

Abstract
Adenosine promotes cytoprotection under condition of infection, ischemic preconditioning and oxidative stress. Previous studies from our laboratory indicate that the expression of the adenosine A1 receptor (A1AR) is induced by oxidative stress via activation of nuclear factor (NF)-κB. The prototypic transcription factor is comprised of homo- or heterodimers of p50 and p65 subunits. To determine the role of NF-κB in the regulation of the A1AR in vivo, we compared the A1AR RNA and protein levels in the brains of mice lacking the p50 subunit of NF-κB (p50−/− mice) and age-matched B6129PF2/J (F2) controls. Radioligand binding assays in the cortex revealed a significantly lower number of A1AR (Bmax) in the cortex of p50−/− mice (151 ± 62 fmol/mg protein) versus 479 ± 181 fmol/mg protein in the F2 (N=5 per strain, p < 0.05), but no change in Kd. Similar reductions in A1AR were measured in the hippocampus, brain stem and hypothalamus and in peripheral tissues, such as the adrenal gland, kidney and spleen. Estimation of the A1AR following purification by antibody affinity columns also indicated reduced A1AR in the p50−/− mice cortex, as compared to the F2 mice. A1AR immunocytochemistry indicates distinct neuronal labeling in the F2 cortex, which was substantially reduced in similar sections obtained from p50−/− mice. p50−/− mice expressed lower levels of A1AR mRNA than F2 mice, as determined by real time PCR. Quantitation of the A1AR transducing G proteins by Western blotting show significantly less Gαi3, no change in Gαi1, but higher levels of Gαo and Gβ in the cortices of p50−/−, as compared to F2 mice. Administration of bacterial lipopolysaccharide (LPS), an activator of NF-κB, increased A1AR expression in the cortices of F2 mice but not p50−/− mice. Cortical neurons cultures prepared from p50−/− mice showed a greater degree of apoptosis, compared to neurons from F2 mice. Activation of the A1AR reduced apoptosis with greater efficacy in cultures from F2 than p50−/− mice. Taken together, these data support a role for NF-κB in determining both the basal and LPS-stimulated A1AR expression in vivo which could contribute to neuronal survival.
Keywords: NF-κB, A1 adenosine receptor, lipopolysaccharide, central nervous system
Introduction
Extracellular purines (adenosine, ADP and ATP) are important signaling molecules that regulate various biological processes, including neurotransmission in peripheral and central nervous systems, sleep homeostasis, and the immune response (Ralevic and Burnstock, 1998). Adenosine exerts its physiologic roles by interacting with seven distinct transmembrane adenosine receptors (ARs), whose actions are in turn mediated by different heterotrimeric G proteins and effector systems (Fredholm et al., 2001). Four subtypes of adenosine receptors, A1, A2a, A2b, and A3, have been identified based on molecular cloning studies, tissue distribution, and pharmacologic profiles (Olah and Stiles, 1992; Tucker and Linden, 1993). The A1AR is the principal AR subtype in the central nervous system, with wide distribution in the cortex, cerebellum, thalamus and hippocampus (Olah and Stiles, 1992; Rivkees et al., 1995). Stimulation of A1AR suppresses the presynaptic release of excitatory amino acids, such as glutamate (Dolphin and Archer 1983). In addition, activation of pre-synaptic A1AR in turn activates K+ conductance (Fredholm and Dunwiddie, 1988) and inhibits Ca2+ influx into the nerve terminals (Rudolphi et al., 1992), thereby leading to neuronal hyperpolarization and reduced neuronal firing rate (Kostopoulos and Phillis, 1977). Activation of the A1AR also inhibits adenylyl cyclase activity and voltage-dependent Ca2+ channels (Olah and Stiles, 1992).
A neuroprotective role of A1AR can be demonstrated during conditions of ischemia, hypoxia, and hypercapnea (Rubio et al., 1975; Van Wylen et al., 1986). Adenosine analogues protect against cerebral ischemia in rats (Evans et al., 1987), whereas the AR antagonist, caffeine, exacerbates ischemic damage. Furthermore, studies from our laboratory indicate that during oxidative stress, the A1AR is induced through activation of NF-κB both in vitro (Nie et al., 1998; Pingle et al., 2004) and in vivo (Ford et al., 1997, Ramkumar et al., 2004) and is protective under these circumstances.
However, the impact of NF-κB on regulation of basal expression of the A1AR is unknown, although the constitutive expression of this receptor has been linked to the activator protein (AP)-1 transcription factor (Ren and Stiles, 1995). In this study, we address this question by studying mice that genetically lack the p50 subunit of NF-κB. The p50 and p65 proteins are the prototypic subunits of NF-κB, which exists as homo- or heterodimers in the central nervous system (Kaltschimdt et al., 1994). Mice that lack the p65 subunit of NF-κB die on day 16 (Beg et al., 1995). In contrast, p50 knockout mice survive to adulthood and have a relatively normal phenotype, albeit with some immune system deficits (Sha et al., 1996).
Our data support a role of NF-κB in determining both the basal and stimulated expression of the A1AR and some of its coupling G protein subunits. Regulating the expression of neuronal A1AR could contribute to the cytoprotective role of NF-κB in vivo.
Materials and Methods
Animal care
Adult male B6129PF2/J (F2) and B6;129P2-Nfkb 1<tm 1 Bal> (p50−/−) mice, were purchased from the Jackson Laboratory (Bar Harbor, ME). The p50−/− mice were homozygous for deletion of the gene for the p50 subunit of NF-κB, and thus are incapable of producing the p50 protein. Since p50−/− mice from the Jackson Laboratory are maintained on a mixed C57BL/6 × 129P3/J, F2 hybrids were used as the control strain, as recommended by the vendor. F2 mice were housed in standard cages and maintained on a 12:12 h light: dark cycle at an ambient temperature of 22 ± 1°C. Rodent laboratory chow (Lab Diet 5001, Nutrition International, Inc., Brentwood, MO) and drinking water were provided ad libitum. Because of subtle immune impairments, p50−/− mice were housed in autoclaved cages, provided with autoclaved food and water, and maintained in separate chambers to reduce exposure to infection. Mice were at least 8−10 weeks of age (22−28 g) at the time of experimental use. All procedures used in this study were approved the Animal Care and Use Committee at Southern Illinois University, School of Medicine.
Chemicals and drugs
HEPES, Tris HCl, soybean trypsin inhibitor, pepstatin, benzamidine, lipopolysaccharide (LPS) (E.coli O111:B4), adenosine deaminase (ADA), polyethyleneimine, 3-[(3-cholamidopropyl) dimethylammonio]-1 propanesulphonate (CHAPS), and 1,3-diisopropyl-8-cyclopentylxanthine (DPCPX) were purchased from Sigma Chemical Company (St. Louis, MO). R-phenylisopropyladenosine (R-PIA) was purchased from Boehringer-Mannheim Biochemicals (Indianapolis, IN). Rat pheochromocytoma PC12 cells were purchased at passage 2 from the American Type Culture Collection (Manassas, VA). Cell culture supplies were obtained from GIBCO BRL (Grand Island, NY). [3H]-DPCPX (160 Ci/mmol) was purchased from Perkin Elmer (Boston, MA). [125I]Na was purchased from Du-Pont New England Nuclear Centre (Boston, MA). Dr. E.M. Schwarz, University of Rochester Medical Center (Rochester, NY), generously provided the adenovirus vector containing the mutant form of IκB-α (mIκB-α).
Sample collection and membrane preparation
For analysis of A1AR in F2 and p50−/− mice, mice were anesthetized under isoflurane and sacrificed by cervical dislocation. A second set of mice of each strain were treated with LPS (10μg) and sacrificed 4 hours later for analysis of A1AR expression. Whole brains were removed and cortex, hippocampus, brain stem, and hypothalamus were dissected under aseptic conditions. In addition, kidneys, spleen, heart and adrenal glands were also dissected from the same animals. The frozen tissues (different brain regions and peripheral tissues) from F2 and p50−/− mice were allowed to thaw on ice and were rinsed with 1 ml of ice cold phosphate-buffered saline (PBS) containing 5 mM EDTA. Tissues were lysed in 1ml of 10 mM Tris HCl buffer, pH 7.4, containing 5mM EDTA, 1mM MgCl2, 10 μg /ml soybean trypsin inhibitor, 10μg/ml benzamidine, and 2 μg /ml pepstatin (Buffer A) and homogenized briefly with a Polytron homogenizer (Brinkmann Instruments, Westbury, NY). Membrane fractions were obtained by differential centrifugation. The homogenates were centrifuged at 1,000 g for 10 min at 4°C, followed by centrifugation of the supernatant at 100,000 g for 15 min at 4°C. The final pellet was resuspended in 50 mM Tris −HCl buffer, pH 7.4, 10mM MgCl2, and 1mM EDTA (containing protease inhibitors, as above, buffer A), subjected to brief sonication for 10 sec to a give a final membrane protein concentration of ∼1 mg/ml. The resuspended fraction containing crude membrane was used for determining receptor densities by radioligand binding after pretreatment with adenosine deaminase (5 units/ml) at 37°C for 10 min to degrade the endogenous adenosine (for radioligand binding studies).
