armadillo repeats of β-catenin. Top: domain architecture of human β-catenin. Bottom: GST-tagged proteins were incubated with equivalent amounts of CARM1-ΔNC or CARM1-N. Loading controls for GST-tagged proteins are shown.
Crystal structures of the catalytic cores of SAH-bound rat PRMT3 (Zhang et al, 2000), PRMT1 bound with SAH and substrate peptide (Zhang and Cheng, 2003), and the apo form of the yeast PRMT1 homologue HMT1 (Weiss et al, 2000) have previously been described. These type I enzymes methylate a plethora of substrates, with target arginines located in RGG or RXR clusters contained within Gly-Arg-rich (GAR) domains (Lin et al, 1996; Tang et al, 1998). CARM1 by contrast, also a type I enzyme, methylates a narrower set of substrates including histone H3 (Schurter et al, 2001), p300/CBP (Xu et al, 2001; Chevillard-Briet et al, 2002; Lee et al, 2005) and several RNA-binding proteins (HuR, HuD, PABP1, TARPP, CA150) (Lee and Bedford, 2002; Li et al, 2002; Fujiwara et al, 2006; Cheng et al, 2007), all of which lack GAR domains or indeed any common consensus sequence motif.
The functional association of p300/CBP and CARM1 in gene transcription, and the close proximity of modified lysine (by p300/CBP) and arginine (by CARM1) residues within histone H3, suggests interplay between lysine acetylation and arginine methylation. Studies using an endogenous oestrogen-responsive promoter (Daujat et al, 2002) and an in vitro reconstituted transcription system (An et al, 2004) demonstrated that H3 acetylation by p300/CBP and H3 methylation by CARM1 are ordered and cooperative processes: CBP acetylation of Lys18 promotes CARM1 methylation of Arg17. This is reminiscent of the observation that phosphorylation of H3 Ser10 promotes acetylation of Lys14 (Cheung et al, 2000; Lo et al, 2000). These examples are part of a growing body of evidence for functional interplay between histone modifications (Fischle et al, 2003), constituting a ‘syntax' for the ‘histone code' (Strahl and Allis, 2000; Jenuwein and Allis, 2001).
Here, we present the crystal structure of the catalytically active core of CARM1 in the apo state, and in complex with the cofactor product SAH refined at 2.7 Å resolution. The structural analysis is complemented by biochemical and kinetic studies, which suggest roles for CARM1's pre-core and post-core regions, and reveal an unusual mechanism for activation of CARM1 by pre-existing modification of vicinal substrate residues.
Results
Mapping activity and interactions
Crystallisation trials with full-length CARM1 were unsuccessful; however, two subconstructs, identified by limited proteolysis, yielded crystals. CARM1-ΔN (aa 147–585) contains the core and post-core regions, whereas CARM1-ΔNC (aa 147–490) contains the core region and 20 residues of the post-core region that we termed as C-extension (Figure 1A). The methyltransferase activities of CARM1-ΔN and CARM1-ΔNC were compared to full-length CARM1 in a gel-based methylation assay, using antibodies specific for asymmetric dimethylated Arg2, Arg17 or Arg26 of histone H3 (Figure 1B). The full-length protein and both subconstructs methylated H3 Arg17, but only the full-length protein, containing the pre-core region, displayed significant activity towards Arg2 and Arg26.
CARM1 has been shown to interact with a C-terminal fragment (aa 1121–1462) of the coactivator GRIP1, encompassing the Gln-rich and AD2 (activation domain 2) domains (Chen et al, 2000; Teyssier et al, 2002). To map the CARM1-GRIP1 interaction further, we constructed a shorter GRIP1 fragment containing only the AD2 domain (aa 1305–1462, GRIPAD2) as a GST-fusion, for use in an in vitro pull-down assay (Figure 1C). GST-GRIP1AD2, but not GST alone, co-precipitated CARM1-full, CARM1-ΔN and CARM1-ΔNC to a similar extent, but did not co-precipitate the isolated pre-core region of CARM1 (CARM1-N). These data show that CARM1-ΔNC is sufficient for a direct interaction with GRIP1AD2.
In addition to GRIP1, CARM1 activates transcription in cooperation with β-catenin (Koh et al, 2002). A similar pull-down analysis, using a variety of GST-β-catenin fusions, showed specific co-precipitation of CARM1-ΔNC, but not CARM-N, with GST-fusions to β-catenin fragments corresponding to aa 21–665 (βcatΔC), 133–665 (βcatARM) and 133–781 (βcatΔN) (Figure 1D). These data indicate that the central region of β-catenin (aa 133–665), corresponding to the armadillo repeats (Huber et al, 1997), is sufficient for a direct interaction with CARM1-ΔNC.
Crystals of CARM1-ΔN and CARM1-ΔNC were obtained in similar conditions. However, only CARM1-ΔNC crystals gave useful diffraction, allowing structure determination by molecular replacement, with PRMT1 as a search model. The structures of CARM1-ΔNC in the apo form (CARM1apo), and a binary complex with SAH (CARM1bin) were both refined at 2.7 Å resolution. For clarity, the CARM1 structure described herein refers to the CARM1bin data, unless stated otherwise.
Overall architecture
The refined structure of CARM1-ΔNC, encompassing amino acids 147–478, consists of an N-domain, C-domain and C-extension (Figure 2A, left). The N-domain (aa 147–288, blue) is formed by three α-helices (αX-αZ) followed by a Rossmann-like α/β sandwich common to all class I SAM-dependent methyltransferases (SAM-MT) (Schluckebier et al, 1995). The C-domain (aa 289–469, red) is a nine-stranded pseudobarrel interrupted by a helix–turn–helix ‘arm' (aa 301–337, yellow), involved in dimerisation. The C-domain is followed by the C-extension (aa 470–478, green), which contains a short β-strand (β16) that forms a β-sheet with strand β7 in the C-domain. The last 12 residues (479–490) of the C-extension are not visible in the electron density map, and are presumably disordered.
Figure 2.

