

Abstract
Trichodiene synthase is a terpenoid cyclase that catalyzes the cyclization of farnesyl diphosphate (FPP) to form the bicyclic sesquiterpene hydrocarbon trichodiene (89%), at least five sesquiterpene side products (11%), and inorganic pyrophosphate (PPi). Incubation of trichodiene synthase with 2-fluorofarnesyl diphosphate or 4-methylfarnesyl diphosphate similarly yields sesquiterpene mixtures despite the electronic effects or steric bulk introduced by substrate derivatization. The versatility of the enzyme is also demonstrated in the 2.85 Å resolution X-ray crystal structure of the complex with Mg2+ 3-PPi and the benzyl triethylammonium cation, which is a bulkier mimic of the bisabolyl carbocation intermediate in catalysis. Taken together, these findings show that the active site of trichodiene synthase is sufficiently flexible to accommodate bulkier and electronically-diverse substrates and intermediates, which could indicate additional potential for the biosynthetic utility of this terpenoid cyclase.
Keywords: terpenoid synthase, farnesyl diphosphate, sesquiterpene, protein crystallography
Sesquiterpene synthases catalyze the cyclization of the universal acyclic precursor, farnesyl diphosphate (FPP)1, to form one or more of over 300 known monocyclic, bicyclic, and tricyclic sesquiterpenes with a wide variety of structures and stereochemistry [1-5]. All these reactions are known to involve a cascade of carbocationic intermediates, and the controlled manipulation of these highly reactive intermediates gives rise to impressive product diversity. Typically, the active site of a terpene cyclase serves as a template that chaperones the conformations and reactions of carbocation intermediates to yield a single or dominant cyclization product. However, some cyclases are more permissive chaperones and can generate a wide array of cyclization products. For example, δ-selinene synthase and γ-humulene synthase from Grand fir (Abies grandis) generate 34 and 52 distinct cyclization products, respectively [6].
Trichodiene synthase from Fusarium sporotrichiodes is an 89 kDa homodimer that catalyzes the cyclization of farnesyl diphosphate to form the bicyclic hydrocarbon trichodiene and the coproduct inorganic pyrophosphate (PPi) [7, 8]. Three Mg2+ ions are required to activate the PPi leaving group and generate a reactive allylic cation that undergoes rapid isomerization to (3R)nerolidyl diphosphate, which subsequently re-ionizes to initiate a complex cyclization cascade [7, 9]. The first cyclic carbocation intermediate in this cascade is the bisabolyl carbocation (Figs 1 and 2a) [8]. The PPi co-product inhibits trichodiene synthase with KI = 0.49 μM [10], and analogues of the bisabolyl cation such as R-azabisabolene (Fig. 2b) require PPi for measurable inhibition with KI = 0.51 μM and an induced inhibition constant of 2.6 μM, with the magnitude of inhibition proportional to the PPi concentration [10]. X-ray crystallographic analyses of azabisabolene complexes with trichodiene synthase mutants show that the binding of these positively charged inhibitors in multiple conformations is stabilized by Mg2+ 3-PPi complexation [11, 12]. A similar Mg2+ 3-PPi cluster also stabilizes the binding of certain aza analogues to the monoterpene synthase, bornyl diphosphate synthase [13].
Fig. 1.

Postulated mechanism for the cyclization of farnesyl diphosphate to trichodiene by trichodiene synthase (OPP = diphosphate, −OPP = inorganic pyrophosphate anion) [8].
Fig. 2.

(a) Bisabolyl carbocation intermediate in trichodiene synthase catalysis. (b) R-Azabisabolene, a cationic analogue of the bisabolyl cation in the trichodiene synthase mechanism, is a strong competitive inhibitor in the presence of inorganic diphosphate (PPi) with Ki for PPi = 0.51 μM and an induced inhibition constant of 2.6 μM [10]. (c) The aza analogue of the α-terpinyl cation is a competitive inhibitor in the presence of inorganic pyrophosphate (PPi) with Ki for PPi = 0.48 μM and an induced inhibition constant of 10.9 μM [10]. (d) The benzyl triethylammonium cation (BTAC) is a phase transfer catalyst for synthetic organic chemistry that partially mimics the bisabolyl carbocation intermediate (a).