SDS-PAGE analysis and Western blotting
To detect the p50 subunit and G-protein subunits, spleen and cortex from each strain were gently homogenized in Buffer A. The homogenates were centrifuged at 1,000 g for 5 min to remove cellular debris. The supernatant was further centrifuged at 100,000 g for 10 min at 4°C and the pellet obtained, consisting of crude membranes, were used for Western blotting analyses. The membrane fraction (∼50 μg protein) was mixed with SDS-PAGE solubilization buffer and resolved on a 12% polyacrylamide gel. Proteins were transferred to nitrocellulose membranes, blocked in Blotto buffer (130 mM NaCl, 2.7 mM KCl, 1.8 mM Na2HPO4, 1.5 mM KH2PO4, 0.1% NaN3, 0.1% Triton-X 100, and 5% low-fat skim milk) for 2 h, and then incubated with specific primary antibody for p50 (sc-1190) (Santa Cruz Biotechnology, Santa Cruz, CA) and for G-proteins (kindly provided by Dr. Tom Gettys, Experimental Obesity Division, Pennington Basic Research Center, Baton Rouge, LA) at 4°C overnight. After five washes in blocking solution, blots were incubated with horseradish peroxidase-labeled secondary antibody (Santa Cruz Biotechnology) for 1 h at room temperature, washed five times with Tris-buffered saline, treated with Enhanced Chemiluminescence Plus reagents (Amersham Pharmacia Biotech, Piscataway, NJ), and then exposed to Kodak XAR film at room temperature. The bands were normalized to an internal control protein (β-actin), and band intensities were determined by densitometry on SynGene Tools software for analysis (Cambridge, England).
Radioligand binding assay
Membrane proteins (∼50 μg) from various regions of F2 and p50−/− mice and 80 μg of whole cell lysates from PC12 cells (see below) were used to evaluate A1AR binding. Radioligand binding experiments were performed using the selective A1AR antagonist [3H]-DPCPX in a concentration of 1 nM for single point binding assays or a range of concentrations (0.25 to 5 nM) for saturation analyses. Radioligand binding assays were performed in a total reaction volume of 250 μl of buffer A, incubated at 37°C for 1 h, in the absence (total binding) or presence of 1 mM theophylline (to define nonspecific binding, which usually ranged from 10 to 40% of total binding (in different tissues). Subsequently, samples were filtered through polyethyleneimine-treated (0.05%) Whatman GF/B glass-fiber filters (Brandel, Gaithersburg, MD) using a cell harvester (Brandel, Gaithersburg, MD) and washed with 9 ml (3 washes) ice-cold Tris buffer, pH 8.26 containing 0.01% CHAPS. The filters were allowed to extract overnight in a toluene-based scintillation fluid before measuring the radioactive content of each filter by a liquid scintillation counter (LS5801) (Beckman Instruments, Fullerton, CA). For competition binding assays, increasing concentrations of R-PIA were used to inhibit the binding of 1.0 nM [3H]-DPCPX. Radioligand binding data was plotted and analyzed using GraphPad Prism 4.0 software (GraphPad Software Inc, San Diego, CA).
Immunoprecipitation of A1AR from pure membrane fraction
Immunoprecipitation was performed according to the protocol contained in the Seize X Mammalian Immunoprecipitation kit (Pierce, Rockford, IL). Briefly, A1AR antibody affinity columns were prepared with 50μg of goat polyclonal A1AR antibody (Santa Cruz Biotechnology) that was cross-linked to immobilized protein G (50% slurry) in mini columns (one column per sample) and incubated overnight to allow binding of antibodies to protein G. Disuccinimidyl suberate (DSS) was then added to cross-link bound antibody to the support. Membrane fractions were prepared by differential centrifugation as described above from cortex of F2 and p50−/− mice (n = 4 per strain) and solubilized with reagent provided in kit and incubated with the A1AR affinity column at 40C overnight for two days to allow ample time for interaction of the A1AR antibody with the membrane fraction. Columns were washed several times with the binding buffer, and the bound A1AR was eluted in 200μl of elution buffer, pH 2.0. The elute containing purified A1AR was neutralized in buffer A and used for single point binding with [3H]-DPCPX as described above, or iodinated and used for SDS-PAGE analysis (described below).
Iodination and quantification of purified cortical A1AR
The purified A1AR was incubated with 0.1 mCi 125I-Na in a total volume of 250 μl of 50 mM potassium phosphate buffer (pH 7.5), containing 10 μg chloramine T, for 1 min. The reaction was stopped by adding 100 μl of sodium metabisulphite (2.4 mg/ml) and the mixture desalted in buffer B (50 mM Tris −HCl buffer, 10mM MgCl2, and 1mM EDTA, pH 7.4 containing 0.01% CHAPS) on Sephadex G25 columns. The eluates were collected as the iodinated A1AR. The labeled purified A1AR was then resolved on a 12% SDS-PAGE gel, loaded with equal amounts of iodinated A1AR. The gel was dried for 1 h at 60°C using a vacuum operated gel dryer and, visualized by exposure to Kodak XAR films. Gel sections corresponding to the A1AR were removed for quantitation of radioactivity in a gamma counter.
Immunohistochemistry
Each mouse was deeply anesthetized with near lethal doses of a mixture of ketamine and xylazine. The chest was opened, a needle was inserted into the left ventricle, and the descending aorta was opened. Perfusion involved sequential injection of 0.9% saline solution (until the blood was washed out), followed by 4% paraformaldehyde for 2 min. The mouse was then decapitated, whole brain was removed, and stored overnight at 4°C in a capped vial containing 4% paraformaldehyde. Sagittal sections were carried out in the midline of the mouse brain and the two hemisections were paraffin-embedded and manually sectioned (10 μm thick) using a Leica RM2125 microtome (Wetzlar, Germany). Sections were placed on Vectabond reagent processed glass slides (Vector Laboratories, Burlingame, CA) and air-dried overnight at room temperature. Sections were de-paraffined in xylene (3 washes), followed by rehydration in decreasing concentrations (100, 95 and 70%) of ethanol.
For immunohistochemistry, sections were rinsed with PBS, blocked for 1 h in 5% normal donkey serum with 0.03% triton X-100 in PBS at room temperature, and then incubated overnight (at 4°C) with a monoclonal A1AR antibody (Ochiishi et al., 1999). Sections were rinsed with PBS and incubated with TRITC conjugated donkey anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) diluted at 1:50 for 2 h at room temperature. After four rinses in PBS, cover slips were mounted on glass microscope slides using Aquamount, and cells were visualized using an Olympus confocal laser scanning microscope (Melville, NY) with krypton laser at 568 nm using a 20X objective.
Nissl staining with cresyl violet was also performed on adjacent sections from the same brain. After sections were de-paraffinized and rehydrated, they were placed in 2.0 g/L of cresyl violet (Sigma-Aldrich) for 5 min. The sections were then rinsed in water to remove any excess cresyl violet, differentiated in acidified 95% ethanol, rinsed in water to stop the differentiation, and dehydrated with increasing concentrations of alcohol (70, 95, and 100%) and xylene rinses. Sections were then mounted on glass microscope slides using permount, allowed to dry over night and visualized using Olympus Optical system (Melville, NY).