Crystal structure of CARM1. (A) Left: crystal structure of CARM1-SAH complex (CARM1bin). A CARM1 monomer consists of the N-domain (blue), C-domain (red), dimerisation arm (yellow) and C-extension (green). Secondary structure elements are labelled. Right-top: the crystallised construct (CARM1-ΔNC) encompasses aa 147–490. Right-bottom: the two active sites (arrow) in a CARM1 dimer face towards the central cavity (dotted circle). (B) Cofactor-binding site of CARM1. The cofactor product SAH is shown in stick representation, together with the 2Fo–Fc electron density map (contoured at 1.5 σ) and residues interacting with SAH. Dotted lines indicate hydrogen bonds. (C) Conformational changes upon SAH binding. Superimposition of CARM1apo (cyan) and CARM1bin (blue) structures show significant conformational differences in the Gly-rich loop and helix αX. SAH is shown in stick representation. (D) Surface representation of the active site in CARM1apo (cyan) and CARM1bin (blue). In CARM1apo, helix αX is disordered and the cofactor pocket is accessible. In CARM1bin, helix αX (dark blue surface) forms a lid covering the cofactor pocket. SAH is only accessible via a narrow opening into the putative arginine pocket.
Dimerisation is necessary for the methyltransferase activity of PRMTs (Weiss et al, 2000; Zhang et al, 2000; Zhang and Cheng, 2003), and CARM1 is a dimer in solution (data not shown) and in the crystals. The dimer interface is formed by the C-domain helix–turn–helix arm of one monomer, and four N-domain helices (αY, αZ, αA, αB) of another monomer (Figure 2A, right). This forms a ring-shaped dimer with a central cavity, which allows access for the substrate and cofactor. In this arrangement, the visible N- and C-termini of the CARM1-ΔNC structure (helix αX and C-extension, respectively) lie within the central cavity, but project away from the dimer plane in opposite directions.
Cofactor binding
The SAH molecule binds to a pocket at the α/β sandwich, in an extended conformation common to type I SAM-MTs (Figure 2B). The adenine ring, ribose moiety and homocysteine carboxylate of SAH are recognised in the cofactor pocket through hydrogen bonds to residues Glu244, Glu215 and Arg169, respectively, all of which are strictly conserved within the PRMT family.
Comparison of CARM1apo and CARM1bin structures reveals SAH-induced conformational changes in two areas of the cofactor pocket (Figure 2C). The first is a conserved Gly-rich loop that packs against helix αY in CARM1apo. In CARM1bin, this loop is displaced by ∼5 Å to accommodate the homocysteine moiety of SAH. The second change occurs in the N-terminal helix αX, which is disordered in CARM1apo, but becomes ordered in the binary complex, and contributes conserved residues Tyr150, Phe151 and Tyr154 to form aromatic interactions with the adenine ring of SAH (Figure 2B). Thus on binding SAH, the ordered helix αX forms a lid covering the cofactor pocket, and together with helix αY forms the upper ridge of a putative substrate-binding groove. As a consequence, the SAH in the cofactor pocket is almost completely buried from the exterior (98% of accessible surface area) (Figure 2D). The sulphur atom in SAH (which is covalently bonded to the methyl donor group in SAM) is thus only accessible to the target substrate arginine in the active site via a narrow opening.
Unique structural features of CARM1
To date, structures of the catalytic cores of three type I PRMTs have been described: rat PRMT1 (and its yeast homologue Hmt1) (Weiss et al, 2000; Zhang and Cheng, 2003), rat PRMT3 (Zhang et al, 2000) and mouse CARM1 (this study). Structural alignment of the four structures highlights significant structural differences between CARM1 and the other three proteins in the C-domain: the helix–turn–helix arm, the loop connecting the last two barrel strands (β14, β15) and the C-extension (Figure 3A, left).
Figure 3.

Unique structural features of CARM1. (A) Left: superimposition of the catalytic cores of mouse CARM1, rat PRMT1, rat PRMT3 and yeast Hmt1. The major structural differences between CARM1 and PRMT1/PRMT3/Hmt1 are highlighted in red. Right: structure-based sequence alignment of CARM1, PRMT1, PRMT3 and Hmt1. Secondary structure elements of CARM1 are coloured as in Figure 2A. Numbers correspond to the CARM1 sequence. Three signature motifs (I, II, III) for class I SAM-MT and two PRMT-specific motifs (VRT and THWY) are underlined. Major structural differences between CARM1 and PRMT1/PRMT3/Hmt1 are indicated in black boxes. Residues 480–490 of the CARM1-crystallised construct (strikethrough) are not visible in the electron density map. (B) CARM1 dimer has a larger central cavity. Left: a CARM1 dimer, rotated 180° along the x-axis relative to the view in Figure 2A, is shown with chains Acrystal (pale blue) and Bcrystal (blue). Right: an enlarged view of the dimer cavity. When chains A and B of the CARM1 dimer are superimposed individually on the equivalent chains of the PRMT1 dimer, chain B of the CARM1 dimer in the crystal (Bcrystal, blue) is >7 Å away (arrow) from the dimer centre relative to the aligned chain B (Baligned, yellow). (C) Surface representation of CARM1 (left) and PRMT1 (right), coloured in a scale of electrostatic potential (kT; positive in blue, negative in red). The active sites in CARM1 and PRMT1 are shown (arrows).
Whereas the catalytic cores of PRMT1, PRMT3 and Hmt1 are highly conserved in sequence (49% sequence identity), CARM1 only shares 34% sequence identity with them. Most of the invariant residues belong to signature motifs for class I SAM-MTs, which are involved in cofactor binding and the catalysis of methyl transfer (Schluckebier et al, 1995) (Figure 3A, right). Other invariant residues conserved within the PRMT subclass include the ‘VRT motif' in helix αZ, containing Arg169, which hydrogen-bonds to SAH, and the ‘THWY loop' in the C-domain, which forms part of the putative substrate-binding groove (Figure 3A, right).