We now report the results of incubation of trichodiene synthase with two analogues of the natural FPP substrate, 4-methylfarnesyl diphosphate (4M-FPP) and the less reactive substrate analogue 2-fluorofarnesyl diphosphate (2F-FPP) (Fig. 1). While cylization of FPP was found to give predominantly trichodiene, accompanied by trace amounts of several aberrant sesquiterpenes previously observed only with trichodiene synthase mutants, cyclization of both FPP analogues gave rise to complex mixtures of sesquiterpene analogues. When crystals of trichodiene synthase were soaked in a buffer solution containing 2FFPP, the resulting crystals were shown by X-ray crystallography to contain bound Mg2+ 3-PPi. We also report the crystal structure of the complex of trichodiene with Mg2+ 3-PPi and the benzyl triethylammonium cation (BTAC), the chloride salt of which is used as a phase transfer catalyst for synthetic organic chemistry; BTAC partially mimics the bisabolyl carbocation or other carbocation intermediates (Fig. 2a). Taken together, these findings suggest that trichodiene synthase, and perhaps many terpene cyclases, may be capable of substantial versatility in binding and catalysis with unnatural substrates.
Materials and Methods
Expression, purification, and crystallization of trichodiene synthase
The plasmid containing the gene encoding for wild-type trichodiene synthase was transformed into Escherichia coli BL21 (DE3); the enzyme was overexpressed, purified, and crystallized by the hanging drop vapor diffusion method as described [9]. The complex between trichodiene synthase and BTAC was prepared by soaking crystals in a buffer solution containing 1 mM MgCl2, 4 mM FPP, and 10 mM BTAC. The substrate turned over in the enzyme crystals, resulting in the formation of a complex between the enzyme, Mg2+ 3-PPi, and a large molecule in the enzyme active site. Initially, we interpreted the electron density in the active site as a trichodiene molecule, but further analysis revealed that this density was better fit by a molecule of BTAC. Enzyme crystals were also soaked in a buffer solution containing 1 mM MgCl2 and 4 mM 2F-FPP to yield the structure of the complex with Mg2+ 3-PPi. Crystals were prepared for data collection by cryoprotection with 25 % ethylene glycol and flash cooling in liquid nitrogen.
Diffraction data for crystals of both complexes were collected at the Cornell High Energy Synchrotron Source (CHESS, beamlines F1 and A1). Data were indexed and merged using the program HKL2000 [14]. Crystals were essentially isomorphous with crystals of the wild-type enzyme (space group P3121, a = b = 122.2 Å, c = 151.2 Å) [9] and each structure was solved by the difference Fourier technique. The programs CNS [15] and O [16] were used for refinement and model building, respectively. Noncrystallographic symmetry constraints were used in the initial stages of refinement and subsequently relaxed into appropriately weighted restraints as judged by Rfree as refinement progressed. Molecular models shown in the figures were prepared with Bobscript version 2.4 [17]. Data collection and refinement statistics are reported in Table 1.
Table I.
Data Collection and Refinement Statistics for Trichodiene Synthase-Ligand Complexes
| Ligands | Mg2+3-PPi-BTAC |
|---|---|
| Resolution Range (Å) | 50-2.85 |
| Reflections (measured/unique) | 127952/27585 |
| Completeness (%) (overall/outer shell) | 94.4/96.7 |
| Rmerge a (overall/outer shell) | 0.088/0.616 |
| <I/σ> (overall/outer shell) | 15.2/2.6 |
| Protein atoms (no.) b | 5717 |
| Solvent atoms (no.) b | 36 |
| Metal ions (no.) b | 3 |
| Ligand atoms (no.) b | 23 |
| Reflections used in refinement (work/free) | 25786/1306 |
| R/Rfree c | 0.216/0.265 |
| r.m.s. deviations | |
| bonds (Å) | 0.008 |
| angles (deg.) | 1.2 |
| dihedral angles (deg.) | 19.1 |
| improper dihedral angles (deg.) | 0.8 |
Rmerge = Σ|Ij-〈Ij〉|/ΣIj, where Ij is the observed intensity for reflection j and 〈Ij〉 is the average intensity calculated for reflection j from replicate data.
per asymmetric unit.
R = Σ||Fo| - |Fc||/Σ|Fo|, where R and Rfree are calculated by using the working and test reflection sets, respectively.
