

Abstract
A serious drawback of tumor necrosis factor α (TNF) as a clinical antitumor agent is that it also has hypotensive activity. To overcome this problem, derivatives of its sister cytokine lymphotoxin (TNF-β or LT) were prepared. One of them, mutein 2 (Mut2) has a deletion of amino acids 1–7 but contains substituted amino acids, Met-Phe-Pro at positions 8–10 of the mature human LT. This mutein has no hypotensive activity at the maximum dose (10 mg/kg) tested on rats. In contrast, a much lower dose (1 mg/kg) of TNF and LT caused a significant blood pressure drop. In vivo studies revealed that Mut2 was more effective than TNF or LT against MethA (a mouse tumor line) as judged by the therapeutic ratio [calculated as LD50 (dose that kills 50% of the animals)/ED50 (dose that reduces the tumor size by 50%)]. With five other different mouse tumors and two different human tumors, Mut2 was also effective and the effectiveness was comparable or superior to that of TNF or LT. These results suggest the possibility that this derivative may be usable as a clinical antitumor agent without the serious side effects associated with TNF.
Various attempts have been made to create muteins of tumor necrosis factor (TNF) and lymphotoxin (LT) to improve their therapeutic values (1). Results of these experiments suggest that up to nine NH2-terminal amino acids of TNF can be removed without losing cytotoxic activity in vitro (2). The replacement of acidic amino acids at the NH2-terminal region with basic amino acids resulted in a better (less toxic with high antitumor activity) TNF (3, 4). Replacement of the COOH-terminal region amino acids (amino acids 141–146) with other amino acids often modified the in vitro cytotoxic activity of TNF. The addition of amino acids to TNF at the NH2-terminal position decreased a side effect of TNF (enhancement of metastasis) without diminishing its anticancer activity (5).
On the other hand, relatively limited information is available for LT because efficient expression in Escherichia coli has become possible only recently (6). In vitro cytotoxic activity was maintained even after removal of the NH2-terminal amino acids residues from LT (7). In contrast, the removal of the COOH-terminal region of LT generally resulted in loss of the cytotoxic activity (8). To the best of our knowledge, LT muteins have never been tested in vivo for their antitumor or hypotensive activities.
TNF is relatively ineffective against human cancers because its hypotensive activity prevents administration of doses that are high enough to cause tumor regression (9, 10). Systemic use of TNF causes hypotension in the experimental animals (11–14). In this paper, we describe an LT mutein (mutein 2, Mut2) that lacks hypotensive effects but has high antitumor activity. The ratio hypotensive dose/therapeutic dose for Mut2 was at least 9-fold higher than that of TNF suggesting the possible future clinical applicability of Mut2.
MATERIALS AND METHODS
Expression of TNF, LT, and Mut2 in E. coli.
TNF and LT genes were cloned from a human leukocyte genomic library by using the standard technique (15). LT mutein genes were constructed from the LT gene by modification of the DNA sequence corresponding to the NH2-terminal region (up to 11th amino acid). Each LT mutein DNA was expressed in E. coli HB101 with the trp promoter and pBluescript SKII(+) expression vector (Stratagene) as described (6).
For purification of the recombinant cytokines, E. coli HB101 containing the cytokine gene (20 g wet weight) were suspended in 500 ml of buffer containing 50 mM Tris·HCl (pH 7.5) and 30 mM NaCl and disrupted with a high-pressure homogenizer. The extract was centrifuged to remove debris, and the nucleic acids were precipitated with 0.75% polyethyleneimine. Protein fractions containing cytokines were then precipitated with 50% saturated (NH4)2SO4 and suspended in 20 mM sodium phosphate (pH 7.5). The protein solution was applied to a CM fast-flow column (26 mm × 50 mm) and the protein fractions containing cytokines were eluted with 0.3 M NaCl in 20 mM sodium phosphate buffer (pH 7.5). Cytokines were further purified by using a Sepharose S-200 column (26 mm × 600 mm) with saline. The purity of the peptide obtained was examined by SDS/polyacrylamide gel electrophoresis on a 15% gel, followed by the Coomassie brilliant blue R-250 staining. As shown in Fig. 1, no significant impurities were observed. The amount of lipopolysaccharide contamination in each sample was less than 1 ng/mg of protein, as determined by the limulus amebocyte lysate assay with commercial reagents (Seikagaku Kogyo, Tokyo). Since our LT and Mut2 were expressed in E. coli, they were not glycosylated whereas the native LT is (8). Sequences of all products were examined by the sequential NH2-terminal analysis (the Edman degradation) and the expected amino acid sequence was confirmed.