Structural alignment of the four PRMT monomers shows a significant structural similarity of CARM1 with PRMT1 (r.m.s.d. 1.35 Å), PRMT3 (r.m.s.d. 1.32 Å) and Hmt1 (1.59 Å). However, the CARM1 dimer as a whole superimposes poorly on its counterparts (overall r.m.s.d.>4 Å). When one chain in the CARM1 dimer is superimposed individually on a chain in the PRMT1 dimer, the second chain in the CARM dimer is >7 Å away from the dimer centre relative to the second chain in PRMT1 (Figure 3B). As a consequence, the CARM1 dimer has a substantially larger central cavity compared to PRMT1/PRMT3/Hmt1 dimers, presumably to accommodate the unique C-extensions in CARM1, which would otherwise clash in the assembled dimer. The increased size of the CARM1 dimer results directly from a unique nine-residue insertion in the helix–turn–helix segment of the dimerisation arm (Figure 3A).
As well as its larger size, the central cavity of CARM1 also has a different charge distribution to the other PRMTs (Figure 3C). The molecular surface of PRMT1, for example, is predominantly negatively charged, due to the presence of 20 aspartate and glutamate residues in the proximity of the dimer cavity and the C-domain barrel (Zhang and Cheng, 2003). This acidic patch, which is proposed to provide an initial binding affinity for basic Arg-rich substrates, is also found in PRMT3 and Hmt1 (Weiss et al, 2000; Zhang et al, 2000). In CARM1, however, only 5 of the 20 PRMT1 acidic residues are retained, providing a more neutral surface.
The C-extension of CARM1, which projects into the central dimer cavity, is unique amongst the PRMTs. It extends from the last barrel strand of the C-domain (β15) and bends to form the short β-strand β16. This strand packs along strand β7 of the C-domain, forming a two-stranded parallel β-sheet (Figure 2A) and the lower ridge of a putative substrate-binding groove (see next section) (Figure 4A). The C-extension contains an aromatic-rich motif (P473FFRY477), strictly conserved among all CARM1 orthologues, in which residues Phe474, Phe475 and Tyr477 form main-chain hydrogen bonds and side-chain van der Waals interactions with Pro339 and Val341 in strand β7. Deletion of the C-extension, which truncates CARM1 to the native C-terminus of the other PRMTs, abrogates the methyltransferase activity of CARM1 towards H3 Arg17 (Figure 4B), demonstrating the importance of this unique feature in conferring the specific activity of CARM1 towards histone H3 Arg17.
Figure 4.

CARM1 active site. (A) The active site forms a putative substrate-binding groove, lined by an upper ridge consisting of helices αX and αY (blue), and a lower ridge contributed by the C-extension PFFRY motif (green). The THWY loop forming part of the groove is also shown (red). (B) Methylation of histone H3-H4 tetramer by CARM1-full, CARM1-ΔNC or a ΔNC construct with the C-extension deleted (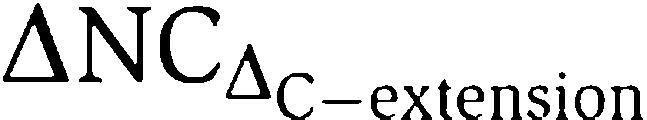 ), probed by anti-H3 dimethyl-Arg17 antibody. (C) Conserved residues in the putative arginine pocket. The upper and lower ridges are coloured blue and green, respectively. This pocket can accommodate the side chain of the target arginine (position indicated by dotted circle).
), probed by anti-H3 dimethyl-Arg17 antibody. (C) Conserved residues in the putative arginine pocket. The upper and lower ridges are coloured blue and green, respectively. This pocket can accommodate the side chain of the target arginine (position indicated by dotted circle).
CARM1 active site
The surface representation of the CARM1 active site (Figure 2D) shows that the cofactor SAH is accessible only via an opening to a narrow pocket in the active site. The dimensions of this pocket can accommodate the side chain of an arginine, so that its guanidino group can be in close proximity with the cofactor at the end of the pocket. The pocket is formed by the helix αZ, the loop connecting β4 and αD in the N-domain (known as the ‘double-E' hairpin; Zhang and Cheng, 2003), as well as the loop connecting β11 and β12 in the C-domain (THWY loop) (Figure 4C). Several residues lining the pocket are strictly conserved among PRMTs. These include two glutamate residues (Glu258, Glu267), Tyr262 and Met269 from the double-E hairpin, His415 from the THWY loop and Asp166 from αZ (Figure 4C). Superimposition of CARM1 with PRMT1/PRMT3/Hmt1 shows that these conserved residues adopt similar positions, with the exception of Glu267. In CARM1 and PRMT3, this glutamate side chain points towards the pocket, whereas in PRMT1 and Hmt1 it points away from the pocket. The acidic nature of Glu267 is essential, as a CARM1 Glu267Gln mutant lacks methyltransferase activity (Lee et al, 2002).
The arginine pocket in turn opens up to a putative substrate-binding groove between two ridges, in which the peptide chain flanking the target arginine could be bound (Figure 4A). The upper ridge consists of the N-terminal helices αX and αY, as observed in other PRMTs, but helix αX is only ordered in the CARMbin structure, suggesting that the upper ridge is only formed once cofactor is bound. The lower ridge is formed by the PFFRY motif of the C-extension, unique to CARM1.
To gain some experimental structural insight into the interaction of histone H3 with CARM1, we sought to determine the structure of a ternary complex (CARM1-cofactor-substrate) by co-crystallisation and/or soaking of H3 peptides containing Arg17. However, despite intense efforts in screening peptides of various lengths and with various post-translational modifications (Supplementary Figure 1), we were unable to observe sufficiently well-ordered electron density for a bound peptide in the active site of the complex structures, to allow model-building and refinement.
In the reported structure of PRMT1 complexed with SAH and a 19-mer Gly-Arg-rich peptide, weak electron density for a target arginine was observed and fitted. Structural alignment of CARM1 with the PRMT1 ternary complex positions the target arginine (of PRMT1) within the CARM1 arginine pocket, albeit clashing with the invariant glutamate Glu267. It is possible that the conformation of the target arginine in CARM1 may be different from that modelled in PRMT1, and/or there is a rearrangement of the double-E hairpin containing Glu267 on substrate binding. However, a well-ordered and fully occupied ternary complex for one or both enzymes will be required to resolve this question.
