

Abstract
Dietary cholesterol absorption contributes to a large part of the circulating cholesterol. However, the mechanism of sterol intestinal uptake is not clearly elucidated. Scavenger receptor class B type I (SR-BI), major component in the control of cholesterol homeostasis, is expressed in the intestine, but its role in this organ remains unclear. We have generated transgenic mice over-expressing SR-BI primarily in the intestine by using the mouse SR-BI gene under the control of intestinal specific “apolipoprotein (apo) C-III enhancer coupled with apo A-IV promoter”. We found SR-BI overexpression with respect to the natural protein along the intestine and at the top of the villosities. After feeding a meal containing [14C] cholesterol and [3H] triolein, SR-BI transgenic mice presented a rise of intestinal absorption of both lipids that was not due to a defect in chylomicron clearance nor to a change in the bile flow or the bile acid content. Nevertheless, SR-BI transgenic mice showed a decrease of total cholesterol, but an increase of triglyceride content in plasma without any change in the HDL apo A-I level. Thus, we describe for the first time a functional role in vivo for SR-BI in cholesterol but also in triglyceride intestinal absorption.
Keywords: Absorption; Animals; Apolipoproteins; chemistry; Bile Acids and Salts; metabolism; Cell Membrane; metabolism; Cholesterol; chemistry; metabolism; Chylomicrons; chemistry; DNA, Complementary; metabolism; Homeostasis; Immunohistochemistry; Intestines; chemistry; metabolism; Lipids; chemistry; Lipoproteins; chemistry; Liver; metabolism; Mice; Mice, Transgenic; Promoter Regions (Genetics); Receptors, Lipoprotein; metabolism; Receptors, Scavenger; chemistry; Scavenger Receptors, Class B; metabolism; Tissue Distribution; Triglycerides; metabolism; Triolein; chemistry
Keywords: SR-BI, intestinal absorption, transgenic mice, cholesterol, triglycerides
Dyslipidemia (e.g. hyperlipidemia or dyslipoproteinemia) plays a key role in the development of obesity, cardiovascular disease or the metabolic syndrome. Intestinal lipid absorption is an important contributor to the overall balance of lipid metabolism, but regulation at the molecular level remains largely unknown. Intestinal lipid absorption involves the luminal lipolysis of triglycerides to monoglycerides and fatty acids and of cholesterol esters to free cholesterol and fatty acids. Free cholesterol from diet or bile and the products of lipid hydrolysis interact with bile salts to form mixed micelles, which pass through an unstirred water layer covering the enterocyte surface (1). The lipid molecules are then released from micelles and enter the enterocyte where they are assembled into chylomicrons before secretion into the circulation via the lymphatic network. They are finally transported to the liver through the chylomicron-remnant pathway (2).
Mechanisms by which enterocyte lipid absorption occurs is still controversial. In addition to a simple passive diffusion, there is strong evidence for the existence of a protein-facilated mechanism (3–6). Due to a significant positive correlation between cholesterol absorption and plasma cholesterol levels (7), cholesterol absorption has been the subject of intense research. Different proteins have successively been implicated in intestinal lipid absorption. For instance, CD 36 - a multiligand scavenger receptor- has been proposed to take up monoglycerides and fatty acids (8), but could also participate in cholesterol absorption (9,10). Recently, a newly discovered protein, the Niemann-Pick C1 Like 1 (NPC1L1), has been shown to play a major role in cholesterol absorption (11) since knock-out mice for this protein decreased intestinal cholesterol absorption by 80%. Also aminopeptidase-N has been implicated in cholesterol absorption (12).
Scavenger receptor class B type I (SR-BI) is also a likely element of intestinal cholesterol absorption. SR-BI is a 82 kDa membrane protein mostly expressed in liver and steroidogenic tissues. It binds native high density lipoproteins (HDL) (13), LDL or oxidized LDL and anionic phospholipids and mediates both the selective uptake of HDL cholesteryl ester by the liver and free cholesterol efflux from cells of peripheral tissues (14). SR-BI is also expressed in enterocyte brush border membranes mainly at the top of intestinal villosities and in the proximal part of intestine where cholesterol absorption mainly occurs (15). However, the importance of SR-BI in intestinal cholesterol absorption is under debate since the disruption of SR-BI gene in mice does not affect cholesterol absorption (16). However, the process seems to be more complex and might depend on the combined actions of transporter proteins involved in uptake and efflux of cholesterol, which could compensate for the lack of SR-BI in the intestine.
In order to get new data on the role of SR-BI in cholesterol absorption, we have generated transgenic mice over-expressing SR-BI primarily in the intestine. The transgene contains the mouse SR-BI gene under the control of the human intestinal specific apolipoprotein (apo) C-III enhancer coupled with the apo A-IV promoter. This construct is the only promoter known to induce a decreasing expression along the gastrocolic axis and an increasing one from the crypt to the top of the villosity (17). Here, we show that SR-BI intestinal overexpression increases intestinal absorption of both cholesterol and triglyceride.
EXPERIMENTAL PROCEDURES
Generation of transgenic mice
The murine SR-BI cDNA was obtained by digestion of pRC/CMV vector by restriction enzyme Hind III and Xba I. The apolipoprotein C-III enhancer (−500/−890 pb)-apolipoprotein A-IV promoter (−700 pb) was excised from pUCSH-CAT plasmids (18) with the restriction enzymes Xba I and Hind III. The construct containing the human apolipoprotein C-III enhancer-apolipoprotein A-IV promoter to the intestinal specificity and the 1.8 kilobase cDNA fragment of SR-BI (19) was cloned into XbaI site of the pc DNA 1.1 vector. A linearized fragment of the construct was obtained by digestion of the vector by the restriction enzymes Sal I and Avr II and used to generate transgenic mice by standard procedures in a B6D2 background. The mice genotyping was performed on DNA extracted from tail biopsies by polymerase chain reaction (PCR) amplification using primers specific for the construct allele, primer 1: 5′ GCTAGGGCTTTGAATAACTAG 3′, primer 2, 5′ GACCAAGATGTTAGGCAGTACAAT 3′. Founder animals were backcrossed to C57/Bl6 for 12 generations to have a homogenous background. The mice were housed at the Institute animal core facility in temperature and humidity controlled room with a 12 h light/dark cycle and fed a basal chow diet. Studies were performed using 8–15 weeks old SR-BI Tg versus control littermates negative for the SR-BI transgene and fasted overnight before the initiation of absorption studies. All experiments were performed with the approval of the Ethical Committee for Animal Experiments of the University of Toulouse (MP/04/11/03/05).