Figure 1.
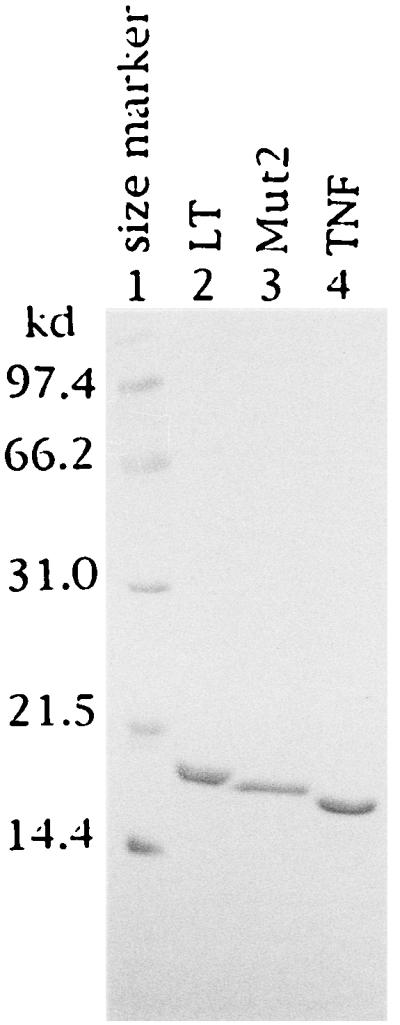
Polyacrylamide gel electrophoresis of purified recombinant LT, TNF, and Mut2. Each sample (3 mg) was suspended in sample buffer containing 5% 2-mercaptoethanol, 3% SDS, and 65 mM Tris·HCl (pH 6.8) and heated at 100°C for 5 min. The samples were subjected to SDS/PAGE (8 × 10 × 0.1 cm) in 15% gels for 2 hr at 150 V and stained with Coomassie brilliant blue R-250.
Blood Pressure Measurement.
Male Sprague–Dawley rats, 350–400 g and 11 weeks old, were obtained from the Charles River Breeding Laboratories. Male Sprague–Dawley rats were anesthetized with diethyl ether (Wako Pure Chemical, Osaka), and polyethylene catheters were implanted in the right femoral artery and vein as described (11). The rats were then left undisturbed for 1 hr to recover from anesthesia. Cytokines were dissolved in saline and LT (1 mg/kg), Mut2 (1–10 mg/kg, depending on the experiments), or TNF (1–10 mg/kg, depending on the experiments) was administered through the femoral venous catheter. The blood pressure was measured with the sensor in the catheter that was connected to a transducer (SBP 105, NEC Medical System, Tokyo) and a thermal array recorder (8M15, NEC Medical System) for 24 hr.
Assay of Antitumor Activities of TNF, LT, and Mut2.
The in vitro cytotoxic activities of each cytokine were measured with actinomycin D-treated murine L-929 cells as described (16). For the in vivo antitumor activity assay, 4- to 5-week-old female mice, C57BL/6, B6C3F1, ICR, BALB/c nu/nu (−), and Balb/c were purchased from CLEA Japan (Osaka) and Charles River Japan (Kanagawa) MethA (17), M5076 (18), B16 melanoma (19), Sarcoma180 (20) cells were maintained intraperitonially (i.p.), whereas Colon26 (21) and Colon38 (21) were kept subcutaneously (s.c.). Human tumors and HeLa S3 (22) and KB cells (23) were maintained in in vitro cell culture with Eagle’s essential medium supplemented with 10% fetal calf serum and antibiotics.
