

Abstract
Folate deficiency causes massive incorporation of uracil into human DNA (4 million per cell) and chromosome breaks. The likely mechanism is the deficient methylation of dUMP to dTMP and subsequent incorporation of uracil into DNA by DNA polymerase. During repair of uracil in DNA, transient nicks are formed; two opposing nicks could lead to chromosome breaks. Both high DNA uracil levels and elevated micronucleus frequency (a measure of chromosome breaks) are reversed by folate administration. A significant proportion of the U.S. population has low folate levels, in the range associated with elevated uracil misincorporation and chromosome breaks. Such breaks could contribute to the increased risk of cancer and cognitive defects associated with folate deficiency in humans.
Folate deficiency (erythrocyte folate <140 ng/ml or plasma folate < 3 ng/ml) is one of the most common vitamin deficiencies (1), occurring in approximately 10% of the U.S. population (2) and, according to two small studies, in nearly half of low-income (mainly African-American) elderly (3) and adolescents (4). Over half of young low-income women have folate intakes below the current Recommended Daily Allowance level (5).
Folate deficiency is associated with increased risk of colon, esophageal, and cervical cancer (6–8), although these epidemiological studies are not definitive. Supplementation with folate reduces the incidence of some precancerous lesions (7, 9–11). Diets high in fruits and vegetables, which are rich sources of folate (12) and other antimutagenic micronutrients (13, 14), are strongly protective against most types of cancer (13–15). The quarter of the population with the lowest fruit and vegetable intake has about twice the risk of developing most types of cancers as the quarter with the highest intake (13–15). Folate deficiency in humans induces extensive chromosome damage (16), fragile site expression (17), micronucleus formation (18, 19), and increased uracil levels in bone marrow cell DNA (20).
Folate is required for transferring one carbon units in the de novo synthesis of nucleotides. Low cytosolic levels of N5,N10-methylenetetrahydrofolate (the folate cofactor for thymidylate synthase) decreases synthesis of thymidylate (dTMP; ref. 21), increasing the cellular dUMP/dTMP ratio and DNA polymerase-mediated dUTP misincorporation into DNA (20, 22–24). Uracil is excised from DNA by uracil-DNA glycosylase and apyrimidinic endonuclease, generating transient single-strand breaks (nicks) that could result in a less repairable and more hazardous double-strand break if two opposing nicks are formed (25). Uracil repair-induced double-strand breaks are the likely cause of genetic deletions and duplications in dUTPase mutant Escherichia coli (26). Nicks are also created by glycosylase repair of lesions generated by oxidants, the major endogenous mutagens (14), so interactions between antioxidant and folate deficiencies are expected (27). Uracil incorporation and removal has been hypothesized to cause chromosome breaks in antifolate-treated tumor cells (22). However, the role of uracil misincorporation into DNA in vivo is unclear; previously reported levels of uracil in DNA vary from artifactually high (20, 24, 28) to undetected levels (29, 30). We have developed a novel technique employing GC/negative chemical ionization mass spectrometry that allows uracil levels in DNA to be accurately determined; the method minimizes artifact by reducing cytosine deamination and RNA hydrolysis to undetectable levels (31). Following a preliminary communication (32), we now report a detailed study on the role of uracil misincorporation in DNA damage induced by folate deficiency.
MATERIALS AND METHODS
Supplies.
Triethylamine, isooctane, and acetonitrile (>99%) were from Fluka; 3,5-bis(trifluoromethyl)benzyl bromide (BTFMBzBr) and HPLC-grade ethanol were obtained from Aldrich; uracil and N-lauroylsarcosine were purchased from Sigma; and phenol, chloroform, isoamyl alcohol, ammonium acetate were from Fisher Scientific. Uracil-DNA glycosylase (1 unit/μl) was purchased from Epicentre Technologies (Madison, WI), and proteinase K (20 units/mg), RNase A (50 units/mg), and RNase T1 (105 units/ml) were from Boehringer Mannheim. Internal standard, [13C,15N2]uracil (99.7% [13C,15N2]), was synthesized as described (32).
Splenectomized Patients and Sampling.
All work with human tissues was approved by the Committee for the Protection of Human Subjects at the University of California, Berkeley. Splenectomized human volunteers were recruited from Kaiser Permanente Hospital (Oakland, CA) through discharge records that indicated splenectomy as the primary discharge diagnosis. Individuals with a history of cancer were excluded. Out of 122 healthy splenectomized subjects, 22 subjects with the highest and lowest values for micronucleated erythrocytes were chosen (33) and categorized by initial erythrocyte folate levels. Folate-deficient and folate-sufficient groups did not differ significantly in age or sex. Individuals with pitted erythrocyte frequencies below 12%, indicative of regeneration of spleen function (34), were omitted from the data set. Blood was drawn by venipuncture into EDTA vacuutainers (Becton Dickinson) and stored at 4°C until processed. Aliquots of the whole blood samples were frozen at −80°C for DNA isolation. Blood samples were taken at the beginning and at 6, 7, and 8 weeks after daily supplementation with 5 mg of folic acid. Additional data were obtained from a splenectomized individual with Crohn disease (19). Frozen blood samples from this individual before and after supplementation with folinic acid (25 mg per day) and folic acid (15 and 5 mg per day) were obtained and analyzed for DNA uracil content.