Incubation of trichodiene synthase with farnesyl diphosphate
Analysis of cyclization products generated from FPP was performed using gas chromatography and mass spectroscopy (GC/MS) as previously described [18, 19]. All glassware was washed and oven-baked prior to use. The FPP substrate (50-500 μM) was incubated with or without purified wild-type or D100E trichodiene synthase (1 mg) in 4 mL buffer (10 mM Tris (pH 7.8), 5 mM MgCl2, 15% glycerol, and 5 mM β-mercaptoethanol) and overlaid with HPLC-grade n-pentane (Fisher P399-1) in a glass test tube at 30 °C for 3-20 hours. Reaction products were extracted with HPLC-grade n-pentane and purified on a 3 cm 230-400 mesh silica gel column. Purified extracts were concentrated on an ice-water mixture under reduced pressure until volume decreased to 50 μL or less, and each resulting concentrate was analyzed by GC/MS. Reaction products were analyzed using a Hewlett–Packard 6890 gas chromatograph (GC) coupled to a 5973 mass selective detector and equipped with an HP-5MS capillary column (0.25 mm i.d. × 30 m with 0.25 μm film) (Agilent Technologies). A total of 2 μL of concentrate was injected in splitless mode into the gas chromatograph-mass spectrometer. The oven temperature was maintained at 35 °C for 1 min, increased at the rate of 5 °C/min to 230 °C, followed by a 20 °C/min increase to 280 °C. The temperature was held at 280 °C for 20 min.
Synthesis of 2-fluorofarnesyl diphosphate
The sample of trans, trans-2-fluorofarnesyl diphosphate (2F-FPP) was obtained by methods similar to those reported for the preparation of 2-fluorogeranyl diphosphate [20] i.e., conversion of trans, trans-2-fluorofarnesol [21] to trans, trans-2-fluorofarnesyl chloride [22] and displacement with tetrabutylammonium pyrophosphate [23]. Procedures and characterization data for trans, trans-2-fluorofarnesyl chloride and 2F-FPP will be published separately.
Incubation of trichodiene synthase with 2-fluorofarnesyl diphosphate
The fluorinated substrate 2F-FPP (50-500 μM) was incubated with or without purified protein (1 mg) in 4 mL buffer (10 mM Tris (pH 7.8), 5 mM MgCl2, 15% glycerol, and 5 mM β-mercaptoethanol) and overlaid with HPLC-grade n-pentane in a glass test tube at 30 °C. In anticipation of a slower reaction rate with a less reactive substrate, 1 mg of enzyme was added to the reaction mixture at 10 h and 20 h reaction time points in order to maximize turnover and product detection. Reaction products were extracted, concentrated, and subject to GC/MS analysis as described above.
Incubation of trichodiene synthase with racemic 4-methylfarnesyl diphosphate
The methylated substrate 4M-FPP [24] (1-2 mg) was incubated with or without purified protein (1 mg) in 4 mL buffer (10 mM Tris (pH 7.8), 5 mM MgCl2, 15% glycerol, and 5 mM β-mercaptoethanol) and overlaid with HPLC-grade n-pentane in a glass test tube at 30 °C. Reaction products were extracted after ∼18 h, concentrated, and subject to GC/MS analysis as described above.
Inhibition of trichodiene synthase by BTAC
For inhibition assays, 1 mL of reaction buffer (40 ng trichodiene synthase, 10 mM Tris (pH 7.8), 5 mM MgCl2, 15% glycerol, and 5 mM β-mercaptoethanol) was overlaid with 0.75-1 mL of HPLC-grade n-pentane and incubated at 30 °C for 7 min. Inhibition of trichodiene synthase with BTAC alone was studied using 3H-FPP (Sigma), diluted with cold FPP (Echelon Biosciences), and pre-washed with n-pentane. Concentrations of 50-1200 nM FPP (∼80 Ci/mol) and 50 μM BTAC were used. The reactions were carried out both with and without BTAC for each substrate concentration. Inhibition of trichodiene synthase in the presence of BTAC and PPi was studied using 1 μM 3H-FPP and a range of BTAC and PPi concentrations. Each reaction was quenched using 75 μL of 0.5 M EDTA (pH 8.0) and vortexed for 25 sec. Products were extracted using an additional 3 × 0.75 mL n-pentane. Extracts were passed over a silica gel column in a Pasteur pipette. The column was washed with an additional 0.75 mL of n-pentane directly into 5 mL scintillation cocktail. Five different concentrations of BTAC ranging from 0-30 μM were used in combination with four different PPi concentrations ranging from 5-20 μM. All reactions were run 3-5 times. Inhibition constants for PPi and BTAC were calculated by fitting all data simultaneously to the general kinetic expression for cooperative competitive inhibition in equation 1 [10, 25],
| (Equation 1), |
where KI and KJ are the inherent inhibition constants of PPi and BTAC, respectively, α is a cooperativity factor, and [BTAC]/KJ ≈ 0 since BTAC is ineffective as an inhibitor in the absence of PPi.