Cytokines were tested with the following combinations of tumor and mice. MethA (5 × 105 cells per mouse) or Colon26 (1 × 106 cells per mouse) were implanted s.c. in BALB/c mice. A fragment (approximately 2 × 2 × 2 mm) of Colon38 was implanted s.c. in B6C3F1 mice. M5076 cells (1 × 106 cells per mouse) were implanted s.c. in B6C3F1 mice. B16 cells (5 × 106 cells per mouse) were implanted s.c. in C57BL/6 mice. Sarcoma180 cells (1 × 106 cells per mouse) were implanted s.c. in ICR mice. HeLa S3 (3 × 106 cells per mouse) or KB (1 × 106 cells per mouse) cells were implanted s.c. in BALB/c nu/nu (−) mice. When the tumors, implanted in the left flank, became 4–8 mm in diameter (3–10 days after tumor implantation), the cytokine treatment was started by injecting various doses of cytokines intravenously, twice a week, for a total of five injections. Initial tumor weight is defined as the weight of the tumor at this point. Control mice received phosphate-buffered saline. Five to 10 mice were used per group. Mean tumor weights were calculated by using data derived from caliper measurements of length (a) and width (b) of tumors in millimeters according to the formula: tumor weight (mg) = a × b2/2 (21). In some cases, data were expressed as relative tumor weight = mean tumor weight at given time/initial mean tumor weight. The antitumor efficacy was expressed by a therapeutic index (TI, or ratio); TI = LD50/ED50. LD50 (the dose that kills 50% of the tumor bearing mice) and ED50 (the dose that causes 50% decrease of tumor weight) were calculated as described (24).
RESULTS
Structure and in Vitro Specific Activity of Mut2.
After extensive studies on in vivo antitumor and hypotensive activities of more than 30 muteins of TNF and LT, we obtained the best derivative Mut2 (MFP12A) that has no hypotensive activity but is still effective against tumors. Examples of unsatisfactory muteins are mutein 4 (MRFP12A) with weak cytotoxic activity in vitro and mutein 5 (MKKFP12A), mutein 13 (M12A), mutein 16 (P12A), mutein 17 (MF12A), and mutein 18 (M29K) with hypotensive activity. Mutein 18 had very high in vitro cytotoxic activity but no in vivo antitumor activity. Details of the structure–activity relationship regarding the sequence of various muteins we synthesized will be reported elsewhere. Table 1 shows the amino acid sequence of Mut2 in comparison with those of TNF and LT. Mut2 has a relatively simple NH2-terminal structure with methionine, phenylalanine, and proline residues at the positions 9–11 of mature human LT. TNF has a specific activity of 1.4 × 108 units/mg, whereas the specific activities of LT and Mut2 are approximately 3- to 4-fold greater than that. Mut2 is only half as active as LT in this assay but the in vivo antitumor activity of Mut2 is better than that of LT as described in the following sections. As noted above with mutein 18, in vitro cytotoxicity does not necessarily reflect the in vivo activity.
Table 1.
In vitro cytotoxic activity of Mut2, LT, and TNF
| Amino acid sequence | Amino acids, n | Specific activity, units/mg | Relative activity vs. TNF | |
|---|---|---|---|---|
| 1 12 172 | ||||
| LT | MLPGVGLTPSAA ----------------L | 172 | 5.80 × 108 | 4.14 |
| Mut2† | MFPA ----------------L | 164 | 3.72 × 108 | 2.66 |
| TNF | MVRSSSRTPSDK ∗∗∗∗∗∗∗∗∗∗∗L | 157 | 1.40 × 108 | 1.00 |
Dashes indicate the amino acid sequence of LT from residues 13 to 171. Asterisks indicate the amino acid sequence of TNF corresponding to residues 13–156.
Registered in DDBJ, AB000737.
Antitumor Effect of Mut2 Against Various Mouse Tumors.
In the experiment described in Fig. 2, 7 days after MethA was implanted, various amounts of Mut2 were injected intravenously. The tumor sizes were followed for 21 days after the tumor inoculation. Mut2 very effectively prevented the growth of tumors in a dose-dependent manner. At the most effective dose (3.75 mg/kg), 95.8% inhibition of the tumor growth was observed 21 days after tumor inoculation. In all the doses tested, except for the lowest dose, the effect was statistically significant as shown in Fig. 2.
Figure 2.

Antitumor effect of Mut2 on MethA. On day 7 after tumor inoculation, Mut2 was injected i.v. as indicated by the arrows. Each group consists of 5–10 mice. Dosages represent the amount per injection. □, Control, no drugs; ▴, 0.06 mg/kg; •, 0.23 mg/kg; ▪, 0.94 mg/kg; ○, 3.75 mg/kg. The t test was used. ∗, P < 0.05; ∗∗, P < 0.01; ∗∗∗, P < 0.005. The arrows represent days of Mut2 injection.