Micronucleus Assay.
Blood (5–10 μl) was spread onto three or more precleaned glass microscope slides, air-dried, fixed in methanol for 5 min, and stained with acridine orange (35). RNA-positive erythrocytes (reticulocytes, 2,000) and RNA-negative erythrocytes (10,000) were scored for micronuclei as described (33). Reticulocyte frequency was estimated by counting the frequency of RNA-positive cells among 10,000 erythrocytes. Serum and erythrocyte folate and B12 levels were determined using a competitive radiobinding assay (Bio-Rad Quantaphase).
Sample Analysis.
DNA was extracted from whole blood samples (2–5 ml) by phenol-chloroform extraction and resuspended in 10 mM Tris·HCl/1 mM EDTA pH 8.0 (TE; ref. 36). Samples were frozen at −20°C until analysis. Uracil levels in whole blood DNA were determined as described (31). DNA (1–50 μg) was treated with 1 unit of uracil-DNA glycosylase in 50 μl of TE buffer for 1 hr at 37°C. After digestion, 300 pg [13C,15N2]uracil was added as an internal standard, and the samples were dried (1 hr) in a speed vac concentrator (Savant). The residue was resuspended in 50 μl of acetonitrile, 10 μl of triethylamine, and 1 μl of BTFMBzBr and shaken at 30°C for 25 min followed by the addition of 50 μl of water. N1,N3-(3,5- Bis[trifluoromethyl]benzyl)uracil, was extracted into 100 μl of isooctane and analyzed by GC/mass spectrometry in negative chemical ionization mode.
Data Analysis.
The log-transformed uracil and micronuclei data were analyzed for significance using a one-sided Student’s t test (assuming unequal variance) in Microsoft excel. The SEMs were calculated using the log-transformed values followed by antilog conversion to arithmetic numbers (thus SEMs cannot be less than 1.0). All values are given as the geometric mean ± SEM.
RESULTS
Chromosomal damage and breakage in erythroid cells can result in fragments of DNA that remain in the mature cell after enucleation (37) and form micronuclei, which can be easily scored. In humans, micronucleated erythrocytes and reticulocytes are normally removed by the spleen, but in splenectomized individuals, micronucleated erythrocytes remain in peripheral circulation and provide an index of chromosomal damage (18, 37). Splenectomy, as the result of traumatic injury, is not known to affect metabolism, and only otherwise healthy subjects were enrolled in this study. Samples for the micronucleus (n = 22 and 19 for pre- and postsupplementation, respectively) and DNA uracil assays (n = 19) were obtained from 22 splenectomized individuals. Of the initial 22 subjects, 19 completed the supplementation portion of the study. DNA uracil levels were also determined in three folate-deficient subjects with normal splenic function. Based on three separate measurements of erythrocyte folate levels over a 6-week baseline period, individuals were assigned to normal (erythrocyte folate >140 ng/ml, n = 14) or deficient (erythrocyte folate <140 ng/ml, n = 8) groups. Individuals were categorized using their presupplementation erythrocyte folate levels since erythrocyte levels are more indicative of long-term folate homeostasis than plasma folate. Blood DNA was analyzed for uracil content (31), and the results of this analysis are presented in Fig. 1A. Folate-deficient individuals had a geometric mean of 3,960,000 ± 507,000 uracils per diploid cell, 8-fold greater (P = 0.003) than the 498,000 ± 315,000 uracils per cell found in controls (n = 14). Bone marrow DNA, isolated from folate-deficient individuals, contained 4,400,000 uracils per cell (n = 3), 9-fold greater (P = 0.004) than the level of 480,000 uracils per cell (n = 7) measured in individuals with normal folate status. Three individuals had normal plasma folate levels and may not have been functionally folate-deficient despite having erythrocyte folate levels in the deficient range (<140 ng/ml). These individuals had the lowest DNA uracil levels in the presupplementation low folate group in Fig. 1A (□) and also had below-average micronucleated reticulocyte and erythrocyte frequencies.
Figure 1.

Uracil levels in DNA and micronuclei frequencies were elevated in folate-deficient subjects and were reduced by folate supplementation. Uracil and micronuclei values were determined in 25 human subjects as described (31, 37). Open and solid symbols represent levels before and after supplementation with 5 mg per day folic acid, respectively. Squares represent individuals with deficient erythrocyte folate levels but borderline plasma folate levels (6 ng/ml ≥ plasma folate ≥4 ng/ml). Triangles are averaged values before and after supplementation of an individual with Crohn disease (19). (A) Uracil levels before and after folate supplementation (5 mg per day). (B and C) Micronuclei values in reticulocytes (B) and erythrocytes (C) before and after folate supplementation.