Results
Incubation of trichodiene synthase with unnatural FPP derivatives
Overnight incubation of 2F-FPP in reaction buffer containing 0.25 mg/ml trichodiene synthase results in the generation of multiple pentane-extractable products (Fig. 3a), the concentrations of which increase considerably by lengthening the incubation time to 36 h and by adding enzyme to the reaction mixture at 0 h, 10 h, and 20 h time points. Control incubations carried out in the absence of enzyme confirm that there is no background formation of these products. The GC/MS data indicate that 8 products exhibit molecular ion peaks of m/z 221 (consistent with the M-1 ions of fluorinated sesquiterpenes), while two additional products exhibit molecular ion peaks of m/z 203 (consistent with the loss of fluorine during the reaction catalyzed by trichodiene synthase or during the GC/MS analysis). Notably, when crystals of wild-type trichodiene synthase are soaked in buffer solutions containing 4 mM 2F-FPP, only the Mg2+ 3-PPi cluster is observed to bind to monomer B of the enzyme (data not shown). The structure of this Mg2+ 3-PPi complex is identical to that determined previously [9] and is consistent with generation of a fluorinated sesquiterpene product that is released from the enzyme active site while the PPi coproduct remains bound.
Fig. 3.

Gas chromatograms of (a) pentane extract resulting from the incubation of trichodiene synthase with 2F-FPP, (b) pentane extract resulting from the incubation of trichodiene synthase with 4M-FPP, and (c) pentane extract resulting from the incubation of trichodiene synthase with FPP.
Overnight incubation of 4M-FPP in reaction buffer containing 0.25 mg/mL enzyme and GC-MS analysis of the n-pentane elutable extract reveal the generation of one major product and 16 minor products (Fig. 3b), each with molecular ion peaks of m/z 217, consistent with the M-1 ions of methylated sesquiterpenes. Unexpectedly, control incubations of wild-type trichodiene synthase with FPP indicate that although trichodiene is the major product (89%), the formation of trichodiene was also accompanied by the generation of 1-2% of each of five additional sesquiterpenes identical by GC/MS with the more abundant aberrant products (total 37%, β-farnesene, α-and β-bisabolene, cuprenene, and isochamigrene) that are known to be produced [18] by mutants of trichodiene synthase (Figure 3c).
Inhibition of trichodiene synthase by BTAC and PPi
Although 50 μM BTAC alone does not inhibit the cyclization of FPP by trichodiene synthase, 5-30 μM BTAC acts as a competitive inhibitor of trichodiene synthase reaction in the presence of 5-20 μM PPi (PPi itself is a strong competitive inhibitor with KI = 0.49 μM [10]). The apparent inhibition constant of PPi in the presence of BTAC is determined to be 0.7 μM while the induced inhibition constant for BTAC in the presence of PPi is 36 μM (Fig. 4). The dependence of BTAC inhibition on the presence of PPi is similar to that previously observed for ammonium analogues of the bisabolyl carbocation intermediate, such as R-azabisabolene, the aza-analogue of the α-terpinyl cation (Fig. 2), and trimethylamine [10] (Table 2).
Fig. 4.