The tight constriction of the arginine pocket that allows access of an arginine side chain to the bound cofactor imposes severe conformational restrictions on the ±1 residues flanking Arg17 (i.e., Pro16, Lys18) such that their side chains must project away from the arginine pocket, towards a nonpolar environment contributed by the C-extension PFFRY motif that forms the bottom ridge of the groove and Tyr417 from the THWY loop (Tyr417 is lysine in all other PRMTs) (Figure 4A). We cannot discern any obvious features within the extended groove, additional to the arginine pocket, that would provide ‘specificity sub-sites' able to selectively recognise residues flanking the target arginine.
Kinetic analysis
In promoters regulated by steroid hormones or p53, pre-acetylation of histone H3 by p300/CBP significantly promotes CARM1 methylation (Daujat et al, 2002; An et al, 2004). As a clear example of syntax within the histone code, we sought to understand the underlying relationship between these two modifications. We first performed an in vitro enzymatic assay using a recombinant GST-tagged histone H3 N-terminal tail (Figure 5A). Equal amounts of GST-H3tail (top panel) were subjected to either CBP acetylation in the presence of Ac-CoA, or a mock reaction in the presence of CoA. Specific acetylation of Lys18, only in the presence of Ac-CoA, was confirmed using a site-specific antibody to acetyl-Lys18 (middle panel, compare lane 2 to lane 1) and a generic antibody to acetyl-lysine (data not shown). Subsequent Arg17 dimethylation of GST-H3tail by CARM1, detected by a site-specific antibody, showed a significantly higher level of Arg17 methylation in the pre-acetylated GST-H3tail (bottom panel, lane 2) compared to the non-acetylated GST-H3tail (bottom panel, lane 1).
Figure 5.

Crosstalk between histone modifications. (A) Lysine pre-acetylation of H3 N-terminal tail potentiates CARM1 methylation in vitro. Equal amounts of GST-H3tail are subjected to either a mock acetylation followed by CARM1-full methylation (lane 1), or CBP acetylation followed by CARM1-full methylation (lane 2). Before CARM1 methylation, GST-H3tail was purified by Glutathione Sepharose to remove CBP and residual CoA/Ac-CoA. Acetylation and methylation are probed by anti-acetyl-Lys18 and anti-dimethyl-Arg17 antibodies, respectively. (B) Top panel: sequences of the H3 peptides used in the kinetic assay. Middle panel: CARM1-full was incubated with K18-peptide (green box), K18Ac-peptide (red diamond) or histone H4-peptide (negative control, cyan circle). Product formation was plotted versus substrate concentration (0.031, 0.063, 0.125, 0.25, 0.5 and 1 mM) and nonlinear regression was performed for determination of Km and kcat values. Inset: Lineweaver–Burk plot of 1/V versus 1/[S]. Bottom panel: table of Km and kcat values. (C) Gel-based methylation assay for K18-peptide and K18Ac-peptide, detected by anti-dimethyl-Arg17 or anti-dimethyl-Arg26 antibodies. Loading controls for CARM1-full and H3 peptides are shown.
To determine whether enhanced Arg17 dimethylation was a specific effect of CBP acetylation of Lys18, rather than any other H3 lysine residue, we analysed the methyltransferase activity of CARM1 on synthetic peptides encompassing histone H3 amino acids 1–29, in which Lys18 was either unmodified (K18) or acetylated (K18Ac) (Figure 5B, top panel). Steady-state kinetic studies (Figure 5B, middle panel) show CARM1 to have a fivefold higher activity towards the K18Ac-peptide than the unmodified K18-peptide. Fitting of the kinetic data to the Michaelis–Menton equation by nonlinear regression or by double-reciprocal plots (Figure 5B, inset) showed the increased activity towards the acetylated K18Ac-peptide was entirely due to an approximately fivefold increase in the catalytic reaction rate (kcat), but with essentially no difference in the affinity (Km) for the two peptides (Figure 5B, bottom panel).
As CARM1 methylates Arg17 and Arg26 in vitro and in vivo, we sought to determine whether prior acetylation of Lys18 could enhance CARM1 methylation at both of these sites, in a gel-based assay using the synthetic peptides (Figure 5C). Consistent with the previous data, the dimethyl-Arg17 antibody detected a higher level of Arg17 methylation for the K18Ac-peptide, compared with the K18-peptide. A parallel experiment for detection using the dimethyl-Arg26 antibody demonstrates that the level of Arg26 methylation is essentially identical in the modified and unmodified peptides, supporting the notion that the increased methylation observed in Figure 5B is fully attributable to Arg17 methylation.
Discussion
Structural evidence for an ordered mechanism
The crystal structure of CARM1 catalytic core provides the first structural view of a PRMT in both apo sp (CARM1apo) and holo state complexed with SAH (CARM1bin), revealing significant conformational changes induced by cofactor binding (Figure 2C and D). Specifically, these involve: (i) rearrangement of a Gly-rich loop to allow cofactor binding, (ii) ordering of helix αX which interacts with and buries the cofactor, and (iii) formation of the upper ridge of a putative substrate-binding groove, defining a narrow pocket which provides restricted access by an arginine side chain to the reactive methyl of the cofactor. These observations suggest that arginine dimethylation by PRMTs proceeds by an ordered mechanism in which cofactor binds first, and in which the intermediate monomethylated arginine substrate must be released from the active site, either transiently to allow replenishment of the cofactor, or to swap to the other molecule in the dimer, for the second methylation reaction to occur.
CARM1 substrate specificity
Although the CARM1 catalytic core adopts a similar two-domain fold and dimeric arrangement to previously characterised type I PRMTs, it displays several unique features that distinguish it from the PRMT1/3 subclass. Most importantly, CARM1 has a unique C-extension, which provides the lower ridge of a defined putative substrate-binding groove connecting to the cofactor and active site (Figure 4A), generates a much more neutral molecular surface charge and is essential for CARM1 methylation of H3 Arg17. These features correlate with functional evidence that CARM1 methylates a different and smaller set of substrates compared to PRMT1/3 in vitro and in vivo (Lee and Bedford, 2002). To date, all characterised CARM1 substrates lack the highly basic GAR-domains, which are the preferred substrates of PRMT1/3 (Lin et al, 1996; Tang et al, 1998).