To establish folate deficiency as the cause of both increased DNA uracil and increased micronucleus frequency, individuals were supplemented with folate (pteroylglutamic acid, 5 mg per day) for 8 weeks. Blood samples were collected at the end of 6, 7, and 8 weeks of supplementation from the 19 folate-replete individuals who completed the protocol. Plasma and erythrocyte folate levels were significantly increased by supplementation, as shown in Table 1. DNA uracil levels were reduced markedly by folate supplementation in individuals with the lowest presupplementation folate levels [before, 3,960,000 uracils per cell (n = 8); after, 186,000 ± 48,000 uracils per cell (n = 5); P = 0.0003; Fig. 1A). This decrease occurred rapidly in the single folate-deficient individual sampled shortly after folate supplementation began; within 6 days, uracil levels dropped from a presupplementation level of 2,906,000 to 489,000 uracils per cell. Supplementation of individuals with erythrocyte folate levels greater than 140 ng/ml significantly reduced the levels of uracil in blood DNA from 498,000 (n = 14) to 134,000 uracils in DNA per cell (n = 14, P = 0.001).
Table 1.
Folate and B12 status before and after folate supplementation
| Supplementation | Circulating
vitamin levels, ng/ml
|
||
|---|---|---|---|
| Plasma folate (n) | Erythrocyte folate (n) | Plasma B12 (n) | |
| Folate-deficient individuals | |||
| Before | 4.3 ± 0.8 (7) | 95.1 ± 9.7 (10) | 0.33 ± 0.07 (8) |
| After | 53.6 ± 9.8** (5) | 486.4 ± 79.3** (5) | 0.40 ± 0.12 (4) |
| Normal individuals | |||
| Before | 11.9 ± 1.3† (16) | 277.6 ± 30.6†† (16) | 0.56 ± 0.07 (16) |
| After | 67.4 ± 7.9** (15) | 457.6 ± 45.0* (15) | 0.52 ± 0.04 (15) |
Work with human tissues was approved by the Committee for the Protection of Human Subjects at the University of California, Berkeley. Folate and B12 levels were determined by competitive radiobinding assay (Bio-Rad). Individuals with erythrocyte folate levels of <140 ng/ml were classified as folate-deficient. Postsupplementation values are the average of values determined 6, 7, and 8 weeks after initiation of daily consumption of 5 mg of folic acid. Individuals with normal levels of folate had higher plasma B12 levels than did the folate deficient group (P = 0.051). Values are expressed as the mean ± SEM of the arithmetic data. Values significantly elevated in the postsupplementation groups compared with values in the presupplementation group are indicated by ∗ (P < 0.005) and ∗∗ (P < 0.0001). Values significantly elevated in normal individuals compared with values in folate-deficient individuals before supplementation are indicated by † (P < 0.001) and †† (P < 0.0001).
Micronucleated reticulocytes and erythrocytes were counted in blood smears (37) from splenectomized individuals of varying folate status. Micronucleus frequencies in reticulocytes and erythrocytes are illustrated in Fig. 1 B and C, respectively, with samples grouped according to the donor’s initial circulating folate levels. The frequency of micronucleated reticulocytes was 3-fold elevated (P = 0.012) in folate-deficient individuals (11.7 ± 1.4 micronucleated reticulocytes per 103 reticulocytes, n = 7) compared with control individuals (3.9 ± 1.1, n = 16). Similarly, the frequency of micronucleated erythrocytes was 3.3-fold elevated (P = 0.047) in folate-deficient individuals (3.7 ± 1.8 micronucleated erythrocytes per 103 erythrocytes, n = 7) compared with normal individuals (1.1 ± 1.2, n = 16). Supplementation of folate-deficient individuals lowered the frequency of micronucleated reticulocytes [before, 11.7 ± 1.4 (n = 7); after, 4.3 ± 1.4 (n = 5); P = 0.03] and erythrocytes [before, 3.7 ± 1.8 (n = 7); after, 1.9 ± 1.6 (n = 5); P = 0.21]. The longer half-life of erythrocytes may partially obscure supplementation-induced changes since many micronucleated erythrocytes formed before supplementation began would remain in circulation after 6–8 weeks (37). Though folate supplementation reduced uracil levels in most individuals, no change in the frequency of micronucleated reticulocytes or erythrocytes was observed in normal individuals with initial erythrocyte folate levels greater than 140 ng/ml [micronucleated reticulocytes per 103 reticulocytes: before, 3.9 ± 0.4 (n = 16); after, 4.5 ± 0.7 (n = 15); micronucleated erythrocytes per 103 erythrocytes: before, 1.1 ± 1.2 (n = 16); after, 1.1 ± 1.2 (n = 15)].
Additional data were obtained both before and after folate supplementation of a splenectomized patient with Crohn disease, a condition that results in the inhibition of dietary folate absorption and transport. Previous study of this individual revealed that he had very high levels of erythrocyte and reticulocyte micronuclei which were reversed by folate and folinic acid supplementation (19). Frozen blood samples from this study were used to investigate the relationship between uracil in DNA and micronuclei (Fig. 2). The basal level of uracil in DNA was significantly elevated (2,910,000 uracils per cell) in this folate-deficient subject (1.9 ng/ml plasma, 70 ng/ml erythrocyte folate), and DNA uracil was reduced by 6 days of folinic acid administration (25 mg per day) to 480,000 uracils per cell. Administration of folic acid (5 mg of pteroylglutamic acid per day) further reduced DNA uracil levels to 51,000 uracils per cell. The decline in DNA uracil levels closely paralleled a decline in micronucleated reticulocytes (before supplementation, 130 per 103 reticulocytes; after supplementation, 3.9 per 103 reticulocytes; Fig. 2). This association is consistent with the hypothesis that uracil in DNA causes double-strand DNA breaks, leading to micronucleus formation. These data, taken together with those illustrated in Fig. 1A, indicate that DNA uracil levels are closely associated with frequencies of micronuclei and that both are minimized by supplementation with folates.