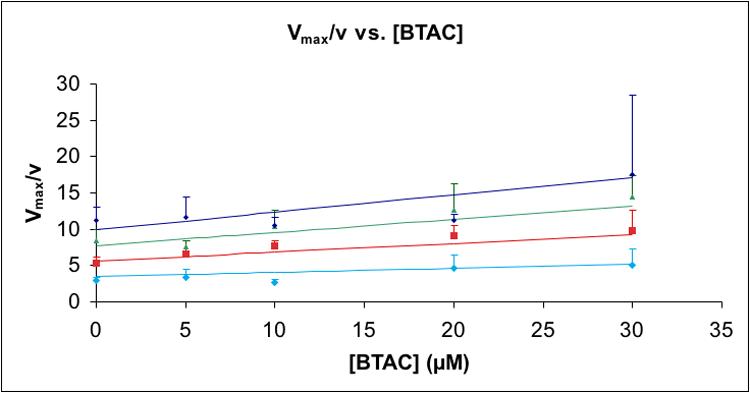
Plot of Vmax/v as a function of [BTAC] at variable [PPi]: (◆) = 5 μM; (■) = 10 μM; (▲) = 15 μM; (●) = 20 μM [PPi]. Positive standard deviations for data are represented by vertical lines of the color corresponding to each [PPi]. The data were fit to equation (1), as described. The calculated inhibition constant Ki for inorganic pyrophosphate is 0.7 μM; the induced inhibition constant for BTAC in the presence of inorganic pyrophosphate is 36 μM.
Table II.
Cooperative competitive inhibition of trichodiene synthase by inorganic pyrophosphate (PPi) and quaternary ammonium analogues of the bisabolyl carbocation intermediate
| inhibitor | PPi | inhibitor |
|---|---|---|
| KI (μM) | αKJa (μM) | |
| PPi b | 0.49 | - |
| trimethylamine b | 0.68 | 172.0 |
| BTAC | 0.7 | 36 |
| α-terpinyl cation analogue b | 0.48 | 10.9 |
| R-azabisabolene b | 0.51 | 2.6 |
| S-azabisabolene b | 0.47 | 2.9 |
Calculated induced inhibition constant of the ammonium analogues (Figure 2) in the presence of PPi, using equation (1) for cooperative competitive inhibition [10, 25].
From reference 10.
Structure of the trichodiene synthase-Mg2+ 3-PPi-BTAC complex
The Mg2+ 3-PPi cluster and BTAC bind to the active site of monomer B of the dimer (Fig. 5). The enzyme undergoes ligand-induced conformational changes similar to those observed in the wild-type enzyme complex with Mg2+ 3-PPi [9]. No ligand binding is observed in monomer A because the ligand-induced conformational changes are hindered by interlattice crystal contacts. The r.m.s. deviation between liganded and unliganded trichodiene synthase structures is 1.3 Å for 350 Cα atoms. The overall conformational changes observed in the BTAC-Mg2+ 3-PPi complex are similar to those observed in the Mg2+ 3-PPi complex, with an r.m.s. deviation of 0.338 Å for 352 Cα atoms between monomers B of the two complexes.
Fig. 5.
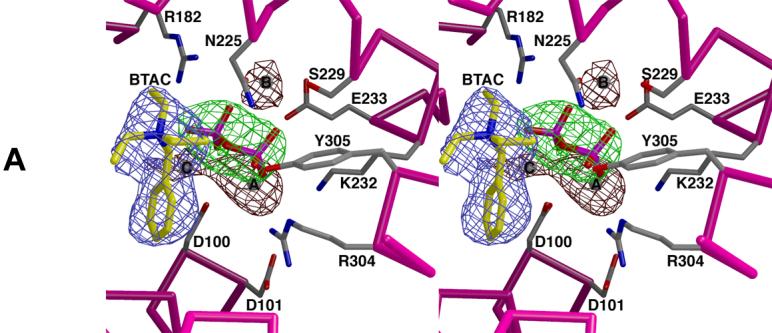
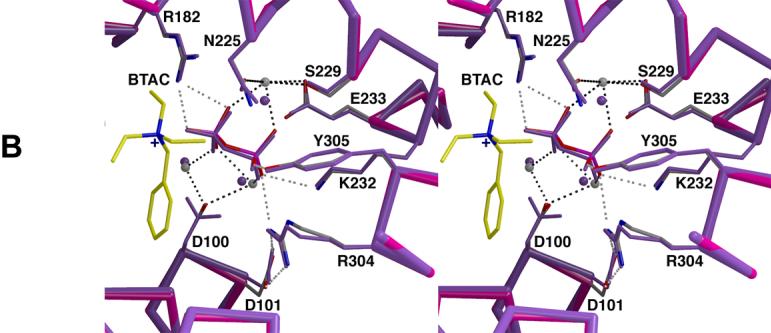
Active site of trichodiene synthase. (a) Simulated annealing omit maps of BTAC (blue, 4.5σ), PPi (green, 7.6σ), and magnesium ions A, B, and C (maroon, 4.1σ). Atoms are color-coded as follows: carbon (protein) = gray, carbon (BTAC) = yellow, oxygen = red, nitrogen = blue, phosphorus = magenta, magnesium = gray. (b) Superposition with the trichodiene synthase-Mg2+3-PPi complex (purple). Hydrogen bond and metal coordination interactions are represented by white and black broken lines, respectively.