To examine other tumors of different origins, we chose two colon tumors (Colon26 and Colon38), a melanoma (B16), one ovary tumor (M5076), and Sarcoma180 (origin unknown). The results are shown in Table 2. The TI of Mut2 appears to be better than that of TNF or LT with almost all the tumors tested. The only exception was Sarcoma180. The reason for the relatively low TI with this tumor is that the LD50 of Mut2 was smaller than that of TNF. The reason for this high toxicity of Mut2 in the mice bearing this tumor is not known.
Table 2.
Antitumor effect of Mut2 on various mouse tumors in comparison with TNF or LT
| Tumor | Mut2
|
TNF
|
LT
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| ED50 | LD50 | TI | ED50 | LD50 | TI | ED50 | LD50 | TI | |
| Colon 26 | 0.3 | 6.9 | 23.0 | 0.3 | 1.5 | 5.0 | 0.3 | 2.3 | 7.7 |
| Sarcoma 180 | 0.2 | 1.2 | 6.0 | 0.2 | >4.0* | >20.0 | 0.3 | 1.2 | 4.0 |
| B16 | 7.3 | >12.0* | >1.6 | >1.3† | 1.2 | <0.9 | 11.9 | >12.0* | >1.0 |
| (ED16) | |||||||||
| M5076 | 0.8 | >12.0* | >15.0 | 0.9 | >4.0† | >4.4 | 3.0 | >12.0* | >4.0 |
| (LD20) | |||||||||
| Colon 38 | 1.4 | >12.0* | >8.6 | >3.0† | >3.0* | >1.0 | 6.6 | >12.0* | >1.8 |
| (ED9) | |||||||||
| Meth A | 0.32 | 7.5 | 23.4 | 0.3 | 1.1 | 3.7 | 0.62 | 4.0 | 6.5 |
Cytokines were given twice a week for a total of five injections. ED50 and LD50 values were expressed as milligrams per kilogram, and represent the dosages given at each injection.
No toxic death was observed with the dose indicated.
ED50 or LD50 was not determined. ED values represent the injection dose that gave maximum inhibition. For example, ED16 means the value that gave 16% inhibition of the tumor growth. Values noted beside LD indicate the percentage of animals that died with the dose administered. For example, LD20 indicates the dose that killed 20% of the animals. On day 1 tumor cells were implanted, and cytokine treatment was started on day 9 for Colon 26, on day 4 for Sarcoma 180, on day 11 for B16, and on day 10 for M5076 and Colon 38. The ED50 value was calculated by using relative tumor weight on day 14 for Colon 26, on day 20 for Sarcoma 180, on day 14 for B16 and M5076, and on day 25 for Colon 38, after the initial cytokine administration.
Some of the TI values in this table are not accurate because of the uncertainty about the values of ED50 or LD50. For example, ED50 of TNF on B16 melanoma could not be determined because under the experimental conditions used, only a 16% reduction in the tumor size was observed (ED16 at a dose of 1.33 mg/kg) even with the maximum dosage. Higher doses could not be given because of its toxicity (LD100 at 4 mg/kg and LD60 at 1.33 mg/kg). In a similar fashion, the LD50 of Mut2 or LT could not be determined with mice bearing M5076 because the maximum dose used (12 mg/kg) did not kill any animal. Therefore, we conclude that the antitumor activity of Mut2 with these varieties of tumors is at least as good as that of LT or TNF if not better.
Antitumor Effect of Mut2 Against Human Tumors.
The effect of Mut2 on human cancer in nude mice was compared with the effects of TNF and LT in Fig. 3. The statistically effective doses of TNF or LT are so toxic to the tumor bearing mice that all of them died within 5 days after administration of these cytokines. In contrast, approximately 60% inhibition of tumor growth was observed by administering 1.6 mg of Mut2 per kg with only a single death occurring out of five animals in the group. These experiments strongly suggest that Mut2 is more effective than LT or TNF against human tumors. Similar results were obtained with another human tumor, HeLa cells, transplanted on nude mice (TI of Mut2 was about 4 whereas the TI values of TNF and LT could not be measured because of their ineffectiveness).
Figure 3.

Antitumor effect of TNF, LT, and Mut2 against human tumor KB. From the day 10 on after tumor inoculation, cytokines were intravenously injected as indicated by the arrows in the figure. Each group consists of five mice. ○, Control, no drugs; ▵, 0.32 mg/kg; □, 1.6 mg/kg; •, 8.0 mg/kg. Doses represent the dose per injection.
Time Course of Blood Pressure Change After Administration of Mut2.