Figure 2.

Uracil levels and micronuclei values were elevated in a subject with Crohn disease and reduced by supplementation of folinic and folic acid. Uracil was measured (31) in DNA isolated from frozen blood samples (19). Uracil levels: ⋄, before supplementation; ♦, after supplementation. Micronucleus frequency (19): ○, before supplementation; •, after supplementation.
DISCUSSION
These results provide strong evidence that folate status can be a key determinant of DNA strand breaks, leading to genetic instability (38) and thus to increased cancer risk (39). The data and analysis presented indicate that uracil misincorporation is a likely mechanism for folate-deficiency-induced cytogenetic damage, although other mechanisms, such as site-specific hypomethylation, could contribute (40, 41). The findings are likely to be applicable to proliferating tissues in general and suggest that, in folate-deficient humans, increased folate intake may decrease the risk of many types of cancer (9–11). Our results support the importance of folate nutriture in degenerative diseases, especially cancer and neurodegeneration.
Consistent with the experimental results on the relationship between uracil levels in DNA and chromosome breaks is a theoretical model of uracil misincorporation leading to double-strand breaks.** This model is based on studies which show that plasmid DNA containing 2 uracils within 14 base pairs on opposite strands is linearized when transfected into cells expressing uracil-DNA glycosylase (25). Our analysis predicts that the frequency of closely spaced uracil residues on opposite DNA strands increases markedly as uracil accumulates in DNA. Therefore, the estimated incidence of 2 closely spaced uracils on opposite strands is 50 times higher in folate-deficient individuals than in normal individuals. Simultaneous removal and strand scission of both closely spaced uracils could cause a double-strand DNA break (25) and would explain the increased levels of micronuclei found in folate-deficient individuals (this time factor is not included in the equation). Since semiconservative DNA replication results in much higher uracil levels in the daughter strands compared with the parental strands, our calculations assume a 1,000-fold difference. Uracil will be present in the parental strands due to carryover following previous rounds of cell division, misincorporation during excision repair and from in situ deamination of cytosine.
Oxidative DNA damage (≤500,000 adducts per diploid cell; unpublished work) should compound the effects of folate deficiency. Spontaneous oxidative damage is prevalent (14) and, unlike uracil misincorporation, is likely to be equally frequent on both strands. Therefore, we predict that increased oxidative damage would act synergistically with elevated levels of uracil in DNA, leading to higher levels of chromosome breaks in individuals deficient in both folate and antioxidants. Indeed, of the five individuals with the highest micronucleus values in this study, three were vitamin C-deficient (all five were folate-deficient; ref. 18). This interaction may be important since 10–15% of men in the United States, particularly African-American populations, have serum ascorbate levels (≤0.3 mg/dl) close to the scurvy threshold (42).
Many other micronutrients in addition to folate and ascorbate are likely to play a significant role in the prevention and repair of DNA damage, and thus to the maintenance of long-term health. Deficiency of vitamin B12 traps folate as N5-methyltetrahydrofolate and thus causes a functional folate deficiency, accumulation of homocysteine (43), and misincorporation of uracil into DNA (20). Niacin contributes to the repair of DNA strand breaks by maintaining nicotinamide adenine dinucleotide levels for the poly(A)DP-ribose protective response to DNA damage (44). As a result, dietary insufficiencies of niacin (15% of some populations are deficient; ref. 45), folate, and antioxidants may act synergistically to adversely affect DNA synthesis and repair. Diets deficient in fruits and vegetables are commonly low in folate, antioxidants (e.g., ascorbate), and many other micronutrients and result in significant amounts of DNA damage and higher cancer rates (12, 13, 15). On the other hand, strict vegetarians are at increased risk of developing a B12 deficiency (43).
In addition to increased cancer risk, inadequate folate intake increases the risk of heart disease. Inadequate folate intake decreases the folate-dependent methylation of homocysteine, resulting in elevated plasma homocysteine levels (46). Homocysteine accumulation is a major risk factor for cerebrovascular and cardiovascular disease (47), including fatal coronary heart disease (48). Elevated homocysteine is thought to be responsible for 10% of the U.S. population’s risk for coronary heart disease. Each 5 μM increase of plasma homocysteine is associated with an increase in coronary artery disease equivalent to a 20 mg/dl increase in cholesterol (49).
Adequate folate is crucial for neural development and function. A woman’s risk of having a child with a neural tube birth defect is associated with early pregnancy erythrocyte folate levels in an inverse dose response relationship; increased risk of birth defects were found at erythrocyte folate levels well into the range currently considered normal (50). Numerous studies have demonstrated that periconceptional folate supplementation reduces the prevalence of neural tube birth defects (51).