Real Time PCR
Total RNA extraction was carried out from cortex of F2 and p50−/− mice using the RNAeasy Mini Kit (Qiagen Science, Valencia, CA) according to manufacturer's instructions. Samples of total RNA (1 μg each) were reverse transcribed to cDNA using iScript cDNA synthesis kit (BioRad, Hercules, CA). The reaction mix contained 1 μg of total RNA, 4 μl of iScript reaction mix, and 1 μl of iScript reverse transcriptase. Nuclease free water was added to bring the total volume to 20ul. The reaction mix was incubated at 25°C for 5 min, 42°C for 30 min and 85°C for 5 min and 1 μl of this reaction volume was used for real time PCR.
Gene transcripts were quantified using iQ™ SYBR Green Supermix kit (BioRad) and the Smart Cycler Real-Time PCR system (Cepheid Inc., Sunnyvale, CA). The final PCR reaction mix (total reaction volume= 25 μl) contained 1 μl of cDNA (diluted 1:5 of the original cDNA extract prepared above), 1.5 μl (∼10 pmol) of each primer sequence, 12.5 μl of iQ SYBR Green Supermix reagent (BioRad) and 8.5 μl of sterile nuclease free water. GAPDH expression was used to normalize the amount of cDNA in each sample. Primers were purchased from Integrated DNA Technologies (Coralville, CA) and were as follows: A1AR primers (Gene Bank accession # NM009629), sense primer: 5’- CATTGGGCCACAGACCTACT-3’, antisense primer: 5’- CAAGGGAGAGAATCCA GCAG-3’; GAPDH primers (Gene Bank accession # BC083149), sense primer: 5’- AACGACCCCTTCATTGAC- 3’, antisense primer: 5’- GAAGACACCAGTAGACT CCAC – 3’. Negative control experiments were performed without the addition of template cDNA. Cycling conditions were: 95°C for 3 min followed by 45 cycles at 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s. On completion of amplification, melting curve analysis was performed by cooling the reaction to 60°C and then heating slowly to 95°C, according to manufacturer's instructions (Cepheid systems). The cycle number at which the sample reaches the threshold fluorescent intensity was termed the cycle threshold (Ct). The relative changes in mRNA levels between basal F2 (1) and p50−/− (2) or between saline- (1) or LPS-treated (2) cortices were measured using the formula: 2(Ct Target gene1-Ct GAPDH1)-(Ct Target gene2-Ct GAPDH2) (Soong et al., 2001). Relative changes in mRNA levels between samples were expressed as a percentage of the mean of control values. End reaction products were visualized on ethidium bromide stained 1% agarose gels to verify correct product sizes.
Cell culture
Pheochromocytoma (PC12) cells
PC12 cells were cultured in RPMI 1640 media supplemented with 10% equine serum (HS), 5% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 units/ml streptomycin. Culture flasks and plates were coated with 0.1 mg/ml poly-D-lysine prior to plating. All cultures were maintained as a monolayer at 370C, in 5% CO2/ 95% ambient air; with the media replaced every 2−3 days. PC12 cells stably transfected with pCMX-IκB-αM or with an empty vector and were cultured in a similar manner, except that 100 μg/ml G418 sulphate was added to the medium. The mutant form of IκB-α has serine-to-alanine substitutions at positions 32 and 36. The mutant IκB-α (mIκB-α) was generated and characterized previously as a dominant negative inhibitor of NF- κB (Van Antwerp et al., 1996; Schwarz et al., 1998; Nie et al., 1999).
Cortical neuronal culture
In brief, neurons were isolated from the cortex of adult (10−12 week old) B6129PF2/J and p50 KO mice according to established procedures (Brewer et al., 1993; Brewer, 1997). Culture media were obtained from GIBCO Laboratories (Grand Island, NY). Before the addition of test substances, neurons were cultured on cover slips for 10 days in B27/NeurobasalA, containing 5 ng/ml fibroblast growth factor and platelet derived growth factor serum-free media, with 0.5 mM glutamine. The cultures contain a mixture of about 80% neurons, 5% microglia, 10% oligodendroglia, and less than 5% astroglia, based on immunostaining (Brewer, 1997). After a complete change to fresh medium, R-PIA (10 μM) and/or DPCPX (1 μM) were added. Cultures were further incubated for 24 h. Another set of neurons were treated similarly in the presence of hydrogen peroxide (10 μM). This procedure does not impose amino acid starvation coincident with the treatment and maintains the neurons in a standard medium that includes vitamin E and three other anti-oxidants. For this reason, toxicity is low as compared to other common protocols (Brewer et al., 1993).
Apoptosis Detection
Apoptosis was detected in cortical neurons using terminal deoxynucleotidyl transferase-mediated digoxigenin-dUTP nick end-labeling (TUNEL) assay, according to the manufacturer's instructions (EMD Biosciences). Following various treatments, neurons were washed with cold PBS, fixed with 4% paraformaldehyde, and then placed in equilibrating buffer and incubated in a reaction buffer containing TdT and dUTP for 60 min at 37°C. After rinsing, the cells were incubated with terminal deoxynucleotidyl transferase (TdT) to catalyze incorporation of fluorescein-dUTP at the free 3'-hydroxyl ends of the fragmented DNA. The percentage of TUNEL-positive cells (bright fluorescent green) were assessed by analysis of digitized images from 5 or more microscopic fields of TUNEL-stained cells from TIFF files (Adobe Photoshop version 7.0).
Protein determination
Protein concentrations were determined by the Bradford protein assay (Bradford et al., 1976) using bovine serum albumin to prepare standard curves.
Statistical analyses
Saturation curves and competition curves were analyzed using Prism (GraphPad Software). Other statistical analyses was performed by Student's t-test and analysis of variance (ANOVA) using Statistical Package for the Social Sciences (SPSS, Chicago, IL).
Results
NF-κB regulates basal level of A1AR expression
The absence of p50 protein in the p50−/− mice was confirmed by Western blotting, performed on the cytosolic fraction of the spleen in the F2 and p50−/− mice (Fig. 1, n=3 for each strain). A band equivalent to the p50 subunit protein was not detected in the p50−/− mice but evident in the F2 mice. p65 subunit protein analyses revealed no change between the strains (data not shown).
Figure 1. p50 protein is absent in the p50−/− mice.

SDS-PAGE analysis and Western blotting of cytosolic fraction of spleen from F2 and p50−/− mice confirmed the absence of the p50 protein in the p50−/− mice. Proteins were normalized to internal control β-actin (n = 3 per strain).
To evaluate the role of NF-κB in A1AR regulation under normal conditions, brain regions from both strains of mice were analyzed for A1AR binding. Quantitation of the A1AR by single point binding assays (using 1 nM [3H]-DPCPX) indicate lower A1AR levels in the cortex, hippocampus, brain stem, and hypothalamus in the p50−/− mice than in the F2 mice. The levels of [3H]-DPCPX binding were 126 ± 9 and 83 ± 5 fmol/mg protein in the cortex obtained from the F2 and p50−/− mice, respectively. The respective changes in F2 and p50−/− mice were 93 ± 1 and 66 ± 6 fmol/mg protein in the hippocampus, 69 ± 3 and 48 ± 1 fmol/mg protein in the brain stem and 33 ± 2 and 22 ± 5 fmol/mg protein in the hypothalamus (Fig. 2A) (n = 4 per strain for each brain region, p<0.05). The difference in radioligand binding was also evident when full saturation curves were performed (Fig. 2B). Maximal binding capacity (Bmax) was significantly lower in cortex from p50−/− mice as compared to F2 mice (151 ± 62 and 479 ± 181 fmol/mg protein, respectively) (Fig.2B; n=5; p < 0.05). The equilibrium dissociation constants (Kd) did not differ between the two strains (Table 1.) (1.2 ± 0.4 and 1.1 ± 0.5 nM for F2 and p50−/− mice, respectively).
Figure 2. Reduction in A1AR in p50−/− mouse brain.