Plasma lipid analysis
Blood samples were collected from mice and plasma HDL were isolated by LDL/VLDL precipitation with phosphotungstate/MgCl2 reagent (Sigma HDL-cholesterol). Total cholesterol, HDL cholesterol and total triglycerides were determined using commercial enzymatic assays (Randox) adapted for micro methods. LDL/VLDL cholesterol was determined from the difference between total and HDL cholesterol.
Plasma lipoprotein analysis
Lipoproteins were separated from equal pool of plasma (400 μl) of transgenic SR-BI and wild type mice by sequential density ultracentrifugation, IDL/VLDL (d=1.006–1.019), LDL (d=1.019–1.055), HDL (d=1.055–1.21). Samples were collected in equal volume and proteins of each fraction were separated by electrophoresis on 4–16 % SDS polyacrylamide gel and stained with Coomassie blue.
Membrane preparation and protein analysis
Animals were sacrificed and different tissues were collected and immediately frozen. Intestine was removed and flushed with cold Phosphate Buffer Saline (PBS), then cut into three equal sections (Proximal-Medium-Distal) on ice; each section was opened and mucosa was scrapped, immediately frozen in liquid nitrogen and stored at −80°C. Tissues were homogenized at 4°C in 1.5 ml PBS containing phenylmethylsulphonylfluoride (PMSF) 0.1mM, Pepstatin 1μM, Iodoacetamide 1mM, Phenanthroline 1mM (from sigma), using a Turax blender, then 20 strokes in a Dounce homogenizer. Lysates were centrifuged at 4°C, 250g, 5 min. to eliminate tissue debris, then membrane proteins were pelleted at 4°C, 200,000g for 15 min. Total membrane proteins were resuspended in PBS with the protease inhibitor cocktail. Proteins were determined using Bradford reagent (Bio-Rad). In some experiments, intestinal epithelial cells were isolated and separated in a villus to crypt gradient according to Weiser (20) and directly used for Western blot analysis. Proteins from the different tissue extracts were separated on 10% SDS-polyacrylamide gel (Bio-Rad) and transferred to nitrocellulose membrane. Immunoblot analysis of SR-BI was performed by chemiluminescence detection (ECL, PerkinElmer Life Sciences) using rabbit antipeptide polyclonal antibodies against mouse SR-BI protein (dilution 1:1000, Abcam, ab3) and horse radish peroxidase coupled-anti rabbit polyclonal secondary antibody (Sigma).
Immunohistochemistry
In some experiments, small pieces of intestinal sections were collected separately and included in paraffin. Paraffin sections were mounted on silanized slides and paraffin was removed with different toluene baths. Sections were regenerated by microwave in citrate buffer pH 6 and biotin and endogenous peroxidase activity were inhibited by the use of Dako Kit. Sections were then incubated 30 min at room temperature with anti-SR-BI primary antibody (1:500, Abcam, anti-SR-BI/BII, ab115) diluted in Tris Buffer Saline (TBS), 0.3% Bovine Serum Albumin (BSA). After washing, sections were incubated with pig anti-goat biotinylated secondary antibody (1:100) for 30 min diluted in TBS with 1/25 pig serum. Sections were washed and incubated with streptavidin-peroxidase complex (Dako) for 30 min. The peroxidase activity was revealed using 3′ diaminobenzidine tetrahydrochloride (DAB) as chromogen (Dako). Samples were observed under a light microscope.
Lipid tissue distribution
Liver and intestinal mucosa divided in 3 parts were homogenized in 1 ml methanol/water (2/1; v/v), EGTA 5 mM (Sigma). Lipids were extracted according to Bligh and Dyer (21) and molecular species of neutral lipids (cholesterol, cholesteryl esters, diacyl- and triacyl-glycerols) were quantitated by gas liquid chromatography (22).
Intestinal absorption of lipids
To determine dietary lipid absorption, mice were fasted overnight then fed by gavage with a mixture of 2 μCi of [4-14C] cholesterol/1 μCi of [9,10-3H(N)] triolein (PerkinElmer Life Sciences) and 2g/l of cholesterol (Sigma) suspended in 100 μl of corn oil. Blood was collected hourly from the tail and plasma was isolated for liquid scintillation counting. Six hours after gavage, mice were sacrificed. Intestines were removed, flushed with PBS, cut in three equal parts and the mucosa was scrapped and rapidly frozen in liquid nitrogen. The gallbladder was removed and the bile was collected for liquid scintillation counting. Livers were harvested and immediately frozen. The different samples were homogenized in methanol/water (2/1; v/v) and tissue distribution of radioactivity was determined by scintillation counting.
In a series of experiments, to gain access to the total intestinal lipid uptake, plasma lipoprotein lipase was inhibited by retro-orbital injection of 100 μl of Triton WR1339-saline (1:6; v/v) prior to gavage (23).
Bile analysis
To determine the excretion rate of cholesterol into bile, animals were anaesthetized by intraperitoneal injection of Hypnorm (fentanyl/fluanisone, 1 ml/kg) and diazepam (10 mg/kg). 100 μl of medium chain triglycerides (Intralipid, Sigma) containing 2 μCi of [14C] cholesterol were injected in the caudal vein;, the common bile duct was ligated close to the duodenum, then the gallbladder was punctured and its content was removed. After 30 min, newly secreted bile was collected for 90 min by gallbladder canulation and the associated [14C] radiolabel was determined by scintillation counting. During bile collection, body temperature was stabilized using a humidified incubator.
In other experiments, bile flow and the bile acid rates were analysed from the bile collected for 60 min via gallbladder canulation (24). Bile flow was determined gravimetrically assuming a density of 1 g/ml for bile. Bile acids were determined by HPLC. Briefly, 2 μl bile were diluted with 15 μl of internal standard (Norcholic acid 6.7 mM, Steraloids, Wilton, NH)) in methanol to precipitate proteins. Samples were centrifuged at 20,000 g for 10 min at 4°C and the supernatant was further diluted in methanol (1/600). Samples (100 μl) were separated by HPLC (DIONEX Sumit) on a LiChrosorb RP-18 analytical column (5 μm particle size, 25 cm × 4.6 mm ID) fitted with a C18 guard column cartridge (1 cm × 4.6 mm, Supelco) and coupled to a light scattering detector (Polymer laboratory ELS 2100, nitrogen flow 1.2 ml/min, evaporating temperature 90°C and nebulisator temperature 70°C). Separation was achieved at a flow rate of 0.7 ml/min using a ternary solvent system (25) modified as following: ammonium acetate pH 5.2 (30mM), methanol, acetonitrile; 35/60/5 for 25 minutes, then 24/53/23 in 5 minutes for 20 minutes.