Folate and vitamin B12 are important for cognitive function. Elderly subjects with low serum folate and B12 concentrations have impaired spatial copying skills (52) and abstraction performance (53), and score worse on tests of nonverbal abstract thinking when compared with age-matched individuals with high serum folate and B12 concentrations (54). Folate-deficient mice also display behavioral abnormalities (55). Folate is found in high concentration in the central nervous system, and some studies have found an association between folate deficiency, psychogeriatric disease (56, 57), depression (58, 59), and the enhanced activity of neurotoxins (60). The effects of folate supplementation on cognitive function and behavior have not been investigated. It is difficult to predict the outcome of such studies as little is known about the reversibility of the damage induced by folate deficiency. Uracil in DNA could contribute to the effects of both folate and B12 deficiencies on brain function (61). Although deficiencies of folate or B12 may not result in uracil misincorporation into genomic DNA of postmitotic neurons, this process could occur in the DNA of dividing glial cells (oligodendrocytes). Loss of these cells might result in demyelination of central nervous system neurons, an analogous condition to that caused by B12 deficiency in peripheral neurons (43). Folate deficiency might also increase uracil misincorporation in neuronal mitochondrial DNA, resulting in an increase in nick formation and DNA deletions (26). This could have an impact on mitochondrial energy production and increase the generation of reactive oxygen species in neurons and other tissues. In addition, folate deficiency might reduce methylation of cellular DNA, proteins, and neurotransmitters (55, 62), and therefore impede neural function.
Measurement of metabolites whose production depends on the different folate pools (e.g., plasma homocysteine or uracil in DNA) provides a more accurate measure of deficiency than do circulating folate levels. Estimates of the potential public health impact of folate deficiency might be expected to rise if such functional folate determinations are used to assess adequate intake. Approximately 30–35% of elderly individuals have metabolic evidence of folate deficiency (elevated homocysteine), with low folate intake as the primary cause (46); supplementation with folate, B6, and B12 reduced homocysteine levels back to the normal range in 92% of those studied (63).
A common polymorphism in methylenetetrahydrofolate reductase, the enzyme responsible for reducing N5,N10-methylenetetrahydrofolate to the N5-methyl form, results in decreased activity in homozygotes and a 2-fold increase in plasma homocysteine. The homozygotes are 5–25% of individuals, depending on the population (64, 65), and appear to have an increased risk of heart disease, stroke (48, 49), and neural tube defects (65, 66). It is likely that this mutation increases N5,N10-methylenetetrahydrofolate at the expense of N5-methyltetrahydrofolate, resulting in decreased DNA uracil levels and increased serum homocysteine. The potential role of uracil misincorporation in human carcinogenesis is supported by two recent studies which demonstrate that individuals homozygous for the mutant alleles of methylenetetrahydrofolate reductase with high plasma folate levels have a 2- to 4-fold lower risk for colon cancer than wild-type controls with low plasma folate levels (67, 68). If the cognitive effects of folate (and B12) deficiency are indeed due to low methylenetetrahydrofolate pool-induced uracil misincorporation into DNA, the methylenetetrahydrofolate reductase polymorphism may have been selected for in populations chronically low in folate to protect the brain at the cost of early heart disease.
Our results indicate that folate-deficient humans have markedly elevated levels of uracil in DNA and chromosome breaks, and that supplementation effectively reduces these lesions. The optimum intake of folate and other micronutrients for long-term health is likely to be greater than that necessary to prevent overt symptoms of deficiency. Further research on larger populations is needed, with careful attention to discovering and avoiding the possible adverse effects of widespread folate supplementation (e.g., masking B12 deficiency). The data at hand clearly indicate that folate intake should be adequate to minimize DNA uracil and plasma homocysteine accumulation, resulting in reduced risk of chromosome breaks, cancer, heart disease, and brain damage. In much of the world, the diet does not meet this standard because of low folate content or poor bioavailability (69). In 1998, U.S. cereal and grain will be fortified with folic acid (140 μg/100 g). This is expected to result in a decrease in the number of elderly with folate intakes below 400 μg per day to 49% of the population; 66% of elderly currently consume less than 400 μg per day (70). If these findings are borne out in larger studies, fortification of the world’s grain with folate would be inexpensive and, in combination with nutritional education programs, might have a major impact on human health and well-being. Food for thought.
Acknowledgments
We thank K. Beckman, A. Bendich, G. Block, C. Butterworth, C. Cooney, D. Freedman, L. Gold, R. A. Jacob, S. J. James, M. Levine, R. Rozen, I. Rosenberg, and W. Willett for helpful comments. B.C.B. thanks the Biomedical Mass Spectrometry Unit at the University of New South Wales for financial support during the writing of a portion of this article. R.B.E. thanks the intramural programs of the National Institute of Environmental Health Sciences and National Health and Environmental Effects Research Laboratory for use of data and specimens gathered while he was with those programs. This work was supported by National Cancer Institute Outstanding Investigator Grant CA39910, by National Institute of Environmental Health Sciences Center Grant ESO1896 (B.N.A.), and by the intramural programs of the National Institute of Environmental Health Sciences and National Health and Enviromnental Effects Research Laboratory (R.B.E.).