Despite intense efforts, we have so far been unable to observe well-ordered density for a bound H3 peptide that would allow crystallographic refinement of a ternary complex. Similar problems were encountered with peptide complexes of PRMT1, and may be related to the high salt conditions required for crystallisation in both cases (Zhang and Cheng, 2003). However, the distinctive surface features of the active site in the CARM1 binary complex, allow some reasonable conclusions to be drawn. The narrow pocket connecting the putative substrate-binding groove of CARM1 to the cofactor-binding site is large enough to accommodate an arginine side chain, and superimposition of the PRMT1 ternary complex on CARM1 positions the bound arginine in this pocket. However, the steric constraints of the mouth of the pocket suggest that unlike PRMT1, the flanking peptide sequence containing the target arginine (Pro16-Arg17-Lys18 in H3) could not adopt an extended conformation, but would have to form a tight β-turn so that the arginine can penetrate the pocket, whereas the side chains of ±1 residues (Pro16, Lys18) are projected away from the pocket and can be accommodated in the groove (Figure 6). Significantly, this stretch of histone H3 adopts just such an ordered β-turn conformation in complex with peptidylarginine deiminase PAD4 (Arita et al, 2006), and suggests that at least part of the specific recognition of this region by modifying enzymes such as CARM1 and PAD4 may be based on its propensity to form turns, favoured by the presence of Pro16. A strong conformational component in CARM1 recognition of its substrates would also go some way to explaining the lack of a consensus amino-acid sequence flanking known CARM1-methylated arginines (Table I), and the lack of clear specificity subsites in the substrate-binding groove.
Figure 6.

Surface features of the CARM1 active site. Features of the active site are coloured: the His415-Asp166 couple (yellow), C-extension (green) and aromatic residues from Tyr262 in the double-E hairpin and the PFFRY/THWY motifs (blue). A peptide sequence of histone H3 (Pro16-Arg17-Lys18) is modelled into the active site with reference to the structures of PRMT1 and PAD4.
Table 1.
Sequences of CARM1 substrates
| CARM1 substrates | Methylated Arg | Sequence |
|---|---|---|
| Histone H3 | R17, R26 | TGGKAPRKQLATKAARKSAPAT |
| PABP1 | R455, R460 | NMPGAIRPAAPRPPFSTM |
| HuR | R206, R217 | YHSPARRFGGPVHHQAQRFRFSPM |
| HuD | R225, R236 | YQSPNRRYPGPLHHQAQRFRLDNL |
| p300 (KIX domain) | R580 R604 | DITQDLRNHLVHK AALKDRRMENLVA |
| CBP (post-KIX domain) | R714 R742 | LPLPVNRMQVSQG QAPMGPRAASPMN |
| R768 | MAISPSRMPQPP | |
| Sequences of CARM1 substrates that have been characterised in vivo. Methylated arginine residues are in bold type. For certain substrates, a primary site (underlined) and other secondary sites have been identified. Histone H3 is the only known substrate with a positively charged residue (italic) at the +1 position. | ||
Hierarchical methylation by CARM1
Our biochemical and structural data (Figures 1B and 4B) demonstrate that the catalytic core of CARM1, together with the C-extension, is sufficient for specific methylation of histone H3 Arg17. However, the observation (Figure 1B) that truncated constructs (CARM1-ΔN, CARM1-ΔNC) still methylate Arg17, but unlike the full-length enzyme, cannot methylate Arg2 or Arg26, has interesting implications. All CARM1 substrates characterised to date, including histone H3, are methylated by CARM1 at more than one site (Table I), raising the possibility that there may be a specific and hierarchical order of methylation, with methylation of secondary sites being dependent on prior methylation of a primary site. At least for the mRNA-binding proteins HuR and HuD, a primary CARM1 methylation site (Arg217 in HuR, Arg236 in HuD) and a secondary site (Arg206 in HuR, Arg225 in HuD) have been identified. Significantly, the secondary Arg206 site was not methylated in a HuR (Arg217Lys) mutation, implying that the methylation of the secondary site is dependent on the primary site (Li et al, 2002; Fujiwara et al, 2006). However, our preliminary in vitro methylation data using H3 Arg17Ala or Arg26Ala mutants (data not shown) do not indicate a hierarchical methylation programme. Together with our observation that H3 Arg17 and Arg26 are differentially methylated by the full-length protein and by constructs lacking the pre-core region, this implicates the pre-core region in providing an additional docking site to make Arg26 a suitable substrate, independent of the methylation status of Arg17. However, further work will be required to delineate the underlying structural and biochemical mechanisms.
Mechanism of crosstalk between lysine acetylation and arginine methylation
The post-translational modification of histone N-terminal tails at different residues, and combinations of these modifications, constitutes a histone code that dictates downstream nuclear processes (Jenuwein and Allis, 2001). The close proximity of the modified residues to each other along the peptide chain, means that modifications present on one residue will inevitably be ‘felt' by an enzyme modifying another residue, and may exert a regulatory effect on that enzyme, thereby enacting a syntax within the histone code.
Modifications of the adjacent histone H3 residues Arg17 and Lys18 are of particular interest, as arginine methylation of H3 by CARM1 in vivo is potentiated by pre-acetylation of H3 Lys18 (Daujat et al, 2002; An et al, 2004). However, the underlying mechanism of this interplay was not understood. Our data (Figure 5) show that acetylation of Lys18 in vitro increases the efficiency of CARM1 methylation by fivefold, and that this enhanced activity on the Lys18-acetylated peptide, attributable to Arg17 methylation, is not due to an increase in affinity of CARM1 for the acetylated peptide, which would be manifest as a decreased Km, but instead results from an increase in the actual rate of the methyltransferase reaction itself (i.e., kcat), and suggests that the chemical nature of the +1 residue can somehow directly influence the efficiency of the catalytic mechanism.