RESULTS
Characterization of the transgenic mice overexpressing SR-BI in the intestine
We have generated transgenic mice over-expressing SR-BI under the control of the apolipoprotein C-III enhancer coupled to the apolipoprotein A-IV promoter and we analyzed the presence of SR-BI in tissue extracts (Fig 1). In control animals, SR-BI protein was classically expressed mostly in the liver among other tissues, whereas transgenic mice displayed a dramatic over-expression of a mature (82 kDa) SR-BI almost totally restricted to the intestine (Fig. 1A and B). Some overexpression was observed in liver extracts from transgenic mice due to the activation of the promoter also in this organ (17). RNA analysis in the two tissues confirmed a 25 fold increase of SR-BI in the intestine and a 3 fold increase in the liver from transgenic mice when compared to control (not shown). The promoter induced an overexpression of SR-BI in accordance to the distribution of endogenous membrane-bound protein, i.e. decreasing from the duodenum (proximal part of the small intestine) to the colon (Fig 1C) and increasing from the crypt to the top of the villosities (Fig. 1D) where intestinal absorption is mainly present. We confirmed the overexpression of SR-BI by immunocytochemistry on different intestinal sections (Fig. 2). We also observed the presence of the protein at the top of the villosities and on the lumenal side of enterocytes in control mice (Fig. 2B, E, H) and overexpression was observed at the same sites in transgenic mice (Fig. 2C, F, I).
Figure 1. Tissue distribution of SR-BI in transgenic mice.
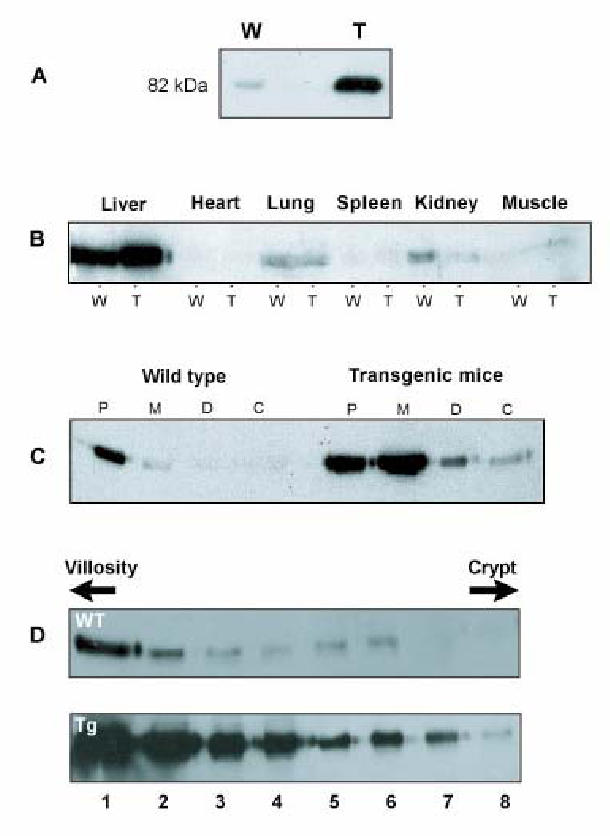
Panel A: 50 μg of membrane protein from total small intestine were analyzed by Western blotting with anti-SR-BI antibodies. W, wild type mice; T, SR-BI transgenic mice.
Panel B: 100 μg of protein from homogenates of different tissues from transgenic and wild type mice were analyzed by Western blotting with anti-SR-BI antibodies.
Panel C: 50 μg of membrane protein from different intestinal segments (duodenum to colon) of wild type and transgenic mice were analyzed by Western blotting with anti-SR-BI antibodies. P, Proximal; M, Medium, D, Distal part of the small intestine; C, Colon.
Panel D, enterocytes from wild type and transgenic mice were separated into 8 different fractions along the villus-crypt axis according to Weiser (20) and homogenized. 50 μg of membrane proteins were analyzed by Western blotting with anti-SR-BI antibodies.
Figure 2. Intestinal localization of SR-BI.
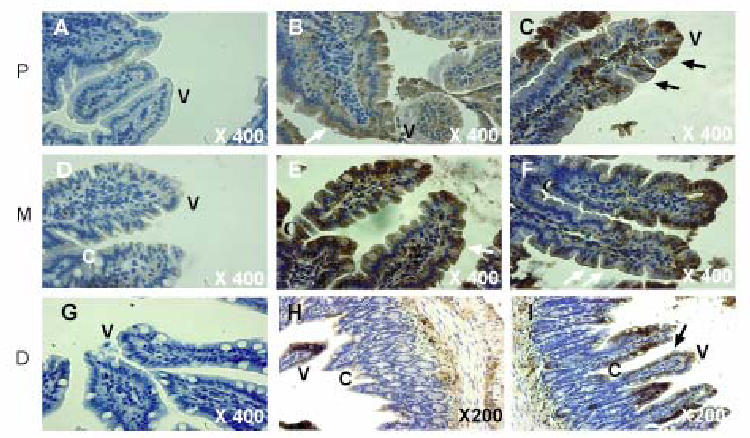
Sections of small intestine from wild type and transgenic mice were incubated with anti-SR-BI antibodies followed by incubation with biotinylated secondary antibodies and with streptavidin-peroxidase complex. Peroxidase activity was revealed using 3′ diaminobenzidine tetrahydrochloride (DAB). A,D,G: control was obtained with an incubation of wild type sections in only the secondary antibodies; B,E,H: wild type mice; C,F,I: SR-BI transgenic mice. P, Proximal; M, Medium; D, Colon. V, Villosity; C, Crypt.