Footnotes
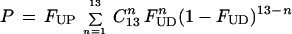 |
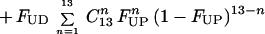 |
References
- 1.Hercberg S, Galan P. Bailliere’s Clin Haematol. 1992;5:143–168. doi: 10.1016/s0950-3536(11)80039-9. [DOI] [PubMed] [Google Scholar]
- 2.Senti F R, Pilch S M. J Nutr. 1985;115:1398–1402. doi: 10.1093/jn/115.11.1398. [DOI] [PubMed] [Google Scholar]
- 3.Bailey L B, Wagner P A, Christakis G J, Araujo P E, Appledorf H, Davis C G, Masteryanni J, Dinning J S. Am J Clin Nutr. 1979;32:2346–2353. doi: 10.1093/ajcn/32.11.2346. [DOI] [PubMed] [Google Scholar]
- 4.Bailey L B, Wagner P A, Christakis G J, Davis C G, Appledorf H, Araujo P E, Dorsey E, Dinning J S. Am J Clin Nutr. 1982;35:1023–1032. doi: 10.1093/ajcn/35.5.1023. [DOI] [PubMed] [Google Scholar]
- 5.U.S. Department of Agriculture. USDA Nationwide Food Consumption Survey: Continuing Survey of Intakes by Individuals II. U.S. Dept. Agriculture; 1986. USDA Report No. 86-4. [Google Scholar]
- 6.Lashner B A. J Cancer Res Clin Oncol. 1993;119:549–554. doi: 10.1007/BF01686465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butterworth C E. In: Micronutrients in Health and Disease. Bendich A, Butterworth C E, editors. New York: Dekker; 1993. pp. 165–185. [Google Scholar]
- 8.Van Helden P D, Beyers A D, Bester A J, Jaskiewicz H. Nutr Cancer. 1987;10:247–255. doi: 10.1080/01635588709513962. [DOI] [PubMed] [Google Scholar]
- 9.Heimburger D C, Alexander C B, Birch R, Butterworth C J, Bailey W C, Krumdieck C L. J Am Med Assoc. 1988;259:1525–1530. [PubMed] [Google Scholar]
- 10.Lashner B A, Heidenreich P A, Su G L, Kane S V, Hanauer S B. Gastroenterology. 1989;97:255–259. doi: 10.1016/0016-5085(89)90058-9. [DOI] [PubMed] [Google Scholar]
- 11.Butterworth C J, Hatch K D, Soong S J, Cole P, Tamura T, Sauberlich H E, Borst M, Macaluso M, Baker V. Ann NY Acad Sci. 1992;669:44–57. doi: 10.1016/0002-9378(92)91337-a. [DOI] [PubMed] [Google Scholar]
- 12.Subar A F, Block G, James L D. Am J Clin Nutr. 1989;50:508–516. doi: 10.1093/ajcn/50.3.508. [DOI] [PubMed] [Google Scholar]
- 13.Block G, Patterson B, Subar A. Nutr Cancer. 1992;18:1–29. doi: 10.1080/01635589209514201. [DOI] [PubMed] [Google Scholar]
- 14.Ames B N, Shigenaga M K, Hagen T M. Proc Natl Acad Sci USA. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ames B N, Gold L S, Willett W C. Proc Natl Acad Sci USA. 1995;92:5258–5265. doi: 10.1073/pnas.92.12.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Menzies R C, Crossen P E, Fitzgerald P H, Gunz F W. Blood. 1966;28:581–594. [PubMed] [Google Scholar]
- 17.Jacky P B, Beek B, Sutherland G R. Science. 1983;220:69–70. doi: 10.1126/science.6828880. [DOI] [PubMed] [Google Scholar]
- 18.MacGregor, J. T., Wehr, C. M., Hiatt, R. A., Peters, B., Tucker, J. A., Langlois, R. G., Jacob, R. A., Jensen, R. H., Yager, J. W., Shigenaga, M. K., Frei, B., Eynon, B. P. & Ames, B. N. (1997) Mutat. Res., in press. [DOI] [PubMed]
- 19.Everson R B, Wehr C M, Erexson G L, MacGregor J T. J Natl Cancer Inst. 1988;80:525–529. doi: 10.1093/jnci/80.7.525. [DOI] [PubMed] [Google Scholar]