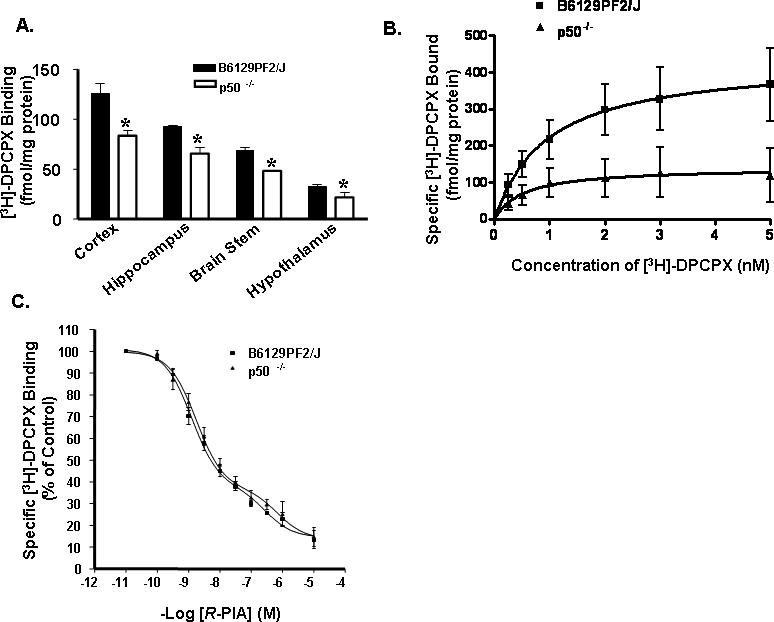
A, A1AR binding in F2 and p50−/− mice (n=4 per strain) was quantified in membrane fractions (50 μg) of cortex, hippocampus, brain stem, and hypothalamus using the specific A1AR antagonist [3H]-DPCPX (1 nM). Values are expressed as fmol/ mg protein and represent mean ± SEM for three independent experiments with samples assayed in triplicate (*, p < 0.05, Student's t-test). B, Saturation binding analysis of cortical membrane for A1AR with [3H]-DPCPX in the absence (total binding) or presence (nonspecific binding) of 0.5 mM theophylline revealed lower maximal binding in the p50−/− mice with no change in affinity. A representative curve representing specific binding is shown for F2 and p50−/− mice. Curves were fitted to a one-site model using GraphPad Prism (n = 5 for F2; n = 4 for p50−/−). C, Competition analyses of cortical A1AR in F2 and p50−/− mice revealed similar affinity in F2 and p50−/− mice. Cortical membranes were incubated with 1nM [3H]-DPCPX and increasing concentrations of AR agonist R-PIA. Data represent percentage of specific binding (n = 4 per strain) and show the mean ± SEM of four independent experiments, with samples assayed in duplicate. The curves were generated using a curve fitting program in GraphPad Prism according to a two state model.
Table 1.
Saturation and Affinity analysis for A1AR in control F2 and p50−/− mice.
| Strain | Bmax fmol | KdnM | KHnM | KLμM | RH % |
|---|---|---|---|---|---|
| F2 | 478.8±181.0 | 1.18±0.44 | 1.30±0.03 | 3.00±0.10 | 0.72 |
| p50 KO | 151.3±61.8 * | 1.08±0.45 | 1.80±0.03 | 7.20±0.10 * | 0.73 |
Bmax and Kd are the maximal binding capacity and the affinity of the receptor for the lignad respectively. KH and KL are the high affinity and low affinity dissociation constants, respectively, calculated assuming a two-state model. RH is the percentage of total receptors in the high affinity state. Values are mean ± SEM. Asterisk represents significant difference between the two strains and indicates p< 0.05 (Student's t test).
To determine whether agonists interact similarly with the A1AR in F2 and p50−/− mouse, we performed competition curves using the agonist R-PIA competing for [3H]-DPCPX binding sites (Fig. 2C). Analyses of competition binding data using Graph Pad Prism software indicate preference of a two-state fit versus a one-state fit. The high affinity (KH) values and the percentage of receptors in the high-affinity state did not differ between the two strains (Table 1). However, the low-affinity constant (KL) of the receptors were statistically different in F2 and p50−/− mice (Table 1) (n = 4 per strain, p < 0.05).
Affinity purification of the A1AR from cortical membranes, followed by radio-iodination and visualization of the purified receptor (∼36 kDa protein) on SDS-PAGE, revealed significantly lower labeling in the cortices obtained from p50−/− versus F2 mice (Fig. 3A) (n=4 in each for each strain). The receptor was visualized as a 36-kDa protein band. In addition, single point radioligand binding assays performed on the affinity-purified receptor fractions from F2 and p50−/− mice revealed ∼30% less binding of the radioligand in the p50−/− mouse (Fig. 3B). To determine whether the decrease in radioligand binding and A1AR protein levels in the p50−/− mice is associated with a decrease in the steady state levels of A1AR mRNA, we performed real time PCR studies on cortical samples. This analysis revealed an ∼35% reduction in A1AR mRNA in p50−/− mice, compared to F2 mice (Fig. 3 C,D) (n=6 per strain; p<0.005, Student's t test).
Figure 3. Purified cortical A1AR and mRNA expression are lower in p50−/− mice.
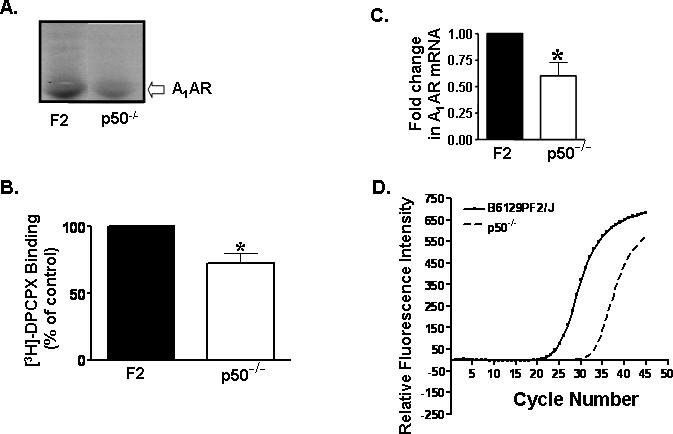
A, Immunopurified A1AR from cortical membrane fractions of F2 and p50−/− mice (n = 4 per strain) were iodinated and analyzed by SDS-PAGE (12%). Equal numbers of counts were loaded onto gels and the A1AR protein was identified as a 36 kDa protein. The level of this protein was lower in the p50−/− mice compared to F2 mice. B, Single point binding assay of immunopurified cortical membrane using [3H]-DPCPX (1nM) revealed 30% less A1AR in p50−/− mice as compared to F2 mice. p50−/− values were expressed as percentage of F2 values and represent the mean ± SEM of four similar experiments (n= 4 per strain; p<0.05). C, Comparative quantitative PCR analysis revealed approximately 35 ± 5.6 % less A1AR mRNA expression in p50−/− mice. Data was normalized to the internal control gene GAPDH and expressed as fold change of A1AR with respect to F2 mice. Data represent mean ± SEM (n= 6 for each strain) (p < 0.01, Student's t test). Asterisk indicates significance of p < 0.05 or p< 0.01 by Student's t-test in the p50−/− mice compared to the F2 mice. D, The threshold for detection of fluorescence occurs at a higher cycle number in p50−/− mice as compared to the F2 mice.
The lower A1AR level in the p50−/− mice was further confirmed by immunocytochemistry using A1AR monoclonal antibody. This antibody revealed specific labeling in cortical layer 2 in the frontal cortex. Sporadic staining was also observed in the layer 3 and 4 of the cortex in both strains of mice. Labeling was not detected in the absence of primary antibody (data not shown). Comparison of similar cortical sections from F2 and p50−/− mice revealed lower staining intensity in all of the cortical layers of the p50−/− mice (Fig. 4B,C) (n=3 per strain). A cresyl violet-stained cortical section from the F2 animal, taken at the level where immunostained sections were obtained, is depicted in Fig. 4A.
Figure 4. A1AR cortical immunohistochemistry in F2and p50−/− mice.

A, Cresyl violet staining in F2 mice. The different layers in the figure are Layer I (molecular), Layer II-III (small pyramidal cells), Layer IV (granular layer), Layer V-VI (infragranular layers). WM represents the white matter. B, Paraformaldehyde stained sections were incubated with monoclonal antibody for the A1AR and visualized using a TRITC labeled mouse secondary antibody. TRITC labeling detected as red fluorescence in layer 2 of the frontal cortex in the F2 and p50−/− mice. C, Fluorescence intensity analysis (n=3 per strain). Asterisk indicates statistical significance between F2 and p50−/− mice by Student's t test).