Apart from a conformational change in the side chain of D100 that appears to weaken coordination interactions with Mg2+A and Mg2+C, the metal ion coordination and hydrogen bond networks formed in the Mg2+ 3-PPi-BTAC complex are similar to those observed in the Mg2+ 3-PPi complex [9]. The positively charged BTAC molecule is presumably stabilized by favorable long-range electrostatic interactions with the PPi anion: the closest PPi oxygen atom is 4.7 Å from the quaternary ammonium nitrogen atom of BTAC and 3.8 Å from the closest N-ethyl carbon atom of BTAC, which is notable insofar that the positive charge of the cation is inductively shared by its neighboring atoms. The phenyl ring of BTAC resides deep within the hydrophobic active site cleft, where it makes van der Waals contacts with the hydrophobic side chains of I70, L97, and F157.
Discussion
The crystal structure of trichodiene synthase complexed with BTAC and Mg2+ 3-PPi provides the first definitive proof that a terpenoid cyclase active site is capable of accommodating a positively charged ligand. Although the structures of trichodiene synthase mutants complexed with the presumably positively charged ligands R-azabisabolene or S-azabisabolene plus Mg2+ 3-PPi have previously been reported [11, 12], the protonation states of their tertiary amino nitrogen atoms cannot be determined in the 2.5-2.95 Ǻ resolution electron density maps of proteins. In contrast, the four hydrocarbon substituents of the quaternary ammonium group of BTAC are readily visible in the electron density map, thereby conclusively confirming that the ligand bound in the active site of trichodiene synthase is positively charged. Therefore, just as the trichodiene synthase active site can accommodate and stabilize a positively charged quaternary ammonium group, so too can it accommodate and stabilize a positively charged carbocation intermediate in catalysis.
In the presence of PPi, trichodiene synthase is inhibited by positively-charged aza compounds that partially mimic the shape and charge of a critical carbocation intermediate in the FPP cyclization reaction (Fig. 2); in the absence of PPi, these compounds are ineffective as inhibitors [10, current studies]. These aza compounds exhibit a 100-fold range of binding affinities in the presence of PPi (Table 2). Structural studies show that both the R- and the S-azabisabolene analogues are bound in a variety of orientations in the active sites of Y305F and R304K trichodiene synthases [11, 12]. Interestingly, while BTAC binds ∼15-fold less tightly than either R- or S-azabisabolene (Table 2, Fig. 4), it is bound in a single, well-defined orientation (Fig. 5), possibly due to the fact that it has three identical alkyl groups.
Structural comparisons of the trichodiene synthase-Mg2+ 3-PPi complex and the trichodiene synthase-Mg2+ 3-PPi complexes with bound ammonium ion analogues R-azabisabolene, S-azabisabolene, or BTAC, reveal that the average PPi-metal ion coordination distances increase from 2.3 Ǻ to 2.5 Ǻ when positively charged ammonium ligands bind in the enzyme active site. Thus, it appears that electrostatic interactions in the Mg2+ 3-PPi cluster are weaker when a positive charge is bound or generated in catalysis. The weakening of electrostatic interactions in the Mg2+ 3-PPi cluster, which “locks” the active site template in the closed conformation, could contribute to the anomalous binding modes observed for R- and S-azabisabolenes in complexes with Y305F and R304K trichodiene synthases [11, 12]. Moreover, it is possible that metal coordination interactions are weakened upon initial carbocation formation in the cyclization cascade. This could “loosen” the active site template and thereby account for the formation of aberrant cyclization products by wild-type and mutant trichodiene synthases. Additionally, this could facilitate the potential function of PPI as a base in the final step of catalysis and also facilitate product release.