In the experiment described in Fig. 4, the time course of mean arterial blood pressure of rats after the intravenous injection of Mut2, LT, or TNF was determined. In confirmation of the previous report that blood pressure does not change within 6 hr after injection of TNF (11), we observed no significant effects of this cytokine up to 10 hr after the injection. However, a significant blood pressure drop was observed at 24 hr after the injection of LT or TNF. The extent of the blood pressure drop for both TNF and LT was almost identical. It is noted in Fig. 4 that the same amount of Mut2 did not change blood pressure at all.
Figure 4.

Time course of blood pressure change after administration of Mut2, TNF, or LT to rats. The blood pressure was measured at various times after administration of cytokines at 1 mg/kg. ○, Control, no drugs; ▪, TNF; □, LT; •, Mut2. ∗∗∗, P < 0.01. MABP, mean arterial blood pressure.
Table 3 shows the effect of various doses of cytokines on blood pressure. At each dose, the time course of the blood pressure change was studied as in Fig. 4, and the hypotensive effect at 24 hr after intravenous injection of cytokines was measured. We observed that Mut2 at 10 mg/kg, which effectively inhibits the growth of MethA (see Table 2), did not reduce the blood pressure at all. On the other hand, LT or TNF gave hypotensive effects even with the dose lower than that which inhibits the growth of MethA (see Table 2). In these experiments, rats were used instead of mice due to the technical difficulty of measuring blood pressure with the tumor-bearing mice. We feel that this is justified because the responses of mice to cytokines are similar to those of rats (25).
Table 3.
Lack of hypoptensive effect of various doses of Mut2
| Sample | Blood pressure change, %
|
||
|---|---|---|---|
| 1 mg/kg | 3 mg/kg | 10 mg/kg | |
| TNF | −16*** | −7.6* | −10.2** |
| LT | −6.9** | NT | NT |
| Mut2 | −1.8 | −1.5 | 0.9 |
Blood pressure changes 24 hr after cytokines injection are indicated as percentage drop (−) or increase (+) in comparison with the blood pressure of the control animals administered with saline. ∗, P < 0.05; ∗∗, P < 0.01; ∗∗∗, P < 0.001; NT, not tested.
As discussed in the Introduction, the most dangerous side effect with TNF in its clinical trial is its hypotensive activity. We therefore calculated the ratio of the minimum dose causing hypotension to the ED50 value. This ratio was called tentatively “safety index” to indicate the degree of safeness for each cytokine as a possible clinical antitumor agent. The safety indices of LT and TNF are 0.32 and 0.67, respectively, whereas that of Mut2 is larger than 6.3. The exact safety index of Mut2 could not be calculated because the amount of Mut2 that causes hypotension could not be measured as doses higher than 10 mg/kg could not be administered due to technical difficulties.
DISCUSSION
In continuing our attempts to create better cytokines that may be useful as antitumor agents (26–28), we present an LT mutein, Mut2, without hypotensive activity. We found that LT and TNF but not Mut2 caused significant hypotension 24 hr after administration to rats. We observed no blood pressure change up to 10 h, but it dropped significantly at 24 h after LT administration, as was observed with sheep at 6 h after LT injection (figure 1 of ref. 29). Previous studies found no pressure change 1 h after LT administration to rats, but blood pressure was not measured at later time periods (30). TNF causes a drop in blood pressure with sheep (29), dogs (12), rats (11), rabbits (13), and humans (9). The TI value (LD50/ED50) of Mut2, was around 24.2 whereas the TI values of TNF and LT were 3.5 and 6.6, respectively. The TI of the commonly used antitumor agent 5-fluoro-5′-deoxyuridine is about 9.1 (31). The high TI of Mut2 is partly due to its high LD50 values. However, loss of hypotensive activity may not be the major reason for the large LD50. Death by LT or TNF in rodents is believed to be due to the increased production of eicosanoids (32), leading to disseminated intravascular coagulation (33). The level of eicosanoids does not appear to be related to hypotension (32).
We postulate that the receptor for LT and TNF produces various cellular responses depending on the ligand, LT or TNF. Thus, LT and TNF exert completely different cell-physiological effects (34–37) despite the fact that these cytokines share the same cellular receptor (38–41). Although this may not apply to the recently reported cytokine LTβ (42, 43), which presumbly functions in complex with LT (44), one must conclude that LT and TNF induce completely different responses of the same cell through the same receptors. The major structural difference between TNF and LT is at the NH2-terminal end (45). We therefore reasoned that modifications of the NH2-terminal end of LT may result in a cytokine that is functionally different from LT. This reasoning was further supported by the x-ray crystallographic studies indicating that the NH2-terminal portions of LT and TNF are very flexible and are situated in the space between the receptor and the ligand (46). Altering the NH2-terminal regions influences these cytokines’ affinity for the receptor (47). Our hypotheses is that Mut2, due to its altered amino acid sequence, fails to yield the cellular signals leading to hypotension, even though it binds to the LT receptors that send signals leading to the antitumor activity.