To further extend our analyses, A1AR binding was evaluated in peripheral tissues and was found to be lower in most of the tissues in the p50−/− vs. F2 mice [(adrenal glands: F2, 8.7 ± 0.8; p50−/−, 4.9 ± 0.4 fmol/mg protein; kidneys: F2, 5.7 ± 0.7; p50−/−, 3.6 ± 0.2 fmol/mg protein; spleen: F2, 5.9 ± 1.5; p50−/−, 4.7 ± 0.5 fmol/mg protein) (n = 4 per strain per region; p,0.05)]. In contrast, binding was slightly greater in the heart of p50−/− mice (F2, 4.3 ± 0.02; p50−/−, 5.8 ± 0. fmol/mg protein) (p<0.05, n=4 per strain).
Clonal cell lines are an ideal model to study changes in AR expression following different in vivo manipulations. To provide additional evidence for a role of NF-κB in the regulation of basal A1AR expression, we compared A1AR binding in normal rat pheochromocytoma (PC12) cells and in cells expressing the mutant form of IκB-α. Previous studies from our laboratory have characterized the A1AR expression in PC12 cells (Jhaveri et al., 2006). Single point binding analysis revealed that binding in normal and mutant PC12 cells was 17.5 ± 2.4 fmol/mg protein and 11.8 ± 1.2 fmol/mg protein, respectively (n=6 per group; p<0.05 by Student's t test).
Cortical G-protein expression is altered in the p50−/− mice
Cortical G-protein expression was evaluated to assess functional aspects of A1AR signaling in the F2 and p50−/− mice. Cortical Gαi1 protein expression was not different between the two strains (F2, 0.8±0.4; p50−/−, 0.9±0.4; n = 6 per strain) (Fig. 5 A,C). Cortical Gαi3 protein was lower in the p50−/− mice (1.7 ± 0.4 normalized units) as compared to 0.9 ± 0.2 normalized units in the F2 mice (Fig. 5 A,C) (p<0.05; n=5 for each strain). On the other hand, the protein expression of both Gαo and Gβ were higher in the p50−/− mice (Fig. 5 B,C) (F2, n = 3; p50−/− = 4), as compared to the F2 mice. The values for cortical Gαo were 0.3 ± 0.1 and 0.7 ± 0.2 (p<0.05) and for Gβ were 0.8 ± 0.4 and 2.4 ± 0.2 normalized units (p<0.05), in the F2 and p50−/− mice, respectively.
Figure 5. Cortical G-protein expression in F2 and p50−/− mice.
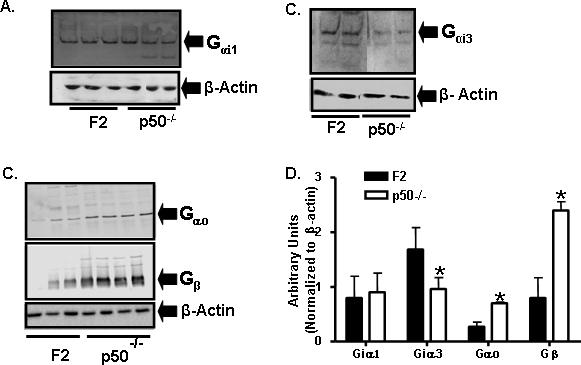
SDS-PAGE analysis and Western blotting of the membrane fraction (40 μg) for different G-protein isoforms in F2 and p50−/− mice revealed different expression between the two strains. Incubation of samples with rabbit polyclonal antibodies for Gαi1, Gαi3, Gαo, and Gβ was carried out. A, Gαi1 and Gαi3 (n = 5 per strain). B, Gαo and Gβ, and C, proteins were normalized to β-actin (F2, n = 3; p50−/− = 4) Asterisk indicates statistical significance between the two strains.
Abrogation of LPS-mediated induction in A1AR in p50−/− mouse
Since LPS is a potent activator of NF- κB in vivo (Glezer et al., 2003), we next determined its impact on A1AR protein and mRNA in cortices of F2 and p50−/− mice. Radioligand binding studies to quantify cortical A1AR 4 h after intraperitoneal administration of LPS (10μg), a time point associated with induction of A1AR in a previous study (Jhaveri et al., 2006). In F2 mice, administration of LPS increased cortical A1AR binding from 100 ± 10 fmol/mg protein (saline controls) to 163 ± 22 fmol/mg protein in the LPS-treated mice (n = 4 per group; p<0.05 by Student's t test). LPS also increased A1AR mRNA expression by 78%± 2.3 in the LPS treated F2 mice as compared to saline treated mice (Fig. 6A,B) (p<0.05; n=4 per group). In contrast, [3H]-DPCPX binding and mRNA expression were not altered by LPS in cortices obtained from p50−/− mice (Fig. 6A,B) (n=4 per group). Similarly, LPS induced A1AR binding in the brain stem of F2 mice (saline treated, 77.8 ± 1.6 fmol/mg protein; LPS treated, 105.7 ± 4.9 fmol/mg protein) (Fig. 6C; n=4 per group; p<0.05). However, A1AR binding was lower in the brain stem of LPS treated p50−/− mice (30.8 ± 3.1 fmol/mg protein) as compared to saline treated p50−/− mice (54.4 ± 5.9 fmol/mg protein) (Fig. 6C; n=4; p<0.05).
Figure 6. Cortical A1AR expression in saline and LPS treated F2 and p50−/− mice.
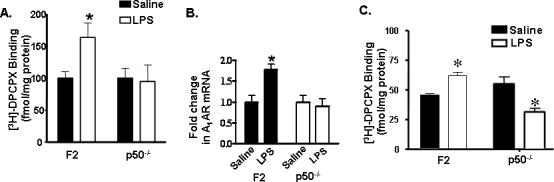
A, A1AR binding in cortical membrane (50 μg) fractions was measured in mice treated with saline (0.2 ml) or LPS (10 μg/0.2 ml). F2 mice showed 63% ± 4.8 more A1AR binding at 4 h after LPS administration, whereas p50−/− mice did not show any change in binding (n = 6 per group). Values represent mean ± SEM of three independent experiments with samples assayed in triplicate. Asterisks represent significance in LPS treated F2 mice compared to saline treated mice (p < 0.05). B, Comparative quantitative PCR analysis revealed approximately 78% ± 2.3 increased A1AR expression in LPS treated F2 mice as compared to saline treated F2 mice while the p50−/− mice did not show any change in A1AR expression between saline and LPS treated mice. Data was normalized to the internal control gene GAPDH and expressed as fold change of A1AR with respect to saline treated mice. Data represent mean ± SEM (n= 4 for each strain) (Asterisk indicates statistical significance from F2 mice, p < 0.01, Student's t test). C,D A1AR binding in brain stem and hypothalamic membrane (50 μg) fractions measured in mice treated with saline or LPS (n = 4 per group per strain). Values represent mean ± SEM of three independent experiments with samples assayed in triplicate. Asterisks represent significance in LPS treated mice compared to saline treated mice (p < 0.05 by Student's t test).
Greater anti-apoptotic efficacy of A1AR agonist in primary cortical neuron cultures from F2 versus p50−/− mice
To determine a potential functional significance of the lower expression of cortical A1AR in p50−/− mice, cortical neurons were prepared from F2 and p50−/− mice and cultured for 6 days. A percentage of the cultured cells demonstrated apoptosis, as evidenced by TUNEL staining (Fig. 7). However, the percentage of TUNEL positive cells was greater in the neurons cultured from p50−/− mice (42±6%) versus F2 mice (11±1%). Incubation of cultures with R-PIA (1 μM) reduced the the percentage of apoptotic cells to 4±1% in F2 mice and to 25±3% in p50−/− mice. This protective action appeared to be mediated via the A1AR since it was reversed by the selective A1AR antagonist DPCPX (apoptotic cells were 11±1% and 32±5%, for F2 and p50−/− mice, respectively) (Fig. 7). As evidenced from these data, A1AR activation conferred a greater degree of protection to cortical neurons obtained from F2 versus p50−/− mice (64±9% inhibition in F2 versus 40±7% inhibition in p50−/− mice). DPCPX added alone enhanced cell killing to 26±4% and 89±9%, for F2 and p50−/− cortical neurons, respectively.
Figure 7. Activation of A1AR reduced apoptosis in cortical neuron cultures obtained from F2 and p50−/− mice.