Plasma total cholesterol but not triglyceride is decreased in transgenic mice
Transgenic mice at 8 weeks were further characterized according to plasma lipid and lipoprotein contents (Fig. 3). Interestingly, we observed a dramatic decrease (2.5 fold) in plasma total cholesterol content that was present both in HDL and LDL/VLDL fractions. By contrast, total triglycerides were mildly but significantly increased, reflecting a differential regulation between cholesterol and triglyceride in the transgenic mice. Analysis of the proteins present in lipoproteins fractions separated by density gradients from the plasma did not show any significant difference in the amount of the apolipoproteins (ApoB100, apoB48, apoE, apoAI) present in VLDL, LDL or HDL fractions between control and transgenic mice (Fig. 4). Thus the size of the lipoprotein particles might be different since the ratio of cholesterol to protein is decreased by more than 2 in the transgenic animals. Analysis of lipid content in tissues extracts from intestine or liver (Table 1) did not reveal any difference between control and transgenic mice for free or esterified cholesterol, triglyceride or diglyceride, indicating that the decrease in cholesterol content observed in plasma was not linked to an accumulation in the major organs. By contrast, free cholesterol was increased by 20 % in bile suggesting an increased catabolism of cholesterol in the transgenic mice. However this was not accompanied by a change in the volume of bile in gallbladder (Table 2). In addition no significant difference in the body mass, the weight of the liver and adipose tissues (inguinal and epididymal representing the peripheral and profound white adipose tissue, respectively) was observed between wild type and SR-BI Tg animals (Table 2). The liver did not show steatosis.
Figure 3. Effect of intestinal over-expression of SR-BI on plasma cholesterol and triglycerides levels.
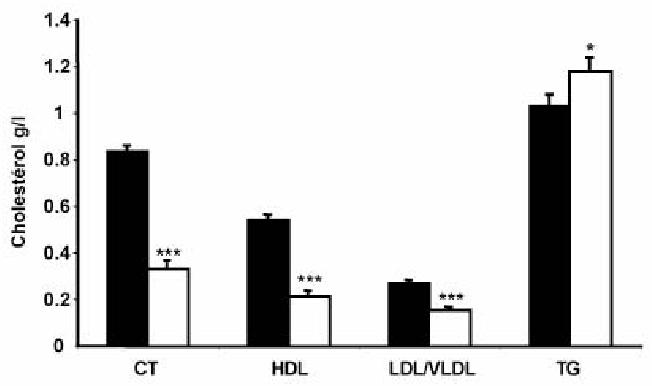
Plasma was collected from 8 weeks aged mice. Total cholesterol, HDL cholesterol, LDL/VLDL cholesterol and triglycerides were determined as described under “Experimental procedures”. Wild type (closed bar), SR-BI Transgenic mice (open bar). Data are presented as mean ± SEM from 10 mice in each group.
Statistical significance between wild type and SR-BI Transgenic mice was determined by Student’s t test.
*** p < 0.01. * p < 0.05
Figure 4. Analysis of plasma apolipoprotein content.
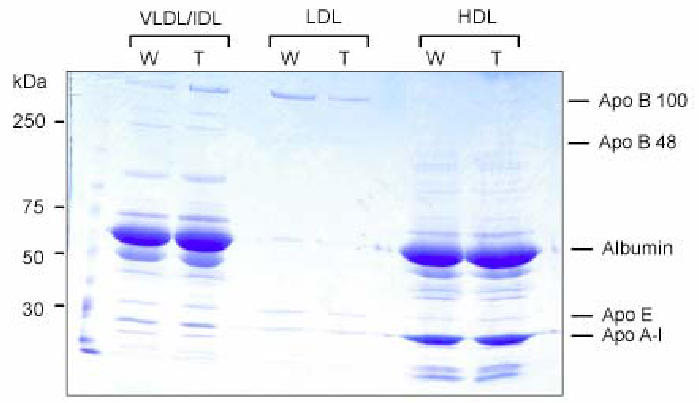
An aliquot of lipoproteins isolated by sequential density ultracentrifugation from equal amount (400 μl) of pooled plasma from 5 mice were loaded onto reducing 4 – 16 % SDS polyacrylamide gel and stained with Coomassie blue. Proteins were identified with respect to the migration of molecular weight standards. W, wild type mice; T, SR-BI transgenic mice.
Table 1. Lipid tissue distribution in wild type and SR-BI Tg mice.
Liver and intestinal mucosa divided in 3 parts were homogenized, then lipids were extracted and quantitated as described under “Experimental procedures”. Values are expressed as nmole/mg of tissue and shown as mean ± SEM, n = 4 for each group. Bile were collected and lipids analyzed as above and expressed as total nmole in collected bile. Statistical significance between wild type and SR-BI Tg mice was determined by Student’s t test.
| Tissue | Genotype | FC | CE | TG | DG |
|---|---|---|---|---|---|
| Proximal Intestine | WT | 24.12±1.12 | 2.37±0.2 | 93.65±29.5 | 16.34±2.24 |
| SR-BI Tg | 23.91±1.63 | 2.08±0.42 | 51.85±26.1 | 10.33±4.76 | |
| Medium Intestine | WT | 19.54±2 | 1.45±0.34 | 394.15±284 | 6.25±1.15 |
| SR-BI Tg | 26.15±3.62 | 1.27±0.20 | 439.80±230 | 19.43±8.16 | |
| Distal Intestine | WT | 14.33±3.04 | 2.14±0.47 | 18.69±3.94 | 37.07±15.46 |
| SR-BI Tg | 17.27±2.89 | 2.40±0.75 | 16.05±6.43 | 33.63±21.14 | |
| Liver | WT | 4.85±0.41 | 3.32±0.22 | 35.45±8.26 | 4.39±2.36 |
| SR-BI Tg | 5.18±0.61 | 2.99±0.44 | 29.52±6.42 | 8.03±4.41 | |
| Bile | WT | 40.02±2.50 | 0 | 11.72±5.21 | 7.27±1.13 |
| SR-BI Tg | 51.72±7.93* | 0 | 3.72±2.34 | 12.90±5.83 |
p < 0.05
Table 2. Characterization of SR-BI Tg mice.