It appears that TNF is part of a defense mechanism against fast-invading foreign “enemies” (32, 48–50). On the other hand, LT may be geared to control troubles such as viral but not bacterial invasions, in addition to other yet unknown disturbances. Consistent with this hypothesis is the fact that lipopolysaccharide (a product of invading bacteria) induces TNF but not LT (51). Lipopolysaccharide gives synergistic lethal and antitumor effects with TNF (ref. 52) but not LT (unpublished observation). TNF and LT have an antiviral effect (53), but only TNF has an antibacterial effect (50). Exogenously administrated antiserum against TNF did not facilitate tumor growth, suggesting that endogenous TNF does not play a major role in the body’s natural defense against tumors (49). Tumor-bearing animals are more prone to be killed by TNF than normal animals (54), but such increased susceptibility to LT in tumor-bearing animals does not occur (unpublished data). These considerations suggest that previous attempts to use TNF as an antitumor agent might have been doomed. On the other hand, Mut2, an LT derivative without hypotensive activity, may be developed to an effective antitumor agent.
ABBREVIATIONS
- TNF
tumor necrosis factor
- LT
lymphotoxin
- Mut2
mutein 2
- LD50
dose that kills 50% of the animals
- ED50
dose that reduces the tumor size by 50%
- TI
therapeutic index
References
- 1.Van Ostade X, Tavernier J, Fiers W. Protein Eng. 1994;7:5–22. doi: 10.1093/protein/7.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Creasey A A, Doyle L V, Reynolds M T, Jung T, Lin L S, Vitt C R. Cancer Res. 1987;47:145–149. [PubMed] [Google Scholar]
- 3.Soma G, Kitahara N, Tsuji Y, Kato M, Oshima H, Gatanaga T, Inagawa H, Noguchi K, Tanabe Y, Mizuno D. Biochem Biophys Res Commun. 1987;148:629–635. doi: 10.1016/0006-291x(87)90923-5. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura S, Kato A, Masegi T, Fukuoka M, Kitai K, Ogawa H, Ichikawa Y, Maeda M, Watanabe N, Kohgo Y, Niitsu Y. Int J Cancer. 1991;48:744–748. doi: 10.1002/ijc.2910480519. [DOI] [PubMed] [Google Scholar]
- 5.Miyata K, Kato M, Shikama H, Nishimura K, Sakae N, Kawagoe K, Nishikawa T, Kuroda K, Yamaguchi K, Aoyama Y, Mitsuishi Y, Yamada N. Clin Exp Metastasis. 1992;10:267–272. doi: 10.1007/BF00133562. [DOI] [PubMed] [Google Scholar]
- 6.Seow H-F, Goh C R, Krishnan L, Porter A G. Bio/Technology. 1989;7:363–368. [Google Scholar]
- 7.Aggarwal B B, Henzel W J, Moffat B, Kohr W J, Harkins R N. J Biol Chem. 1985;260:2334–2344. [PubMed] [Google Scholar]
- 8.Gray P W, Aggarwal B B, Benton C V, Bringman T S, Henzel W J, Jarrett J A, Leung D W, Moffat B, Ng P, Svedersky L P, Palladino M A, Nedwin G E. Nature (London) 1984;312:721–724. doi: 10.1038/312721a0. [DOI] [PubMed] [Google Scholar]
- 9.Saks S, Rosenblum M. Tumor Necrosis Factors: Structure, Function, and Mechanism of Action. New York: Marcel Dekker; 1991. pp. 567–587. [Google Scholar]
- 10.Spriggs D R, Yates S W. Tumor Necrosis Factors: The Molecules and Their Emerging Role in Medicine. New York: Raven; 1992. pp. 383–406. [Google Scholar]
- 11.Turner C R, Esser K M, Wheeldon E B, Slivjak M, Smith E F., III Circ Shock. 1989;28:369–384. [PubMed] [Google Scholar]