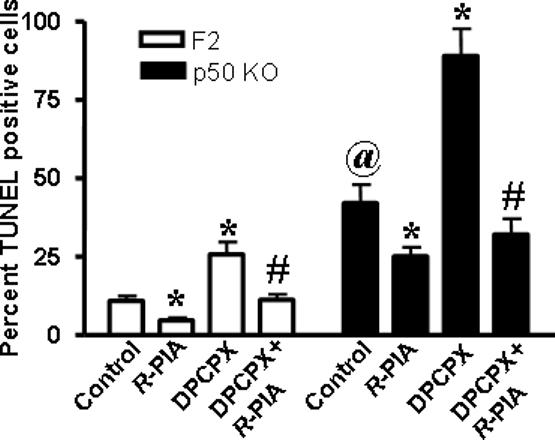
Cortical neurons were obtained from mice and cultured for 6 days. Apoptotic cells were visualized by TUNEL assay, according to the manufacturer's instructions (EMD Biosciences). The percentage of TUNEL-positive cells was assessed by analysis of digitized images from 5 or more microscopic fields of stained cells. The data are presented as the percent mean ± SEM of TUNEL positive cells in cultures treated with R-PIA (1 μM) and/or DPCPX (1 μM). Asterisks (*) indicates statistically significant difference from F2 control group, (#) indicates statistically significant difference from R-PIA treated group, and ‘@’ indicates statistical difference in p50 KO control group from F2 control group (p<0.05), respectively.
Discussion
The major finding of this study is that mice which lack the p50 subunit of NF-κB show reduced A1AR expression in various brain regions and organs, as compared to genetically intact F2 mice. This finding indirectly suggests constitutive basal NF-κB activity influences A1AR expression in both central nervous system (CNS) and peripheral tissues. Thus, A1AR expression could be regulated by NF- κB not only under conditions of oxidative stress and infectious challenge (Nie et al., 1998; Jajoo et al., 2006), which lead to increased NF-κB activity, but also under normal physiological conditions.
Previous studies suggest that oxidative stress induced by chemotherapeutic agents, including cisplatin and anthracyclines, increases A1AR expression and promoter activity via NF-κB activation (Nie et al., 1998). A consensus sequence for NF-κB was detected 623 bases upstream of promoter A transcription start site in plasmid construct pBLPnif/PmtA (Ren and Stiles, 1995) which regulates transcription in response to reactive oxygen species production. A second NF-κB consensus sequence located 306 bases upstream of the second start site of the human A1AR gene did not confer significant NF-κB activity (Nie et al., 1998). NF-κB activation and subsequent induction of A1AR in the cochlea is also observed in response to noise exposure (Ramkumar et al., 2004) and in rat kidney by cisplatin (Bhat et al., 2002). NF-κB has also been implicated in mediating sleep rebound after sleep deprivation (Basheer et al., 2001). AP-1 transcription sites have also been detected in the A1AR promoter region (Ren and Stiles, 1995). However, inhibition of the MAP kinase pathway, an upstream activator of AP-1 transcription factors, did not inhibit A1AR expression (Ramkumar et al., unpublished data), strongly indicative of a crucial role of NF-κB in the regulation of A1AR expression. Our data show that A1AR levels in several brain regions and organs are lower in the p50−/− mice, as compared to F2 mice. The lower cortical protein expression is paralleled by a corresponding decrease in A1AR mRNA, suggesting the possibility that transcription of the A1AR gene is reduced in p50−/− mice. However, an alternative possibility is that the reduced amount of A1AR transcript could reflect decreased stability of this mRNA in the p50−/− mice. Defective receptor transport, endosomal sorting, and insertion into the membrane could also contribute to reduced A1AR binding in various regions.
The genetic absence of p50 appears to significantly affect kainate-induced κB DNA binding activity in the hippocampus (Yu et al., 1999) and possibly in other brain regions, suggesting functional deficit in NF-κB in the p50−/− mice. NF-κB plays a central role in maintaining neuronal survival (Barger et al., 1995; Maggirwar et al., 1998). Hippocampal pyramidal neurons from p50−/− mice demonstrate greater kainate excitotoxicity than do normal wild type mice (Yu et al., 1999). p50−/− mice also exhibit increased auditory nerve degeneration and sensitivity to noise-induced hearing loss (Lang et al., 2006). Furthermore, under normal physiological conditions, NF-κB activity is constitutive in the brain and appears to be essential for neuronal survival during development and in the adult (Bhakar et al., 2002). However, the signals responsible for the maintenance of constitutive NF-κB are not clearly defined. These signals could result from constitutive paracrine or autocrine activation loops (Bhakar et al, 2002). Constitutive activity could also derive from retrograde signaling after activation of NF-κB at the nerve terminal by neurotransmitters. Such retrograde transport of NF-κB has been demonstrated in neurons after stimulation with various agents (Wellman et al., 2001). The implication of these findings is that neuronal survival might be intricately linked to synaptic activity and NF-κB activity.
Our data suggest that constitutive NF-κB activity could be important, at least in part, in regulating the expression level of the A1AR under normal physiological conditions. This was evident in a number of brain regions (cortex, hippocampus, brain stem and hypothalamus and in non-neuronal tissues such as the kidney, spleen, and adrenal gland. Some brain regions (e.g. cortex and hippocampus) have high constitutive NF-κB activity, which could be related to the reduced in A1AR expression in these regions in the p50−/− mice (Bakalkin et al., 1993). However, NF-κB activity has not been determined systematically in other brain regions in a manner that would allow us to conclude that the regions showing reduced A1AR expression in the p50−/− mice were also those with high constitutive activity in the F2 mice. It is also not clear whether the different tissue levels of A1AR expression grossly defined by single concentration radioligand binding studies reflect differences in the activity of NF-κB intrinsic to these tissues. Certainly, other factors, such as subunit composition of the NF-κB homodimers could be important determinants of transcriptional efficacy. Different brain regions might contain NF-κB of different subunit composition, which could in turn mediate different specific functions. As in other cell types, p50/p65 is the most commonly occurring heterodimer combination in the CNS (Bakalkin et al., 1993; Meffert and Baltimore, 2005) although other subunit compositions (e.g. p50 homodimers, p50/c-Rel, and p65/c-Rel) have also been detected (Bakalkin et al., 1993). Nonetheless, the relative preponderance of p50/p65 heterodimers in the adult brain may account for the generalized reduction in A1AR that we measured in different brain regions of p50−/− mice, as well as the higher number of these receptors in the various brain regions compared to other tissues assessed in the present study.
In addition to NF-κB, another factor which determines the levels of A1AR produced by a cell is posttranslational mRNA processing. Tissues which express low levels of the A1AR express transcripts containing exons 4, 5 and 6, while those which express high levels of A1AR (such as brain, kidney, testis) express transcripts containing exons 3, 5 and 6 and exon 4, 5 and 6 (Ren and Stiles, 1994). The inhibitory role of exon 4 is determined by the presence of two upstream ATG start codons (Ren and Stiles, 1994).
Our data also reveal that levels of G protein α and β subunits are also altered in the cortex of the p50−/− mice as compared to the wild type mice. More specifically, levels of the Gαi3 proteins were significantly lower in the cortices of p50−/− mice, while both cortical Gαo and Gβ were higher in these mice. Moreover, levels of another prominent isoform Gαi1 was similar between the two strains of mice. Whether the expression of these G protein subunits are regulated by NF-κB or by other factors is not clear. Based on our data, the decrease in Gαi3 could reflect a positive regulation of this G protein subunit via a p50 containing NF-κB complex. In contrast, the levels of Gαo and Gβ could be negatively regulated by this complex. NF-κB. Based on our data, if these subunits are regulated by NF-κB, then they are regulated differentially. Regulation of two heterotrimeric G protein subunits by NF-κB has recently been described. These reports show induction of Gαi2 in K562 cells (Arinze and Kawai, 2005) and repression of Gαs proteins in human myometrium (Chapman et al., 2005) and raise the possibility that other G protein subunits are similarly regulated by NF-κB. An alternative hypothesis is that the changes in G protein subunits could reflect changes in G protein coupled receptor expression and regulation of downstream signaling. For example, previous studies have shown coordinate changes in receptor and G protein expression after receptor desensitization and sensitization, suggesting that the levels of the G proteins are directly influenced by receptor function (for review see Stiles, 1992). However, the divergent regulation of the different Gαi and β subunits would discount such a possibility.