The currently accepted catalytic mechanism for PRMTs (Zhang et al, 2000) involves two invariant glutamates from the double-E hairpin (Glu258 and Glu267 in CARM1), which polarise a guanidine nitrogen atom of the substrate arginine for nucleophilic attack on the sulphur–methyl bond of SAM. The attacking guanidine nitrogen is deprotonated via a proton transfer to an invariant ‘His-Asp' dyad (His415 from the THWY loop and Asp166 from helix αZ in CARM1), and in common with other SN2 reactions, the availability of the deprotonated nucleophile is likely to be rate limiting. The efficiency of this essential proton transfer depends on the pKa of the His–Asp couple, which will be very sensitive to the local electrostatic environment, as will the stability of the transition state itself, in the nucleophilic reaction. Thus, the overall charge environment in the active site can directly influence the catalytic efficiency of the enzyme. Binding of a histone H3 tail containing an unmodified Lys18 would introduce a positive charge within a few angstroms of the active site, elevating the pKa of the His–Asp couple, and destabilising the positively charged transition state of the nucleophilic reaction. By contrast, the side chain of an acetylated Lys18 is neutral and would have far less effect on the electrostatics of the catalytic apparatus. Thus, the modification status of the adjacent residue could directly influence the catalytic efficiency of Arg17 methylation in histone H3, as we observe, by an electrostatic communication mechanism that encapsulates a specific syntactical feature of the histone code. Consistent with this idea, we note that the +1 residue is uncharged in all identified CARM1 substrates with the exception of histone H3 (Table I).
Conclusion
Histone arginine methylation has been shown to play essential roles in the regulation of transcription, and is functionally coupled to other histone modifications such as lysine acetylation. Like lysine acetylation, arginine methylation appears to be dynamically regulated. However, unlike acetyl-lysine, enzymes for the reversal of arginine methylation as part of dynamic regulation, and general modules for the specific recognition of asymmetric dimethyl-arginine in histones, have not been identified (Kouzarides, 2007), so that the biochemical mechanism by which arginine methylation regulates transcription remains to be described.
Materials and methods
Materials
Peptides were prepared by solid-phase synthesis, HPLC-purified, and verified by mass-spectrometry. All antibodies were purchased from AbCam and Upstate with the exception of anti-H3 dimethyl-Arg2, which is a gift from Professor Tony Kouzarides.
Expression and purification of various constructs
Various Mus musculus CARM1 constructs (CARM1full, aa 3–585;  , aa 147–585;
, aa 147–585; 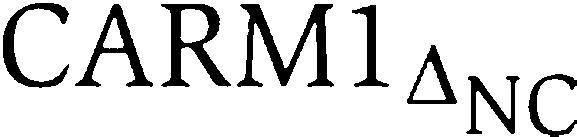 , aa 147–490 and
, aa 147–490 and 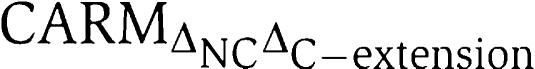 , aa 147–471) were amplified from the I.M.A.G.E. consortium cDNA clone (ID: 4935077), and subcloned into the pFastbac-HTa vector (Invitrogen) in frame with an N-terminal His6-tag and a TEV protease cleavage site. His-CARM1 fusion proteins were expressed in Sf9 cells, purified by metal-affinity chromatography (Talon resin), subjected to His-tag removal by TEV protease and further purified to homogeneity by size-exclusion (Superdex S200) and anion exchange (Source Q) chromatography.
, aa 147–471) were amplified from the I.M.A.G.E. consortium cDNA clone (ID: 4935077), and subcloned into the pFastbac-HTa vector (Invitrogen) in frame with an N-terminal His6-tag and a TEV protease cleavage site. His-CARM1 fusion proteins were expressed in Sf9 cells, purified by metal-affinity chromatography (Talon resin), subjected to His-tag removal by TEV protease and further purified to homogeneity by size-exclusion (Superdex S200) and anion exchange (Source Q) chromatography.
For pull-down studies, the following proteins were expressed as GST-fusions using the pGEX-6P1 vector: CARM1N (aa 25–154), GRIP1AD2 (aa 1236–1392), β-cateninN (aa 21–133), β-cateninΔC (aa 21–665), β-cateninARM (aa 133–665), β-cateninΔN (aa 133–781) and histone H3tail (aa 1–44). Fusion proteins were purified by anion exchange (Q-Sepharose), affinity (Glutathione Sepharose 4B) and size-exclusion chromatography (Superdex 200).
Crystallisation, data collection and refinement
Crystals of apo-CARM1 were grown at 295 K in hanging drops with equal volumes of protein (CARM1-ΔNC, 18 mg/ml) and mother liquor containing 1.6 M diammonium hydrogenphosphate and 100 mM HEPES, pH 7.5. To crystallise a binary complex, protein was pre-incubated with fourfold molar excess of SAH. Data sets (CARM1apo, CARM1bin) were collected on beamline ID14-2 at ESRF Grenoble, and processed using the CCP4 packages (CCP4, 1994). Space group determination was not unambiguous. CARM1apo crystals belong to an I-centred orthorhombic crystal form (2 CARM1 copies per asymmetric unit), whereas CARM1bin crystals belong to a primitive orthorhombic crystal form (4 copies per asymmetric unit), and exhibit pseudo-I222 symmetry as indicated by native Patterson peaks. The pseudosymmetry hinders the assignment of screw-axes through examination of systematic absences. The structure of CARM1bin was first solved by molecular replacement, using PRMT1 structure (PDB entry 1ORI) to search all possible space groups in the 222 point group using Phaser (McCoy et al, 2005). Convincing rotation and translation function solutions were obtained for the P21212 space group, giving an interpretable electron density map in which the majority of the structure was built using Coot (Emsley and Cowtan, 2004). Calculation of difference Fourier maps showed clear electron density corresponding to the cofactor SAH. The structure of CARM1apo was subsequently solved by molecular replacement with the CARM1bin structure. Both structures were refined using CNS (Brunger et al, 1998) and REFMAC5 (Murshudov et al, 1997). Crystallographic statistics are presented in Table II. Coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers 2V7E (CARM1apo) and 2V74 (CARM1bin).
Table 2.