The finding that wild-type trichodiene synthase generates 89% trichodiene and 11% side products (13 total) contrasts with our earlier observation, obtained with less sensitive instrumentation, indicating that cyclization of FPP generates trichodiene as the exclusive product [18, 19]. This low but real rate of formation of aberrant products that is enhanced in active site mutants is reminiscent of the properties of other wild-type sesquiterpene cyclases that have been found to generate multiple products. For example, aristolochene synthase from Penicillum roqueforti generates (S)-germacrene-A (4%) and (−)-valencene (2%) in addition to aristolochene, although aristolochene synthase from Aspergillus terreus generates 100% aristolochene [26]. The promiscuity of product formation exhibited by a terpene synthase can therefore vary even from one species to another. Strikingly, γ-humulene synthase from Abies grandis generates 52 products from FPP [6]. However, a relatively small number of mutations (5 or less) of the residues that define the active site contour of γ-humulene synthase can substantially enhance the proportion of a single cyclization product while decreasing the relative abundance of the remaining sesquiterpene coproducts, without significant decrease in the overall rate of product formation [27]. The occurrence of minor side products in the reaction catalyzed by a terpenoid cyclase therefore indicates the evolutionary potential of the cyclase to generate a significantly different product or product array. As noted by Petsko and colleagues [28], low levels of alternative catalytic activity provide a “toe hold” for the evolution and optimization of enzyme function toward such alternative activity.
The cyclization of farnesyl diphosphate by trichodiene synthase is initiated by the departure of the diphosphate group from C1, resulting in the generation of an allylic carbocation intermediate with the positive charge delocalized over C1-C3 (Fig. 1). The electron-withdrawing effect of the C2 fluorine atom in 2F-FPP is expected to destabilize the allylic cation and therefore retard if not abolish the cyclization reaction. This would be the case regardless of whether the enzyme utilizes the mechanism in Figure 1 or the mechanism recently proposed by Hong and Tantillo [29] for FPP cyclization. Nonetheless, 2F-FPP is converted to a mixture containing predominantly fluorinated sesquiterpene derivatives. The fact that 4M-FPP can also serve as a surrogate substrate further illustrates the ability of the trichodiene synthase active site to accommodate derivatives of the natural FPP substrate. Future work will focus on determining the structures of the sesquiterpene analogues that are generated from such alternative substrates and the kinetics of their formation. Coupled with binding studies of unnatural carbocation analogues such as BTAC, the current work sets a foundation for studying diversity in ligand binding and catalysis by trichodiene synthase.
Acknowledgements
We thank the Cornell High Energy Synchrotron Source and Elettra Synchrotron Light Source for access to X-ray crystallographic data collection facilities, we thank Dr. Barry Cooperman for the use of the scintillation counter, and we thank Kevin Jude, Tatiana Zakharian, and Xin Lin for helpful scientific discussions.
Footnotes
This work was supported by NIH grants GM56838 (D.W.C.), GM30301 (D.E.C.), and GM13956 (R.M.C.).
Atomic coordinates of wild-type trichodiene synthase complexed with BTAC and with inorganic pyrophosphate, resulting from the reaction with 2-fluorofarnesyl diphosphate, have been deposited in the Research Collaboratory for Structural Bioinformatics (http://www.rcsb.org/pdb) with the following accession codes: 2Q9Y and 2Q9Z, respectively.