Despite changes in Gα subunits, we observed no change in agonist high affinity (G protein-coupled) state of the A1AR. This could suggest that the increases in Gαi1 and Gαo subunits could cancel out any functional deficits in Gαi3. However, this explanation is purely speculative since Freissmuth et al. (1992) showed, at least for the recombinant Gαi and Gαo subunits, a 10-fold higher affinity of the A1AR for Gαi3 than for the other Gαi or Gαo subunits. Coordinate regulation of G protein-coupled receptors and and heterotrimeric G protein subunits by NF-κB appears to be an intriguing idea since it provides an additional level of control of receptor signaling. Coordinate regulation of the AR2AAR and Gαs and Gαolf proteins was similarly observed in striata of p50−/− mice and leads to increased activation of A2AAR signaling cascade (Xie et al., 2006). The reduced ability of R-PIA to decrease apoptosis in neuron cultures obtained from p50−/− versus F2 mice could reflect the lower levels of A1AR expression, rather that their ability to couple with G proteins, since receptor-G rotein coupling (as defined by agonist high affinity binding) was not altered. The enhancement in apoptosis induced by DPCPX would support a role of endogenously produced adenosine in mediating protection under normal culture conditions. Furthermore, the increased apoptosis could reflect a decreased ability of the p50−/− neurons to adapt to the stress of in vitro culturing, better than the F2 mice. In spite of the apparent difference in F2 and p50−/− response to R-PIA, our data highlights the importance of A1AR activation in mediating cell viability under normal culture conditions, as evidenced by the finding that DPCPX significantly increased apoptosis in neuronal cultures obtained from both F2 and p50−/− mice.
Our data do not rule out the possibility of compensation by other members of the κB family in response to deletion of the p50 subunit. p50−/− mice can form alternative p52:p65 dimers, but this dimer does not compensate functionally for p50 (Kunsch et al., 1992; Franzoso et al., 1998; Hoffmann et al., 2003). The interaction of both p50 and p65 components is required for normal NF-κB binding and transcriptional activation of specific genes, and the lone availability of either of these subunits may be insufficient for binding of or transcriptional activation by NF-κB (Kunsch et al., 1992). Finally, not only does the transcription of individual genes require a specific NF-κB dimer combination (Pizzi et al., 2002; Hoffmann et al., 2003), but even single nucleotide differences in the κB sites of the gene can significantly alter specific NF-κB dimer requirements (Leung et al., 2004). Thus, each transcriptional event requires recruitment of different dimer combinations. Moreover, our study highlights the fact that even members of the κB family that lack transactivation domains (such as p50) can be highly relevant in transcriptional regulation of a gene as prominent as A1AR.
In conclusion, A1AR expression and binding are modulated by NF-κB in various brain regions and peripheral tissues. Although this study focused on the impact of the p50 subunit of NF-κB, a future challenge is to delineate the involvement of other members of the κB family and associated co-activator proteins that underlie receptor regulation by pathologic and non-pathologic stimuli. A genetic approach using such knockout mice models provides a powerful tool to characterize the regulation of A1AR in the brain.
Acknowledgement
This work was supported by NIH grant # R01-7543 and by the Excellence in Academic Medicine program of the SIU School of Medicine.
Glossary
Abbreviations
- A1AR
Adenosine A1 receptor
- ADA
Adenosine deaminase
- AR
Adenosine receptor
- Bmax
Maximal binding capacity
- CHAPS
Polyethylneimine and 3-[(3-cholamidopropyl) dimethylammonio]-1 propanesulphonate
- CNS
Central nervous system
- GAPDH
Glyceraldehyde phosphate 3-dehydrogenase
- GPCR
G-protein coupled receptor
- [3H]-DPCPX
([3H]8-cyclopentyl-1,3-di[2’,3’-3H] propylxanthine
- Kd
Equilibrium dissociation constant
- LPS
Lipopolysaccharide
- mI κB-α
Mutant I κB-α
- NF- κB
Nuclear factor – kappa B
- PBS
Phosphate buffered saline
- PC12 cells
Rat pheochromocytoma cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arinze IJ, Kawai Y. Transcriptional activation of the human Gαi2 gene promoter through nuclear factor-κB and antioxidant response elements. J Biol Chem. 2005;280:9786–9795. doi: 10.1074/jbc.M414006200. [DOI] [PubMed] [Google Scholar]
- Bakalkin G, Ya, Yakovleva T, Terenius L. NF-κB-like factors in the murine brain. Developmentally-regulated and tissue-specific expression. Brain Res Mol Brain Res. 1993;20:137–146. doi: 10.1016/0169-328x(93)90119-a. [DOI] [PubMed] [Google Scholar]
- Barger SW, Horster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP. Tumor necrosis factors alpha and beta protect neurons against amyloid beta- peptide toxicity: evidence for involvement of a κB-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci U SA. 1995;92:9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basheer R, Rainnie DG, Porkka-Heiskanen T, Ramesh V, McCarley RW. Adenosine, prolonged wakefulness, and A1-activated NF-κB DNA binding in the basal forebrain of the rat. Neuroscience. 2001;104:731–739. doi: 10.1016/s0306-4522(01)00111-7. [DOI] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- Bhakar AL, Tannis LL, Zeindler C, Russo MP, Jobin C, Park DS, MacPherson S, Barker PA. Constitutive nuclear factor- κB activity is required for central neuron survival. J Neurosci. 2002;22:8466–8475. doi: 10.1523/JNEUROSCI.22-19-08466.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat SG, Mishra S, Mei Y, Nie Z, Whitworth CA, Rybak LP, Ramkumar V. Cisplatin up-regulates the adenosine A1 receptor in the rat kidney. Eur J Pharmacol. 2002;442:251–264. doi: 10.1016/s0014-2999(02)01510-8. [DOI] [PubMed] [Google Scholar]
- Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brewer G, Torricelli J, Evege E, Price P. Optimized survival of hippocampal neurons in B27 supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;32:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Brewer G. Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods. 1997;71:143–155. doi: 10.1016/s0165-0270(96)00136-7. [DOI] [PubMed] [Google Scholar]
- Chapman NR, Smyrnias I, Anumba DOC, Europe-Finner GN, Robson SC. Expression of the GTP-binding protein (Gαs) is repressed by nuclear factor κB RelA subunit in human myometrium. Endocrinology. 2005;146:4994–5002. doi: 10.1210/en.2005-0533. [DOI] [PubMed] [Google Scholar]
- Dolphin AC, Archer ER. An adenosine agonist inhibits and a cyclic AMP analogue enhances the release of glutamate but not GABA from slices of rat dentate gyrus. Neurosci Lett. 1983;43:49–54. doi: 10.1016/0304-3940(83)90127-1. [DOI] [PubMed] [Google Scholar]
- Evans MC, Swan JH, Meldrum BS. An adenosine analogue, 2-chloroadenosine, protects against long term development of ischaemic cell loss in the rat hippocampus. Neurosci Lett. 1987;83:287–292. doi: 10.1016/0304-3940(87)90101-7. [DOI] [PubMed] [Google Scholar]
- Ford MS, Nie Z, Whitworth C, Rybak LP, Ramkumar V. Up-regulation of adenosine receptors in the cochlea by cisplatin. Hear Res. 1997;111:143–152. doi: 10.1016/s0378-5955(97)00103-2. [DOI] [PubMed] [Google Scholar]
- Franzoso G, Carlson L, Poljak L, Shores EW, Epstein S, Leonardi A, Grinbery A, Tran T, Scharton-Kersten T, Anver M, Love P, Brown K, Siebenlist U. Mice deficient in nuclear factor (NF)–κB/p52 present with defcts in humoral responses, germinal centre reactions, and splenic microarchitecture. J Exp Med. 1998;187:147–159. doi: 10.1084/jem.187.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Dunwiddie TV. How does adenosine inhibit transmitter release? Trends Pharmacol Sci. 1988;9:130–134. doi: 10.1016/0165-6147(88)90194-0. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- Freissmuth M, Schutz W, Linder ME. Interactions of the bovine brain A1-adenosine receptor with recombinant G protein a-subunits. J Biol Chem. 1991;266:17778–17783. [PubMed] [Google Scholar]