Crystallographic statistics
| Data set | CARM1apo | CARM1bin |
|---|---|---|
| Data collection | ||
| Resolution limits | 25.0–2.7 Å | 30–2.7 Å |
| Cell parameters | a=74.14 Å | a=74.72 Å |
| b=98.02 Å | b=98.99 Å | |
| c=206.92 Å | c=206.70 Å | |
| α=β=γ=90° | α=β=γ=90° | |
| Spacegroup | I222 | P21212 |
| Rmerge | 0.066 (0.21) | 0.09 (0.38) |
| I/σ | 8.5 (5.1) | 16.8 (3.0) |
| Completeness (%) | 99.8 (99.4) | 99.2 (99.2) |
| Multiplicity | 5.8 | 6.6 |
| Refinement | ||
| Unique reflections | 19 941 | 40 542 |
| Rcryst/Rfree | 0.227/0.276 | 0.223/0.273 |
| No of protein atoms | 5039 | 10 467 |
| r.m.s.d. bond length (Å) | 0.008 | 0.009 |
| r.m.s.d. bond angle (deg) | 1.140 | 1.247 |
| In the asymmetric unit of CARM1apo, there are two molecules of CARM1 (chains A and B). In the asymmetric unit of CARM1bin, there are four molecules each of CARM1 (chains B, D, F, H) and SAH (chains B1, D1, F1, H1). | ||
Gel-based methylation assay
The recombinant H3–H4 tetramer used in the assay was expressed in Escherichia coli, purified and reconstituted as described previously (Luger et al, 1997). A 2 μg aliquot of histone H3–H4 was incubated with 1 mM SAM and the indicated CARM1 construct in 20 μl HMT buffer (25 mM Tris, pH 8.0, 200 mM NaCl, 0.5 mM EDTA, 2 mM DTT) for 1 h at 37°C. Reactions were stopped by addition of SDS loading buffer and analysed by SDS–PAGE and immunoblotting. Acetylation was performed using a baculovirus-expressed mouse CBP construct (aa 1231–1711), in the presence of Ac-CoA for 1 h at 37°C.
Kinetic measurement
CARM1-full (1 μM) was incubated with different concentrations of peptide substrates (31 μM–1 mM), [methyl-3H]SAM (1 μCi), and an excess of cold SAM in 20 μl HMT buffer. The H3-K18, H3-K18Ac and H4 peptides have a C-terminal lysine conjugated to biotin. Concentrations of different peptides were normalised by absorbance at 230 nm. Reactions were initiated by substrate addition, incubated at 37°C for 15 min and terminated by addition of 5% acetonitrile. Peptides were bound by Sep-pak reverse-phased C18 columns (Waters), washed three times, eluted with 40% acetonitrile and subjected to liquid scintillation counting. The nanomolar [methyl-3H] incorporated for each data point (in duplicates) were fitted to Michaelis–Menton and Lineweaver–Burk plots. Values for the binding constant Km and catalytic turnover kcat were calculated from nonlinear regression analysis using the proFit package.
In vitro protein interaction assay
Equal amounts (50 μg) of indicated protein and pre-bound GST-tagged protein were incubated in 50 μl Glutathione Sepharose resin pre-equilibrated with PD buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.1% NP-40, 0.1 mM SAM and 1 mg/ml BSA). Binding was performed overnight in 200 μl PD buffer at 4°C. The washed resin was diluted 50-fold, resolved by SDS–PAGE, electroblotted and probed with antibody as indicated.
Supplementary Material
Supplementary Figure 1
Acknowledgments
We thank Jacky Metcalfe for peptide synthesis, Anselm Oberholzer for β-catenin constructs, Valerie Good for assistance in baculovirus expression, Jon Wilson for helpful discussions and the European Synchrotron Research Facility for access to synchrotron beam time. This work was supported by a programme grant from Cancer Research UK (LHP).
References
- An W, Kim J, Roeder RG (2004) Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell 117: 735–748 [DOI] [PubMed] [Google Scholar]
- Arita K, Shimizu T, Hashimoto H, Hidaka Y, Yamada M, Sato M (2006) Structural basis for histone N-terminal recognition by human peptidylarginine deiminase 4. Proc Natl Acad Sci USA 103: 5291–5296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer UM, Daujat S, Nielsen SJ, Nightingale K, Kouzarides T (2002) Methylation at arginine 17 of histone H3 is linked to gene activation. EMBO Rep 3: 39–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford MT, Richard S (2005) Arginine methylation an emerging regulator of protein function. Mol Cell 18: 263–272 [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Chen D, Huang SM, Stallcup MR (2000) Synergistic, p160 coactivator-dependent enhancement of estrogen receptor function by CARM1 and p300. J Biol Chem 275: 40810–40816 [DOI] [PubMed] [Google Scholar]
- Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR (1999) Regulation of transcription by a protein methyltransferase. Science 284: 2174–2177 [DOI] [PubMed] [Google Scholar]
- Chen SL, Loffler KA, Chen D, Stallcup MR, Muscat GE (2002) The coactivator-associated arginine methyltransferase is necessary for muscle differentiation: CARM1 coactivates myocyte enhancer factor-2. J Biol Chem 277: 4324–4333 [DOI] [PubMed] [Google Scholar]
- Cheng D, Cote J, Shaaban S, Bedford MT (2007) The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol Cell 25: 71–83 [DOI] [PubMed] [Google Scholar]
- Cheung P, Tanner KG, Cheung WL, Sassone-Corsi P, Denu JM, Allis CD (2000) Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol Cell 5: 905–915 [DOI] [PubMed] [Google Scholar]
- Chevillard-Briet M, Trouche D, Vandel L (2002) Control of CBP co-activating activity by arginine methylation. EMBO J 21: 5457–5466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook JR, Lee JH, Yang ZH, Krause CD, Herth N, Hoffmann R, Pestka S (2006) FBXO11/PRMT9, a new protein arginine methyltransferase, symmetrically dimethylates arginine residues. Biochem Biophys Res Commun 342: 472–481 [DOI] [PubMed] [Google Scholar]
- Covic M, Hassa PO, Saccani S, Buerki C, Meier NI, Lombardi C, Imhof R, Bedford MT, Natoli G, Hottiger MO (2005) Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-kappaB-dependent gene expression. EMBO J 24: 85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daujat S, Bauer UM, Shah V, Turner B, Berger S, Kouzarides T (2002) Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr Biol 12: 2090–2097 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Allis CD (2003) Histone and chromatin cross-talk. Curr Opin Cell Biol 15: 172–183 [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Mori Y, Chu DL, Koyama Y, Miyata S, Tanaka H, Yachi K, Kubo T, Yoshikawa H, Tohyama M (2006) CARM1 regulates proliferation of PC12 cells by methylating HuD. Mol Cell Biol 26: 2273–2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AH, Nelson WJ, Weis WI (1997) Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell 90: 871–882 [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074–1080 [DOI] [PubMed] [Google Scholar]