Wild type and SR-BI Tg mice were analyzed at 10 weeks of age. After weighting, animals were sacrificed and the different organs were examined and weighted. iWAT and eWAT: inguinal and epididymal white adipose tissues. Values are expressed as mean ± SEM, n = 5 for each group.
| Body mass (g) | Liver (g) | iWAT (g) | eWAT (g) | Bile (μl) | |
|---|---|---|---|---|---|
| Wild type | 27.65±0.50 | 1.24±0.05 | 0.23±0.02 | 0.61±0.03 | 23±1.8 |
| SR-BI Tg | 27.85±0.44 | 1.16±0.06 | 0.19±0.03 | 0.51±0.03 | 23±1.3 |
Intestinal lipid absorption in transgenic mice
In order to assess the importance of SR-BI intestinal overexpression on intestinal lipid absorption, control or transgenic mice were force-fed with a mixture of [14C] cholesterol and [3H] fatty acid-labeled triglycerides in corn oil, then plasma radioactivities were determined in a time dependent manner. Interestingly, we observed a significant increase for both [14C] (Fig. 5A) and [3H] (Fig. 5B) labels in the plasma up to 4 hours following gavage (corresponding to a post-prandial status). This indicated a net and similar increase in the intestinal absorption of cholesterol and at least the fatty acyl part of triglyceride in response to the overexpression of SR-BI in the intestine. Moreover, when experiment was performed after inhibiting of lipoprotein lipase (infusion with Triton WR1339), the levels of radiolabels in plasma were much higher, but the same difference between wild type and SR-BI Tg mice was observed for both [14C] (Fig. 5C) and [3H] (Fig. 5D) labels, indicating that increased absorption rates were not due to a defective chylomicron remnant clearance.
Figure 5. Lipid absorption rates in wild type and SR-BI Tg mice.
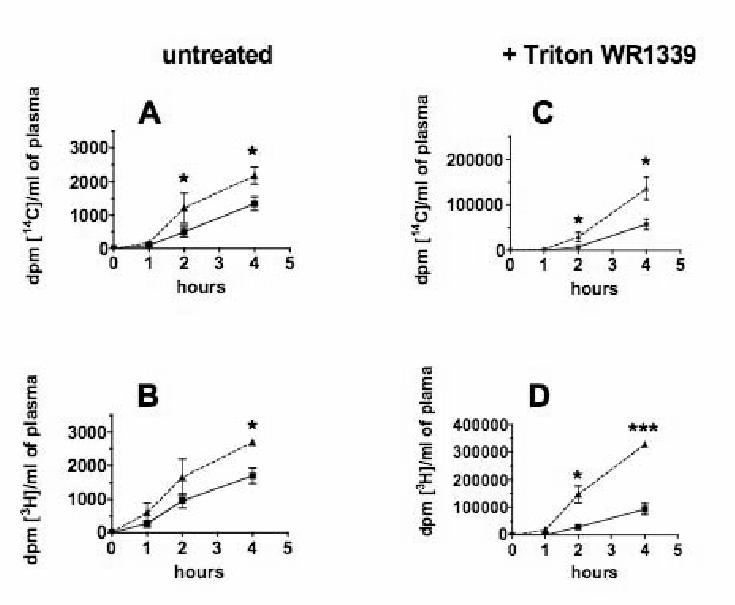
Wild type (square) and SR-BI Tg (triangle) mice were fed by stomach gavage with a 100 μl test meal containing 2 μCi [14C] cholesterol, 1μCi [3H] triolein and 2 g/l cholesterol suspended in corn oil, without (panels A and B) or after (panels C and D) retro-orbitaly injection of 0.1ml Triton WR 1339 in NaCl 0.9 % (1:6, v/v). Blood samples were collected up to 4 h and radioactivity was measured by liquid scintillation counting. Cholesterol (A, C) and triglycerides (B, D) absorption rates are presented as mean ± SEM from 6 mice in each group (A, B) and mean ± SEM from 3 mice (C, D).
* Statistical significance, p < 0.05 between wild type and SR-BI Tg mice as determined by Student’s t test.
*** p < 0.01.
Radioactive cholesterol also significantly accumulated in the intestinal tissue and in the liver (Fig. 6A), indicating a redistribution of the absorbed cholesterol in the organism. Also, radioactivity related to the triglyceride fraction was increased in different organs (Fig. 6B), that was in agreement with the observed increase in plasma total triglycerides (Fig. 3).
Figure 6. Tissue distribution of [14C] and [3H] radiolabels in wild type and SR-BI Tg mice following gavage.
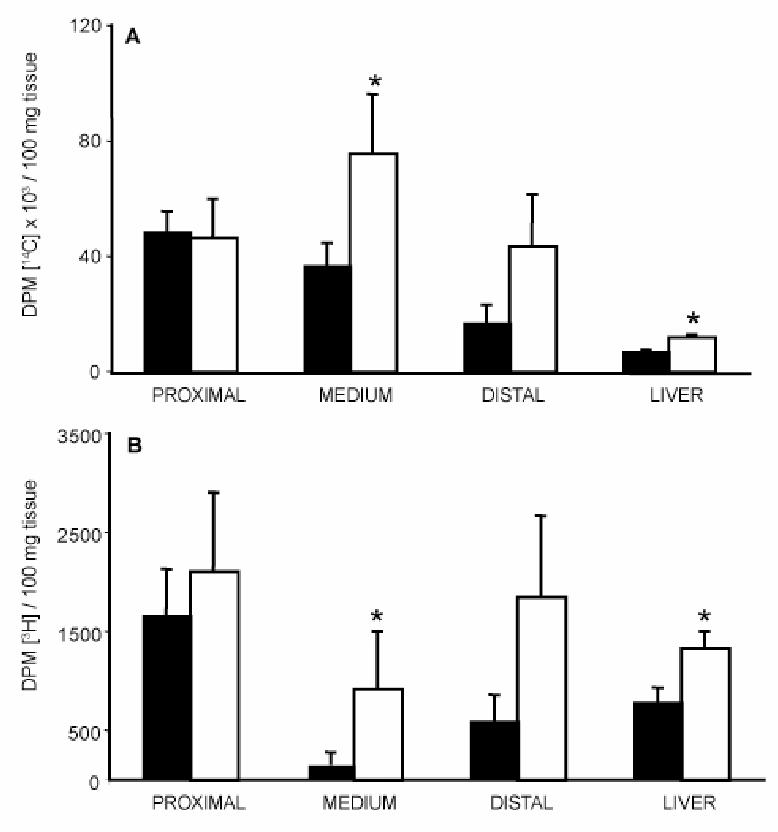
On the same animals as in Figure 5 (A and B), liver and intestinal mucosa (divided in 3 parts) were harvested 6 h after gavage and homogenized as described under “Experimental procedures”. Tissue distribution of [14C] cholesterol (A) and [3H] radiolabel (B) were determined by scintillation counting. Wild type (closed bar), SR-BI Transgenic mice (open bar). Data are presented as mean ± SEM from 6 mice in each group.