- 20.Wickramasinghe S N, Fida S. Blood. 1994;83:1656–1661. [PubMed] [Google Scholar]
- 21.Das K C, Herbert V. Am J Hematol. 1989;31:11–20. doi: 10.1002/ajh.2830310103. [DOI] [PubMed] [Google Scholar]
- 22.Goulian M, Bleile B, Tseng B Y. Proc Natl Acad Sci USA. 1980;77:1956–1960. doi: 10.1073/pnas.77.4.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reidy J A. Mutat Res. 1988;200:215–220. doi: 10.1016/0027-5107(88)90085-1. [DOI] [PubMed] [Google Scholar]
- 24.Luzzatto L, Falusi A O, Joju E A. N Engl J Med. 1981;305:1156–1157. doi: 10.1056/NEJM198111053051918. [DOI] [PubMed] [Google Scholar]
- 25.Dianov G L, Timchenko T V, Sinitsina O I, Kuzminov A V, Medvedev O A, Salganik R I. Mol Gen Genet. 1991;225:448–452. doi: 10.1007/BF00261686. [DOI] [PubMed] [Google Scholar]
- 26.Sedwick W D, Brown O E, Glickman B W. Mutat Res. 1986;162:7–20. doi: 10.1016/0027-5107(86)90066-7. [DOI] [PubMed] [Google Scholar]
- 27.Smith M T. Eur J Haematol. 1996;57:107–110. doi: 10.1111/j.1600-0609.1996.tb01655.x. [DOI] [PubMed] [Google Scholar]
- 28.Ramsahoye B H, Burnett A K, Taylor C. Blood. 1996;87:2065–2070. [PubMed] [Google Scholar]
- 29.Green D A, Deutsch W A. Anal Biochem. 1984;142:497–503. doi: 10.1016/0003-2697(84)90495-0. [DOI] [PubMed] [Google Scholar]
- 30.Kirsh M E, Cutler R G, Hartman P E. Mech Aging Dev. 1986;35:71–77. doi: 10.1016/0047-6374(86)90067-9. [DOI] [PubMed] [Google Scholar]
- 31.Blount B C, Ames B N. Anal Biochem. 1994;219:195–200. doi: 10.1006/abio.1994.1257. [DOI] [PubMed] [Google Scholar]
- 32.Blount B C, Ames B N. Bailliere’s Clin Haematol. 1995;8:461–478. doi: 10.1016/s0950-3536(05)80216-1. [DOI] [PubMed] [Google Scholar]
- 33.Smith D F, MacGregor J T, Hiatt R A, Hooper N K, Wehr C M, Peters B, Goldman L R, Yuan L A, Smith P A, Becker C E. Cancer Res. 1990;50:5049–5054. [PubMed] [Google Scholar]
- 34.Pearson H A, Johnston D, Smith K A, Touloukian R J. N Engl J Med. 1978;298:1389–1392. doi: 10.1056/NEJM197806222982504. [DOI] [PubMed] [Google Scholar]
- 35.Hayashi M, Sofuni T, Ishidate M J. Mutat Res. 1983;120:241–247. doi: 10.1016/0165-7992(83)90096-9. [DOI] [PubMed] [Google Scholar]
- 36.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidmann J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. New York: Wiley; 1989. [Google Scholar]
- 37.Schlegel R, MacGregor J T, Everson R B. Cancer Res. 1986;46:3717–3721. [PubMed] [Google Scholar]
- 38.Branda R F, McCormack J J, Perlmutter C A, Mathews L A, Robison S H. Cancer Res. 1988;48:4529–4534. [PubMed] [Google Scholar]
- 39.Solomon E, Borrow J, Goddard A D. Science. 1991;254:1153–1160. doi: 10.1126/science.1957167. [DOI] [PubMed] [Google Scholar]
- 40.Mason J. In: Folate in Health and Disease. Bailey L B, editor. Vol. 1. New York: Dekker; 1995. pp. 361–378. [Google Scholar]
- 41.Kim Y-I, Pogribny I P, Basnakian A G, Miller J W, Selhub J, James S J, Mason J. Am J Clin Nutr. 1997;65:46–52. doi: 10.1093/ajcn/65.1.46. [DOI] [PubMed] [Google Scholar]
- 42.National Center for Health Statistics (1982) Vital Health Statistics, Series II (232) (U. S. Gov. Printing Office, Washington, DC), DHHS Publ. No. 83-1682.