- Glezer I, Munhoz CD, Kawamoto EM, Marcourakis T, Avellar MC, Scavone C. MK-801 and 7-Ni attenuate the activation of brain NF-κB induced by LPS. Neuropharmacology. 2003;45:1120–1129. doi: 10.1016/s0028-3908(03)00279-x. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-κB/Rel transcription factors defines functional specificities. EMBO. 2003;22:5530–5539. doi: 10.1093/emboj/cdg534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jajoo S, Mukherjea D, Pingle SC, Sekino Y, Ramkumar V. Induction of adenosine A1 receptor expression by pertussis toxin via an ADP ribosylation independent pathway. J Pharmacol Exp Ther. 2006;317:1–10. doi: 10.1124/jpet.105.096255. [DOI] [PubMed] [Google Scholar]
- Jhaveri KA, Toth LA, Sekino Y, Ramkumar V. Nitric oxide serves as an endogenous regulator of brain A1 adenosine receptor. J. Neurochem. 2006;99:42–53. doi: 10.1111/j.1471-4159.2006.04095.x. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle PA. Constitutive NF-κB activity in neurons. Mol Cell Biol. 1994;14:3981–3992. doi: 10.1128/mcb.14.6.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostopoulos GK, Phillis JW. Purinergic depression of neurons in different areas of the rat brain. Exp Neurol. 1977;55:719–724. doi: 10.1016/0014-4886(77)90296-5. [DOI] [PubMed] [Google Scholar]
- Kunsch C, Ruben SM, Rosen CA. Selection of optimal κB/Rel DNA-binding motifs: interaction of both subunits of NF- κB with DNA is required for transcriptional activation. Mol Cell Biol. 1992;12:4412–4421. doi: 10.1128/mcb.12.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang H, Schulte BA, Zhou D, Smythe N, Spicer SS, Schmiedt RA. Nuclear factor-κB deficiency is associated with auditory nerve degeneration and increased noise-induced hearing loss. J Neurosci. 2006;26:3541–3550. doi: 10.1523/JNEUROSCI.2488-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung TH, Hoffmann A, Baltimore D. One nucleotide in a κB site can determine cofactor specificity for NF-κB dimers. Cell. 2004;118:453–464. doi: 10.1016/j.cell.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Maggirwar SB, Sarmiere PD, Dewhurst S, Freeman RS. Nerve growth factor-dependent activation of NF-κB contributes to survival of sympathetic neurons. J Neurosci. 1998;18:10356–10365. doi: 10.1523/JNEUROSCI.18-24-10356.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meffert MK, Baltimore D. Physiological functions for brain NF-κB. Trends Neurosci. 2005;28:37–43. doi: 10.1016/j.tins.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Nie Z, Mei Y, Ford M, Rybak L, Marcuzzi A, Ren H, Stiles GL, Ramkumar V. Oxidative stress increases A1 adenosine receptor expression by activating nuclear factor-κB. Mol Pharmacol. 1998;53:663–669. doi: 10.1124/mol.53.4.663. [DOI] [PubMed] [Google Scholar]
- Nie Z, Mei Y, Malek RL, Marcuzzi A, Lee NH, Ramkumar V. A role of p75 in NGF-mediated down-regulation of the A2A adenosine receptors in PC12 cells. Mol Pharmacol. 1999;56:947–954. doi: 10.1124/mol.56.5.947. [DOI] [PubMed] [Google Scholar]
- Ochiishi T, Chen L, Yukawa A, Saitoh Y, Sekino Y, Arai T, Nakata H, Miyamoto H. Cellular localization of adenosine A1 receptors in rat forebrain: immunohistochemical analysis using adenosine A1 receptor-specific monoclonal antibody. J Comp Neurol. 1999;411:301–316. [PubMed] [Google Scholar]
- Olah ME, Stiles GL. Adenosine receptors. Annu Rev Physiol. 1992;54:211–225. doi: 10.1146/annurev.ph.54.030192.001235. [DOI] [PubMed] [Google Scholar]
- Pingle S, Mishra S, Marcuzzi A, Bhat S, Sekino Y, Rybak L, Ramkumar V. Osmotic diuretics induce adenosine A1 receptor expression and protect renal proximal tubular epithelial cells against cisplatin-mediated apoptosis. J. Biol. Chem. 2004;279:43157–43167. doi: 10.1074/jbc.M405666200. [DOI] [PubMed] [Google Scholar]
- Pizzi M, Goffi F, Boroni F, Benarese M, Perkins SE, Liou HC, Spano P. Opposing roles for NF-κB/Rel factors p65 and c-Rel in the modulation of neuron survival elicited by glutamate and interleukin-1β. J Biol Chem. 2002;277:20717–20723. doi: 10.1074/jbc.M201014200. [DOI] [PubMed] [Google Scholar]
- Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- Ramkumar V, Whitworth CA, Pingle SC, Hughes LF, Rybak LP. Noise induces A1 adenosine receptor expression in the chinchilla cochlea. Hear Res. 2004;88:47–56. doi: 10.1016/S0378-5955(03)00344-7. [DOI] [PubMed] [Google Scholar]
- Ren H, Stiles GL. Posttranslational mRNA processing as a mechanism for regulation of human A1 adenosine receptor expression. Proc Natl Acad Sci USA. 1994;91:4864–4866. doi: 10.1073/pnas.91.11.4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Stiles GL. Separate promoters in the human A1 adenosine receptor gene direct the synthesis of distinct messenger RNAs that regulate receptor abundance. Mol Pharmacol. 1995;48:975–980. [PubMed] [Google Scholar]
- Rivkees SA, Price SL, Zhou FC. Immunohistochemical detection of A1 adenosine receptors in rat brain with emphasis on localization in the hippocampal formation, cerebral cortex, cerebellum, and basal ganglia. Brain Res. 1995;677:193–203. doi: 10.1016/0006-8993(95)00062-u. [DOI] [PubMed] [Google Scholar]
- Rubio R, Berne RM, Bockman EL, Curnish RR. Relationship between adenosine concentration and oxygen supply in rat brain. Am J Physiol. 1975;228:1896–1902. doi: 10.1152/ajplegacy.1975.228.6.1896. [DOI] [PubMed] [Google Scholar]
- Rudolphi KA, Schubert P, Parkinson FE, Fredholm BB. Neuroprotective role of adenosine in cerebral ischemia. Trends Pharmacol Sci. 1992;13:439–445. doi: 10.1016/0165-6147(92)90141-r. [DOI] [PubMed] [Google Scholar]
- Schwarz EM, Badorff C, Hiura TS, Wessely R, Badorff A, Verma IM, Knowlton KU. NF-κB-mediated inhibition of apoptosis is required for encephalomyocarditis virus virulence: a mechanism of resistance in p50 knockout mice. J Virol. 1998;72:5654–5660. doi: 10.1128/jvi.72.7.5654-5660.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha WC, Liou HC, Tuomanen EI, Baltimore D. Target disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- Soong R, Beyser K, Basten O, Kalbe A, Rueschoff J, Tabiti K. Quantitative reverse transcription – polymerase chain reaction detection of cytokeratin 20 in noncolorectal lymph nodes. Clin Cancer Res. 2001;7:3423–3429. [PubMed] [Google Scholar]
- Stiles GL. Adenosine receptors. J Biol Chem. 1992;267:6451–6454. [PubMed] [Google Scholar]
- Tucker AL, Linden J. Cloned receptors and cardiovascular responses to adenosine. Cardiovasc Res. 1993;27:62–67. doi: 10.1093/cvr/27.1.62. [DOI] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Van Wylen DG, Park TS, Rubio R, Berne RM. Increases in cerebral interstitial fluid adenosine concentration during hypoxia, local potassium infusion, and ischemia. J Cereb Blood Flow Metab. 1986;6:522–528. doi: 10.1038/jcbfm.1986.97. [DOI] [PubMed] [Google Scholar]
- Wellmann H, Kaltschmidt B, Kaltschmidt C. Retrograde transport of transcription factor NF-κB in living neurons. J Biol Chem. 2001;276:11821–11829. doi: 10.1074/jbc.M009253200. [DOI] [PubMed] [Google Scholar]
- Xie X, Toth LA, Ramkumar V. Adenosine A2A receptor downstream signaling molecules are increased in striata of mice lacking nuclear factor (NF)-κB p50 subunit. Society for Neuroscience. 2006 Abstract 721.2. [Google Scholar]
- Yu Z, Zhou D, Bruce-Keller AJ, Kindy MS, Mattson MP. Lack of the p50 subunit of nuclear factor-κB increases the vulnerability of hippocampal neurons to excitotoxic injury. J Neurosci. 1999;19:8856–8865. doi: 10.1523/JNEUROSCI.19-20-08856.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]