Statistical significance between wild type and SR-BI Transgenic was determined by Student’s t test. * p < 0.05
Bile content in [14C] label was also increased two fold in the transgenic animals (not shown), suggesting a stimulation of the cholesterol secretion that could be attributed to the over-expression of SR-BI in liver. To confirm this hypothesis, labeled cholesterol was directly injected in the caudal vein and the distribution of cholesterol label was analyzed in bile and plasma (Fig. 7). A net increase of the bile to plasma cholesterol ratio was observed in transgenic mice indicating that the radioactive cholesterol injected in blood was rapidly excreted into bile, supporting the increased secretion of cholesterol from liver of SR-BI transgenic animals.
Figure 7. contribution of hepatic SR-BI over-expression to the catabolism of cholesterol.
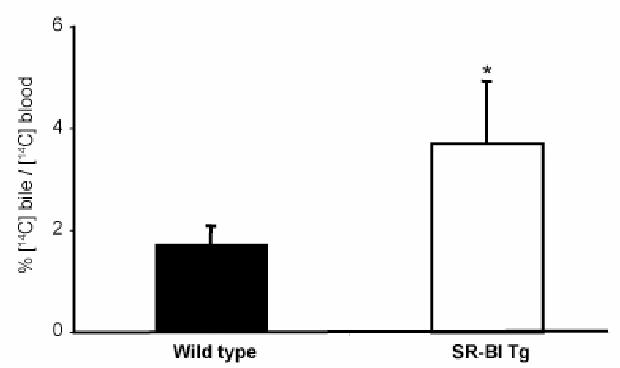
Mice were injected intravenously with 100 μl of intralipid containing 2 μCi [14C] cholesterol, then biles were collected from 30 min to 120 min (see “Experimental procedures”). [14C] radiolabel excreted into bile was determined by scintillation counting. Data are presented as the percentage of [14C] radiolabel in the bile per [14C] radiolabel in plasma ± SEM from 5–6 mice in each group. Wild type (closed bar), SR-BI Tg (open bar). Statistical significance between wild type and SR-BI Tg was determined by Student’s t test. * p < 0.05
However, in order to evaluate a possible role of the bile acid pool in the increased lipid absorption observed in SR-BI-Tg mice, we performed gallbladder canulation followed by bile collection up to 1 hour to access the bile acid pool and bile flow delivered to the intestine. No significant difference was observed in the bile flow rate (4.2±0.7 and 2.5±1.1 μl/min/100g of body weight) nor in the two major bile acids, taurocholic acid (198±41 versus 134±64 nmole/min/100g of body weight) and tauromuricholic acid (166±39 and 138±49 nmole/min/100g of body weight) in wild type (n=5) and SR-BI-Tg (n=4), respectively.
DISCUSSION
We generated transgenic mice over-expressing SR-BI mainly in the small intestine using a specific part of the Apo C-III/Apo A-IV promoter sequence. This promoter induced also a mild over-expression in the liver as previously described using CAT gene reporter (17). This transgenic mouse strain is up to now the only model of intestinal over-expression of SR-BI respecting the natural expression of the protein, i.e. at the apical site of enterocytes, increasing from crypt to villus and with a cephalo-caudal decrease. Thus, SR-BI is expressed where intestinal cholesterol absorption mainly occurs in SR-BI transgenic mice as in wild type littermate.
This study evidences for the first time that SR-BI plays a role in vivo in intestinal cholesterol absorption. Indeed, the intestinal cholesterol uptake determined by radioactive acute phase measurement of plasma cholesterol was doubled at 3 hours after gavage in the transgenic mice. However, this is in contradiction with recent results obtained on KO SR-BI mice, which indicated only a minor effect of SR-BI in intestinal cholesterol uptake (16). Nevertheless, numerous studies evidenced that intestinal absorption mechanisms involved the combined actions of different transporters which could compensate for the lack of SR-BI in the knock-out model, such as ABCA1 (26), ABCG-5/ABCG-8 (27), NPC1L1 (11,28) and the aminopeptidase N (29). Moreover, the increased intestinal absorption of lipids could not be attributed to a defect in the clearance of chylomicrons in SR-BI Tg mice, since inhibition of lipoprotein lipase did not change the differences in the absorption rates between wild type and transgenic mice.
Interestingly, in contrast to the increased intestinal cholesterol uptake, we observed an unexpected decrease of plasma cholesterol content in all lipoprotein fractions (around 50%), which resembles to the major phenotype observed in animals over-expressing SR-BI in the liver (30–33). All of these studies concluded to a major role for hepatic SR-BI in the clearance of HDL cholesterol as well as in the increased bile cholesterol secretion. From our transgenic model, we show the contribution of SR-BI overexpression both in the intestine and the liver. In the intestine, the observed increase of the radiolabeled cholesterol in plasma following acute phase might reflect an increase of chylomicrons secretion. Indeed, a net increase of tocopherol, a specific marker for chylomicron synthesis (34), is measured in the plasma of our transgenic mice following gavage with tocopherol (Reboul and Borel, personal communication). But the rise of intestinal cholesterol absorption is likely balanced by the moderate over expression of SR-BI in the liver inducing an increase of cholesterol catabolism shown by the bile cholesterol secretion enhancement (Fig. 7 and Table 1) as well as an increase of chylomicrons catabolism (35). Nevertheless, the increased intestinal uptake is not due to a simple rise in the bile flow or in the bile acid pool that remained unchanged in the wild type and SR-BI Tg mice.
Also contradictory is the status of apo AI, the major apolipoprotein of HDL; indeed, liver SR-BI overexpression presented a decrease of cholesterol associated with a decrease of apo A-I in correlation with the expression levels of SR-BI (30–33). Surprisingly, in our transgenic model, we do not observed such a diminution in plasma apo AI that remained constant, whereas HDL cholesterol was decreased by two fold. Apo A-I can be synthesized by the liver through HDL formation or by the intestine associated with chylomicron secretion. Since, up-regulation of apo AI synthesis by the liver has not been observed in the liver SR-BI over-expressing models (31), our results suggest that the intestine could participate in the regulation of apo AI synthesis through an increase of chylomicron secretion.