Abbreviations: FPP, farnesyl diphosphate; PPi, inorganic pyrophosphate; 2FFPP, 2-fluorofarnesyl diphosphate; 4M-FPP, 4-methylfarnesyl diphosphate; BTAC, benzyl triethylammonium cation; GC, gas chromatography; MS, mass spectroscopy; r.m.s., root mean square.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cane DE. Acc. Chem. Res. 1985;18:220–226. [Google Scholar]
- 2.Croteau R, Cane DE. Methods in Enzymology. Steroids and Isoprenoids. Part A. Vol. 110. Academic Press; New York: 1985. pp. 383–504. [Google Scholar]
- 3.Cane DE. Chem. Rev. 1990;90:1089–1103. [Google Scholar]
- 4.Wendt KU, Schulz GE. Structure. 1998;6:127–133. doi: 10.1016/s0969-2126(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 5.Christianson DW. Chem. Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 6.Steele CL, Crock J, Bohlmann J, Croteau R. J. Biol. Chem. 1998;273:2078–2089. doi: 10.1074/jbc.273.4.2078. [DOI] [PubMed] [Google Scholar]
- 7.Hohn TM, VanMiddlesworth F. Arch. Biochem. Biophys. 1986;251:756–761. doi: 10.1016/0003-9861(86)90386-3. [DOI] [PubMed] [Google Scholar]
- 8.Cane DE, Swanson S, Murthy PPN. J. Am. Chem. Soc. 1981;103:2136–2138. [Google Scholar]
- 9.Rynkiewicz MJ, Cane DE, Christianson DW. Proc. Natl. Acad. Sci. U.S.A. 2001;98:13543–13548. doi: 10.1073/pnas.231313098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cane DE, Yang G, Coates RM, Pyun H-J, Hohn TM. J. Org. Chem. 1992;57:3454–3462. [Google Scholar]
- 11.Vedula LS, Rynkiewicz MJ, Pyun H-J, Coates RM, Cane DE, Christianson DW. Biochemistry. 2005;44:6153–6163. doi: 10.1021/bi050059o. [DOI] [PubMed] [Google Scholar]
- 12.Vedula LS, Cane DE, Christianson DW. Biochemistry. 2005;44:12719–12727. doi: 10.1021/bi0510476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whittington DA, Wise ML, Urbansky M, Coates RM, Croteau RB, Christianson DW. Proc. Natl. Acad. Sci. U.S.A. 2002;99:15375–15380. doi: 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Otwinowski Z, Minor W. Methods in Enzymology. Macromolecular Crystallography. Part A. Vol. 276. Academic Press; San Diego: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 15.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 16.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 17.Merritt EA, Bacon D. Methods in Enzymology. Macromolecular Crystallography. Part B. Vol. 277. Academic Press; San Diego: 1997. pp. 505–524. [DOI] [PubMed] [Google Scholar]
- 18.Cane DE, Xue Q, Fitzsimons BC. Biochemistry. 1996;35:12369–12376. doi: 10.1021/bi961344y. [DOI] [PubMed] [Google Scholar]
- 19.Cane DE, Shim JH, Xue Q, Fitzsimons BC, Hohn TM. Biochemistry. 1995;34:2480–2488. doi: 10.1021/bi00008a011. [DOI] [PubMed] [Google Scholar]
- 20.Hyatt DC, Youn B, Zhao Y, Santhamma B, Coates RM, Croteau RB, Kang CH. Proc. Natl. Acad. Sci. USA. 2007;104:5360–5365. doi: 10.1073/pnas.0700915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin Y, Williams DC, Croteau R, Coates RM. J. Am. Chem. Soc. 2005;127:7834–7842. doi: 10.1021/ja050592r. [DOI] [PubMed] [Google Scholar]
- 22.Meyers AI, Collington EW. J. Org. Chem. 1971;36:3044–3045. [Google Scholar]
- 23.Woodside AB, Huang Z, Poulter CD. Org. Synth. CV. 1993;8:616–620. [Google Scholar]
- 24.Ohnuma S, Ito M, Koyama T, Ogura K. Tetrahedron. 1989;45:6145–6160. [Google Scholar]
- 25.Cleland WW. In: Enzymes. Boyer PD, editor. Vol. 2. Academic Press; New York: 1970. pp. 1–65. [Google Scholar]
- 26.Felicetti B, Cane DE. J. Am. Chem. Soc. 2004;126:7212–7221. doi: 10.1021/ja0499593. [DOI] [PubMed] [Google Scholar]
- 27.Yoshikuni Y, Ferrin TE, Keasling JD. Nature. 2006;440:1078–1082. doi: 10.1038/nature04607. [DOI] [PubMed] [Google Scholar]
- 28.Petsko GA, Kenyon GL, Gerlt JA, Ringe D, Kozarich JW. Trends Biochem. Sci. 1993;18:372–376. doi: 10.1016/0968-0004(93)90091-z. [DOI] [PubMed] [Google Scholar]
- 29.Hong YJ, Tantillo DJ. Org. Lett. 2006;8:4601–4604. doi: 10.1021/ol061884f. [DOI] [PubMed] [Google Scholar]