- 12.Nakatsuji K, Kii Y, Fujitani B, Ito T. Arzneim-forsch/Drug Res. 1990;40:218–225. [PubMed] [Google Scholar]
- 13.Weinberg J R, Wright D J M, Guz A. Clin Science (London) 1988;75:251–255. doi: 10.1042/cs0750251. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi K, Ando K, Ono A, Shimosawa T, Ogata E, Fujita T. Life Sci. 1992;50:1437–1444. doi: 10.1016/0024-3205(92)90262-n. [DOI] [PubMed] [Google Scholar]
- 15.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 16.Mosmann T. J Immunological Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 17.Carswell E A, Old L J, Kassel R L, Green S, Fiore N, Williamson B. Proc Natl Acad Sci USA. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Talmadge J E, Key M E, Hart I R. Cancer Res. 1981;41:1271–1280. [PubMed] [Google Scholar]
- 19.Goldin A, Venditti J M, MacDonald J S, Muggia F M, Henney J E, Devita V T., Jr Eur J Cancer. 1981;17:129–142. doi: 10.1016/0014-2964(81)90027-x. [DOI] [PubMed] [Google Scholar]
- 20.Foley G E, Drolet B P, McCarthy R E, Goulet K A, Dokos J M, Filler D A. Cancer Res. 1960;20:930–939. [PubMed] [Google Scholar]
- 21.Corbett T H, Griswold D P, Jr, Roberts B J, Peckham J C, Schabel F M., Jr Cancer. 1977;40:2660–2680. doi: 10.1002/1097-0142(197711)40:5+<2660::aid-cncr2820400940>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 22.Puck T T, Marcus P I, Cieciura S J. J Exp Med. 1956;103:273–284. doi: 10.1084/jem.103.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eagle H. Proc Soc Exp Biol Med. 1955;89:362–364. doi: 10.3181/00379727-89-21811. [DOI] [PubMed] [Google Scholar]
- 24.Chou T-C. J Theor Biol. 1976;59:253–276. doi: 10.1016/0022-5193(76)90169-7. [DOI] [PubMed] [Google Scholar]
- 25.Shimizu M, Uchiya N, Inoue A, Udaka K. The Clinical Report. 1985;19:7–8. [Google Scholar]
- 26.Matsuyama N, Okawa N, Tsukii Y, Endo T, Kaji A. FEBS Lett. 1992;302:141–144. doi: 10.1016/0014-5793(92)80425-g. [DOI] [PubMed] [Google Scholar]
- 27.Ito R, Matsumoto H, Uchida K, Kubo T, Tsukii Y, Endo T, Kaji A. Biochim Biophys Acta. 1991;1096:245–252. doi: 10.1016/0925-4439(91)90012-x. [DOI] [PubMed] [Google Scholar]
- 28.Morishige H, Ohkuma T, Kaji A. Biochim Biophys Acta. 1993;1151:59–68. doi: 10.1016/0005-2736(93)90071-7. [DOI] [PubMed] [Google Scholar]
- 29.Kreil E A, Greene E, Fitzgibbon C, Robinson D R, Zapol W M. Circ Res. 1989;65:502–514. doi: 10.1161/01.res.65.2.502. [DOI] [PubMed] [Google Scholar]
- 30.Ulich T R, del Castillo J, Keys M, Granger G A. Am J Pathol. 1987;128:5–12. [PMC free article] [PubMed] [Google Scholar]
- 31.Ishitsuka H, Miwa M, Takemoto K, Fukuoka K, Itoga A, Maruyama H B. Gann. 1980;71:112–123. [PubMed] [Google Scholar]
- 32.Fletcher J R, Collins J N, Graves E D, Luterman A, Williams M D, Izenberg S D, Rodning C B. Ann Surg. 1993;217:668–675. doi: 10.1097/00000658-199306000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tracey K J, Beutler B, Lowry S F, Merryweather J, Wolpe S, Milsark I W, Hariri R J, Fahey T J, III, Zentella A, Albert J D, Shires G T, Cerami A. Science. 1986;234:470–474. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- 34.Locksley R M, Heinzel F P, Shepard H M, Agosti J, Eessalu T E, Aggarwal B B, Harlan J M. J Immunol. 1987;139:1891–1895. [PubMed] [Google Scholar]