Our transgenic model also highlights a discrepancy between cholesterol and TG metabolism. Indeed, by contrast to cholesterol, the plasmatic increase of radioactivity coming from diet TG paralleled a rise in plasma TG content. It has been already demonstrated that SR-BI was able to uptake TG in vitro using SR-BI transfected COS cell (36), adrenal Y1 and hepatic HepG2 cells (37). Our data are then the first in vivo evidence for a role of SR-BI in intestinal uptake of either TG-hydrolysis products or direct TG. In fact, we cannot completely exclude that some TG molecules could be directly taken up through SR-BI, although diet TG are almost completely hydrolyzed in vivo by gastric and pancreatic lipases (38). Our in vivo observations are however in contradiction with the recent paper of Bennekum et al (10), which showed that SR-BI as well as CD36 in transfected COS cells did not play any role in fatty acid uptake, whereas it was clearly demonstrated that CD36 deficient enterocytes accumulate lipids, reflecting abnormal lipid processing (8). One hypothesis is that SR-BI may act as a micelle sensor to favor the uptake of micellar lipids in cooperation with other transporters. Interestingly, Hansen et al (39) showed that SR-BI was mainly localized in the microvillar membrane in a fasting state and evidenced that during absorption of dietary fat, SR-BI is endocytosed from the enterocyte brush border as a mean to regulate SR-BI availability at the surface. This original mechanism may explain contradictory results concerning the role of SR-BI since major works were done in a non-fasting state. Moreover, in macrophages, SR-BI is implicated in bidirectional fluxes of cholesterol and phospholipids (19,40); thus SR-BI could also modulate bidirectional fluxes of lipids in function of the lipids status of enterocytes.
All these results demonstrate that SR-BI acts in vivo as a multi-ligand transporter in the small intestine. SR-BI is responsible for the uptake of cholesterol and triglyceride hydrolysis products (present work) as well as non-polar vitamins such as beta-carotene (10) and vitamin E (Reboul and Borel, companion paper). Genetic factors have been proposed to explain the human inter-individual variability in the intestinal absorption of lipids (41). SR-BI may contribute to these observations since a recently described polymorphism at the exon 1 has been implicated in a lower postprandial lipemic response (42). Thus, this receptor may represent a new target in order to modulate intestinal lipid absorption.
Acknowledgments
We thank Pr. J. Chambaz and Pr. P. Cardot for the gift of the apo CIII-apo AIV construct and Pr. A. Tall for providing the SR-BI cDNA.
Grant support: This work was in part granted by AIC Nutrition INSERM/INRA (ER 66) and funded by ANRS (R03038BS). The postdoctoral fellowship (D.Y) was supported by the French government; the doctoral fellowship (F.B) was supported by the French Atherosclerosis society (NSFA) and the French Nutrition society (SFN). This project was partly supported by Genopole Toulouse Midi-Pyrénées/Platforms of physiopathological exploration of genome/Experimental histopathology technical plateau (Florence Capilla) and Lipid mediator analysis technical facilities (Véronique Roques and Justine Bertrand-Michel).
Abbreviations
- Apo
apolipoprotein
- DG
diacylglycerol
- CT
total cholesterol
- CE
cholesteryl ester
- EGTA
ethylene glycol-bis (2-aminoethylether)-N,N,N‘, N‘-tetraacetic acid
- FC
free cholesterol
- HDL
High Density Lipoprotein
- LDL
Low Density Lipoprotein
- SR-BI
Scavenger Receptor; Class B Type 1
- TG
triglyceride
- VLDL
Very Low Density Lipoprotein
References
- 1.Dawson PA, Rudel LL. Curr Opin Lipidol. 1999;10:315–320. doi: 10.1097/00041433-199908000-00005. [DOI] [PubMed] [Google Scholar]
- 2.Ros E. Atherosclerosis. 2000;151:357–379. doi: 10.1016/s0021-9150(00)00456-1. [DOI] [PubMed] [Google Scholar]
- 3.Compassi S, Werder M, Boffelli D, Weber FE, Hauser H, Schulthess G. Biochemistry. 1995;34:16473–16482. doi: 10.1021/bi00050a031. [DOI] [PubMed] [Google Scholar]
- 4.Thurnhofer H, Hauser H. Biochemistry. 1990;29:2142–2148. doi: 10.1021/bi00460a026. [DOI] [PubMed] [Google Scholar]
- 5.Schulthess G, Compassi S, Boffelli D, Werder M, Weber FE, Hauser H. J Lipid Res. 1996;37:2405–2419. [PubMed] [Google Scholar]
- 6.Hauser H, Dyer JH, Nandy A, Vega MA, Werder M, Bieliauskaite E, Weber FE, Compassi S, Gemperli A, Boffelli D, Wehrli E, Schulthess G, Phillips MC. Biochemistry. 1998;37:17843–17850. doi: 10.1021/bi982404y. [DOI] [PubMed] [Google Scholar]
- 7.McGill HC., Jr Am J Clin Nutr. 1979;32:2664–2702. doi: 10.1093/ajcn/32.12.2664. [DOI] [PubMed] [Google Scholar]
- 8.Drover VA, Ajmal M, Nassir F, Davidson NO, Nauli AM, Sahoo D, Tso P, Abumrad NA. J Clin Invest. 2005;115:1290–1297. doi: 10.1172/JCI21514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Werder M, Han CH, Wehrli E, Bimmler D, Schulthess G, Hauser H. Biochemistry. 2001;40:11643–11650. doi: 10.1021/bi0109820. [DOI] [PubMed] [Google Scholar]
- 10.van Bennekum A, Werder M, Thuahnai ST, Han CH, Duong P, Williams DL, Wettstein P, Schulthess G, Phillips MC, Hauser H. Biochemistry. 2005;44:4517–4525. doi: 10.1021/bi0484320. [DOI] [PubMed] [Google Scholar]