- 43.Herbert V. In: Present Knowledge in Nutrition. Ziegler E E, Filer L J, editors. Washington, DC: Int. Life Sci. Inst. Press; 1996. pp. 191–205. [Google Scholar]
- 44.Zhang J Z, Henning S M, Swendseid M E. J Nutr. 1993;123:1349–1355. doi: 10.1093/jn/123.8.1349. [DOI] [PubMed] [Google Scholar]
- 45.Jacobson E L. J Am Coll Nutr. 1993;12:412–416. doi: 10.1080/07315724.1993.10718330. [DOI] [PubMed] [Google Scholar]
- 46.Selhub J, Jacques P F, Wilson P W, Rush D, Rosenberg I H. J Am Med Assoc. 1993;270:2693–2698. doi: 10.1001/jama.1993.03510220049033. [DOI] [PubMed] [Google Scholar]
- 47.Landgren F, Israelsson B, Lindgren A, Hultberg B, Andersson A, Brattstrom L. J Intern Med. 1995;237:381–388. doi: 10.1111/j.1365-2796.1995.tb01190.x. [DOI] [PubMed] [Google Scholar]
- 48.Morrison H I, Schaubel D, Desmeules M, Wigle D T. J Am Med Assoc. 1996;275:1893–1896. doi: 10.1001/jama.1996.03530480035037. [DOI] [PubMed] [Google Scholar]
- 49.Boushey C J, Beresford S A, Omenn G S, Motulsky A G. J Am Med Assoc. 1995;274:1049–1057. doi: 10.1001/jama.1995.03530130055028. [DOI] [PubMed] [Google Scholar]
- 50.Daly L E, Kirke P N, Molloy A, Weir D G, Scott J M. J Am Med Assoc. 1995;274:1698–1702. doi: 10.1001/jama.1995.03530210052030. [DOI] [PubMed] [Google Scholar]
- 51.Milunsky A, Jick H, Jick S S, Bruell C L, MacLaughlin D S, Rothman K J, Willett W. J Am Med Assoc. 1989;262:2847–2852. doi: 10.1001/jama.262.20.2847. [DOI] [PubMed] [Google Scholar]
- 52.Riggs K M, Spiro A, Tucker K, Rush D. Am J Clin Nutr. 1996;63:306–314. doi: 10.1093/ajcn/63.3.306. [DOI] [PubMed] [Google Scholar]
- 53.La Rue A, Koehler K M, Wayne S J, Chiulli S J, Haaland K Y, Garry P J. Am J Clin Nutr. 1997;65:20–29. doi: 10.1093/ajcn/65.1.20. [DOI] [PubMed] [Google Scholar]
- 54.Goodwin J S, Goodwin J M, Garry P J. J Am Med Assoc. 1983;249:2917–2921. [PubMed] [Google Scholar]
- 55.Gospe S M, Jr, Gietzen D W, Summers P J, Lunetta J M, Miller J W, Selhub J, Ellis W G, Clifford A J. Physiol Behav. 1995;58:935–941. doi: 10.1016/0031-9384(95)00156-d. [DOI] [PubMed] [Google Scholar]
- 56.Reynolds E H. Clin Haematol. 1976;5:661–696. [PubMed] [Google Scholar]
- 57.Ortega R M, Manas L R, Andres P, Gaspar M J, Agudo F R, Jimenez A, Pascual T. J Nutr. 1996;126:1992–1999. doi: 10.1093/jn/126.8.1992. [DOI] [PubMed] [Google Scholar]
- 58.Carney M W, Sheffield B F. Psychol Med. 1978;8:139–144. doi: 10.1017/s0033291700006711. [DOI] [PubMed] [Google Scholar]
- 59.Abou-Saleh M T, Coppen A. J Psychiatr Res. 1986;20:91–101. doi: 10.1016/0022-3956(86)90009-9. [DOI] [PubMed] [Google Scholar]
- 60.Manzo L, Locatelli C, Candura S M, Costa L G. Neurotoxicology. 1994;15:555–565. [PubMed] [Google Scholar]
- 61.Mazzarello P, Focher F, Verri A, Spadari S. Int J Neurosci. 1990;50:169–174. doi: 10.3109/00207459008987169. [DOI] [PubMed] [Google Scholar]
- 62.Botez M I, Young S N, Bachevalier J, Gauthier S. Nature (London) 1979;278:182–183. doi: 10.1038/278182a0. [DOI] [PubMed] [Google Scholar]
- 63.Naurath H J, Joosten E, Riezler R, Stabler S P, Allen R H, Lindenbaum J. Lancet. 1995;346:85–89. doi: 10.1016/s0140-6736(95)92113-3. [DOI] [PubMed] [Google Scholar]
- 64.Frosst P, Blom H J, Milos R, Goyette P, Sheppard C A, Matthews R G, Boers G J, den Heijer M, Kluijtmans L A, van den Heuvel L P, Rozen R. Nat Genet. 1995;10:111–113. doi: 10.1038/ng0595-111. [DOI] [PubMed] [Google Scholar]
- 65.Whitehead A S, Gallagher P, Mills J L, Kirke P N, Burke H, Molloy A M, Weir D G, Shields D C, Scott J M. Q J Med. 1995;88:763–766. [PubMed] [Google Scholar]
- 66.Posey D L, Khoury M J, Mulinare J, Adams M J, Jr, Ou C Y. Lancet. 1996;347:686–687. [PubMed] [Google Scholar]
- 67.Chen J, Giovannucci E, Kelsey K, Rimm E B, Stampfer M J, Colditz G A, Spiegelman D, Willett W C, Hunter D J. Cancer Res. 1996;56:4862–4864. [PubMed] [Google Scholar]
- 68.Ma, J., Stampfer, M. J., Giovannucci, E., Artigas, C., Hunter, D. J., Fuchs, C., Willett, W. C., Selhub, J., Hennekens, C. H. & Rozen, R. (1997) Cancer Res., in press. [PubMed]
- 69.Gregory J F. In: Folate in Health and Disease. Bailey L, editor. Vol. 1. New York: Dekker; 1995. pp. 195–235. [Google Scholar]
- 70.Tucker K L, Mahnken B, Wilson P W, Jacques P, Selhub J. J Am Med Assoc. 1996;276:1879–1885. doi: 10.1001/jama.1996.03540230029031. [DOI] [PubMed] [Google Scholar]