- Koh SS, Li H, Lee YH, Widelitz RB, Chuong CM, Stallcup MR (2002) Synergistic coactivator function by coactivator-associated arginine methyltransferase (CARM1) and beta-catenin with two different classes of DNA-binding transcriptional activators. J Biol Chem 277: 26031–26035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T (2007) Chromatin modifications and their function. Cell 128: 693–705 [DOI] [PubMed] [Google Scholar]
- Lee J, Bedford MT (2002) PABP1 identified as an arginine methyltransferase substrate using high-density protein arrays. EMBO Rep 3: 268–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Coonrod SA, Kraus WL, Jelinek MA, Stallcup MR (2005) Regulation of coactivator complex assembly and function by protein arginine methylation and demethylimination. Proc Natl Acad Sci USA 102: 3611–3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Koh SS, Zhang X, Cheng X, Stallcup MR (2002) Synergy among nuclear receptor coactivators: selective requirement for protein methyltransferase and acetyltransferase activities. Mol Cell Biol 22: 3621–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Park S, Kilburn B, Jelinek MA, Henschen-Edman A, Aswad DW, Stallcup MR, Laird-Offringa IA (2002) Lipopolysaccharide-induced methylation of HuR, an mRNA-stabilizing protein, by CARM1. Coactivator-associated arginine methyltransferase. J Biol Chem 277: 44623–44630 [DOI] [PubMed] [Google Scholar]
- Lin WJ, Gary JD, Yang MC, Clarke S, Herschman HR (1996) The mammalian immediate-early TIS21 protein and the leukemia-associated BTG1 protein interact with a protein-arginine N-methyltransferase. J Biol Chem 271: 15034–15044 [DOI] [PubMed] [Google Scholar]
- Lo WS, Trievel RC, Rojas JR, Duggan L, Hsu JY, Allis CD, Marmorstein R, Berger SL (2000) Phosphorylation of serine 10 in histone H3 is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol Cell 5: 917–926 [DOI] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, Flaus AJ, Waye MM, Richmond TJ (1997) Characterization of nucleosome core particles containing histone proteins made in bacteria. J Mol Biol 272: 301–311 [DOI] [PubMed] [Google Scholar]
- Ma H, Baumann CT, Li H, Strahl BD, Rice R, Jelinek MA, Aswad DW, Allis CD, Hager GL, Stallcup MR (2001) Hormone-dependent, CARM1-directed, arginine-specific methylation of histone H3 on a steroid-regulated promoter. Curr Biol 11: 1981–1985 [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr D 61: 458–464 [DOI] [PubMed] [Google Scholar]
- Miao F, Li S, Chavez V, Lanting L, Natarajan R (2006) Coactivator-associated arginine methyltransferase-1 enhances nuclear factor-kappaB-mediated gene transcription through methylation of histone H3 at arginine 17. Mol Endocrinol 20: 1562–1573 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Schluckebier G, O'Gara M, Saenger W, Cheng X (1995) Universal catalytic domain structure of AdoMet-dependent methyltransferases. J Mol Biol 247: 16–20 [DOI] [PubMed] [Google Scholar]
- Schurter BT, Koh SS, Chen D, Bunick GJ, Harp JM, Hanson BL, Henschen-Edman A, Mackay DR, Stallcup MR, Aswad DW (2001) Methylation of histone H3 by coactivator-associated arginine methyltransferase 1. Biochemistry 40: 5747–5756 [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403: 41–45 [DOI] [PubMed] [Google Scholar]
- Tang J, Gary JD, Clarke S, Herschman HR (1998) PRMT 3, a type I protein arginine N-methyltransferase that differs from PRMT1 in its oligomerization, subcellular localization, substrate specificity, and regulation. J Biol Chem 273: 16935–16945 [DOI] [PubMed] [Google Scholar]
- Teyssier C, Chen D, Stallcup MR (2002) Requirement for multiple domains of the protein arginine methyltransferase CARM1 in its transcriptional coactivator function. J Biol Chem 277: 46066–46072 [DOI] [PubMed] [Google Scholar]
- Teyssier C, Ou CY, Khetchoumian K, Losson R, Stallcup MR (2006) Transcriptional intermediary factor 1alpha mediates physical interaction and functional synergy between the coactivator-associated arginine methyltransferase 1 and glucocorticoid receptor-interacting protein 1 nuclear receptor coactivators. Mol Endocrinol 20: 1276–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Padilla ME, Parfitt DE, Kouzarides T, Zernicka-Goetz M (2007) Histone arginine methylation regulates pluripotency in the early mouse embryo. Nature 445: 214–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss VH, McBride AE, Soriano MA, Filman DJ, Silver PA, Hogle JM (2000) The structure and oligomerization of the yeast arginine methyltransferase, Hmt1. Nat Struct Biol 7: 1165–1171 [DOI] [PubMed] [Google Scholar]
- Xu W, Chen H, Du K, Asahara H, Tini M, Emerson BM, Montminy M, Evans RM (2001) A transcriptional switch mediated by cofactor methylation. Science 294: 2507–2511 [DOI] [PubMed] [Google Scholar]
- Yadav N, Lee J, Kim J, Shen J, Hu MC, Aldaz CM, Bedford MT (2003) Specific protein methylation defects and gene expression perturbations in coactivator-associated arginine methyltransferase 1-deficient mice. Proc Natl Acad Sci USA 100: 6464–6468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cheng X (2003) Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure 11: 509–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhou L, Cheng X (2000) Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. EMBO J 19: 3509–3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zika E, Fauquier L, Vandel L, Ting JP (2005) Interplay among coactivator-associated arginine methyltransferase 1, CBP, and CIITA in IFN-gamma-inducible MHC-II gene expression. Proc Natl Acad Sci USA 102: 16321–16326 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1