- 11.Altmann SW, Davis HR, Jr, Zhu LJ, Yao X, Hoos LM, Tetzloff G, Iyer SP, Maguire M, Golovko A, Zeng M, Wang L, Murgolo N, Graziano MP. Science. 2004;303:1201–1204. doi: 10.1126/science.1093131. [DOI] [PubMed] [Google Scholar]
- 12.Kramer W, Girbig F, Corsiero D, Burger K, Fahrenholz F, Jung C, Muller G. Biochim Biophys Acta. 2003;1633:13–26. doi: 10.1016/s1388-1981(03)00068-4. [DOI] [PubMed] [Google Scholar]
- 13.Acton SL, Rigotti A, Landschulz K, Xu S, Hobbs HH, Krieger M. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 14.Stangl H, Hyatt M, Hobbs HH. J Biol Chem. 1999;274:32692–32698. doi: 10.1074/jbc.274.46.32692. [DOI] [PubMed] [Google Scholar]
- 15.Cai SF, Kirby RJ, Howles PN, Hui DY. J Lipid Res. 2001;42:902–909. [PubMed] [Google Scholar]
- 16.Mardones P, Quinones V, Amigo L, Moreno M, Miquel JF, Schwarz M, Miettinen HE, Trigatti B, Krieger M, VanPatten S, Cohen DE, Rigotti A. J Lipid Res. 2001;42:170–180. [PubMed] [Google Scholar]
- 17.Le Beyec J, Chauffeton V, Kan HY, Janvier PL, Cywiner-Golenzer C, Chatelet FP, Kalopissis AD, Zannis V, Chambaz J, Pincon-Raymond M, Cardot P. J Biol Chem. 1999;274:4954–4961. doi: 10.1074/jbc.274.8.4954. [DOI] [PubMed] [Google Scholar]
- 18.Ogami K, Hadzopoulou-Cladaras M, Cladaras C, Zannis VI. J Biol Chem. 1990;265:9808–9815. [PubMed] [Google Scholar]
- 19.Ji Y, Jian B, Wang N, Sun Y, Moya ML, Phillips MC, Rothblat GH, Swaney JB, Tall AR. J Biol Chem. 1997;272:20982–20985. doi: 10.1074/jbc.272.34.20982. [DOI] [PubMed] [Google Scholar]
- 20.Weiser MM. J Biol Chem. 1973;248:2536–2541. [PubMed] [Google Scholar]
- 21.Bligh EG, Dyer WJ. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 22.Vieu C, Jaspard B, Barbaras R, Manent J, Chap H, Perret B, Collet X. J Lipid Res. 1996;37:1153–1161. [PubMed] [Google Scholar]
- 23.Kirby RJ, Howles PN, Hui DY. J Lipid Res. 2004;45:89–98. doi: 10.1194/jlr.M300148-JLR200. [DOI] [PubMed] [Google Scholar]
- 24.Kruit JK, Plosch T, Havinga R, Boverhof R, Groot PH, Groen AK, Kuipers F. Gastroenterology. 2005;128:147–156. doi: 10.1053/j.gastro.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Torchia EC, Labonte ED, Agellon LB. Anal Biochem. 2001;298:293–298. doi: 10.1006/abio.2001.5379. [DOI] [PubMed] [Google Scholar]
- 26.Drobnik W, Lindenthal B, Lieser B, Ritter M, Christiansen Weber T, Liebisch G, Giesa U, Igel M, Borsukova H, Buchler C, Fung-Leung WP, Von Bergmann K, Schmitz G. Gastroenterology. 2001;120:1203–1211. doi: 10.1053/gast.2001.23250. [DOI] [PubMed] [Google Scholar]
- 27.Yu L, Li-Hawkins J, Hammer RE, Berge KE, Horton JD, Cohen JC, Hobbs HH. J Clin Invest. 2002;110:671–680. doi: 10.1172/JCI16001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis HR, Jr, Zhu LJ, Hoos LM, Tetzloff G, Maguire M, Liu J, Yao X, Iyer SP, Lam MH, Lund EG, Detmers PA, Graziano MP, Altmann SW. J Biol Chem. 2004;279:33586–33592. doi: 10.1074/jbc.M405817200. [DOI] [PubMed] [Google Scholar]
- 29.Kramer W, Girbig F, Corsiero D, Pfenninger A, Frick W, Rhein M, Wendler W, Lottspeich F, Hochleitner EO, Orso E, Schmitz G. [Google Scholar]
- 30.Ueda Y, Royer L, Gong E, Zhang J, Cooper PN, Francone O, Rubin EM. J Biol Chem. 1999;274:7165–7171. doi: 10.1074/jbc.274.11.7165. [DOI] [PubMed] [Google Scholar]
- 31.Wang N, Arai T, Ji Y, Rinninger F, Tall AR. J Biol Chem. 1998;273:32920–32926. doi: 10.1074/jbc.273.49.32920. [DOI] [PubMed] [Google Scholar]
- 32.Ji Y, Wang N, Ramakrishnan R, Sehayek E, Huszar D, Breslow JL, Tall AR. J Biol Chem. 1999;274:33398–33402. doi: 10.1074/jbc.274.47.33398. [DOI] [PubMed] [Google Scholar]
- 33.Kozarsky KF, Donahee MH, Rigotti A, Iqbal SN, Edelman ER, Krieger M. Nature. 1997;387:414–417. doi: 10.1038/387414a0. [DOI] [PubMed] [Google Scholar]
- 34.Traber MG, Burton GW, Ingold KU, Kayden HJ. J Lipid Res. 1990;31:675–685. [PubMed] [Google Scholar]
- 35.Out R, Kruijt JK, Rensen PC, Hildebrand RB, de Vos P, Van Eck M, Van Berkel TJ. J Biol Chem. 2004;279:18401–18406. doi: 10.1074/jbc.M401170200. [DOI] [PubMed] [Google Scholar]
- 36.Thuahnai ST, Lund-Katz S, Williams DL, Phillips MC. J Biol Chem. 2001;276:43801–43808. doi: 10.1074/jbc.M106695200. [DOI] [PubMed] [Google Scholar]
- 37.Greene DJ, Skeggs JW, Morton RE. J Biol Chem. 2001;276:4804–4811. doi: 10.1074/jbc.M008725200. [DOI] [PubMed] [Google Scholar]
- 38.Hui DY, Howles PN. Semin Cell Dev Biol. 2005;16:183–192. doi: 10.1016/j.semcdb.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 39.Hansen GH, Niels-Christiansen LL, Immerdal L, Danielsen EM. Gut. 2003;52:1424–1431. doi: 10.1136/gut.52.10.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de La Llera-Moya M, Connelly MA, Drazul D, Klein SM, Favari E, Yancey PG, Williams DL, Rothblat GH. J Lipid Res. 2001;42:1969–1978. [PubMed] [Google Scholar]
- 41.Wang DQ, Paigen B, Carey MC. J Lipid Res. 2001;42:1820–1830. [PubMed] [Google Scholar]
- 42.Perez-Martinez P, Lopez-Miranda J, Ordovas JM, Bellido C, Marin C, Gomez P, Paniagua JA, Moreno JA, Fuentes F, Perez-Jimenez F. J Mol Endocrinol. 2004;32:237–245. doi: 10.1677/jme.0.0320237. [DOI] [PubMed] [Google Scholar]