- 35.Seregina T M, Mekshenkov M I, Turetskaya R L, Nedospasov S A. Mol Immunol. 1989;26:339–342. doi: 10.1016/0161-5890(89)90089-8. [DOI] [PubMed] [Google Scholar]
- 36.Buck C, Digel W, Schoniger W, Stefanic M, Raghavachar A, Heimpel H, Porzsolt F. Leukemia. 1990;4:431–434. [PubMed] [Google Scholar]
- 37.De Togni P, Goellner J, Ruddle N H, Streeter P R, Fick A, Mariathasan S, Smith S C, Carlson R, Shornick L P, Strauss J-S, Russell J H, Karr R, Chaplin D D. Science. 1994;264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 38.Hohmann H-P, Remy R, Poschl B, van Loon A P G M. J Biol Chem. 1990;265:15183–15188. [PubMed] [Google Scholar]
- 39.Schoenfeld H-J, Poeschil B, Frey J R, Loetscher H, Hunziker W, Lustig A, Zulauf M. J Biol Chem. 1991;266:3863–3869. [PubMed] [Google Scholar]
- 40.Schall T J, Lewis M, Koller K J, Lee A, Rice G C, Wong G H W, Gatanaga T, Granger G A, Lentz R, Raab H, Kohr W J, Goeddel D V. Cell. 1990;61:361–370. doi: 10.1016/0092-8674(90)90816-w. [DOI] [PubMed] [Google Scholar]
- 41.Aggarwal B B, Eessalu T E, Hass P E. Nature (London) 1985;318:665–667. doi: 10.1038/318665a0. [DOI] [PubMed] [Google Scholar]
- 42.Browning J L, Ngam-ek A, Lawton P, DeMatinis J, Tizard R, Chow E P, Hession C, O’Brine-Greco B, Foley S F, Ware C F. Cell. 1993;72:847–856. doi: 10.1016/0092-8674(93)90574-a. [DOI] [PubMed] [Google Scholar]
- 43.Crowe P D, VanArsdale T L, Walter B N, Ware C F, Hession C, Ehrenfels B, Browning J L, Din W S, Goodwin R G, Smith C A. Science. 1994;264:707–710. [PubMed] [Google Scholar]
- 44.Browning J L, Dougas I, Ngam-ek A, Bourdon P R, Ehrenfels B N, Miatkowski K, Zafari M, Yampaglia A M, Lawton P, Meier W, Benjamin C P, Hession C. J Immunol. 1995;154:33–46. [PubMed] [Google Scholar]
- 45.Pennica D, Nedwin G E, Hayflick J S, Seeburg P H, Derynck R, Palladino M A, Kohr W J, Aggarwal B B, Goeddel D V. Nature (London) 1984;312:724–729. doi: 10.1038/312724a0. [DOI] [PubMed] [Google Scholar]
- 46.Banner D W, D’Arcy A, Janes W, Gentz R, Schoenfeld H-J, Broger C, Loetscher H, Lesslauer W. Cell. 1993;73:431–445. doi: 10.1016/0092-8674(93)90132-a. [DOI] [PubMed] [Google Scholar]
- 47.Wakabayashi T, Asada M, Nagasu T, Iijima A, Hasegawa Y, Shikata Y, Kitoh K. J Biol Chem. 1990;265:7604–7609. [PubMed] [Google Scholar]
- 48.Kolls J, Peppel K, Silva M, Beutler B. Proc Natl Acad Sci USA. 1994;91:215–219. doi: 10.1073/pnas.91.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sherry B A, Gelin J, Fong Y, Marano M, Wei H, Cerami A, Lowry S F, Lundholm K G, Moldawer L L. FASEB J. 1989;3:1956–1962. doi: 10.1096/fasebj.3.8.2721856. [DOI] [PubMed] [Google Scholar]
- 50.Havell E A. J Immunol. 1989;143:2894–2899. [PubMed] [Google Scholar]
- 51.Lieberman A P, Pitha P M, Shin H S, Shin M L. Proc Natl Acad Sci USA. 1989;86:6348–6352. doi: 10.1073/pnas.86.16.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rothstein J L, Schreiber H. Proc Natl Acad Sci USA. 1988;85:607–611. doi: 10.1073/pnas.85.2.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong G H W, Goeddel D V. Nature (London) 1986;323:819–822. doi: 10.1038/323819a0. [DOI] [PubMed] [Google Scholar]
- 54.Asher A, Mule J J, Reichert C M, Shiloni E, Rosenberg S A. J Immunol. 1987;138:963–974. [PubMed] [Google Scholar]