

Abstract
As in many other types of cells, retinal pigment epithelial (RPE) cells have an active ubiquitin—proteasome pathway (UPP). However, the function of the UPP in RPE remains to be elucidated. The objective of this study is to determine the role of the UPP in controlling the levels and activities of transcription factors hypoxia-inducible factor (HIF) and NF-κB. We inhibited the UPP with proteasome-specific inhibitors and determined the activation of HIF and NF-κB as well as the expression and secretion of pro-angiogenic factors. HIF-1α was not detectable in ARPE-19 cells under normal culture conditions. However, when proteasome activity was inhibited, HIF-1α accumulated in RPE in a time-dependent manner. Consistent with accumulation of HIF-1α in the cells, levels of mRNA for vascular endothelial growth factor (VEGF) and angiopoietin-2 (Ang-2) in RPE were up to 7-fold higher upon inhibition of the proteasome. Proteasome inhibition was also associated with a 2-fold increase in levels of mRNA for angiopoietin-1 (Ang-1). ARPE-19 cells secrete significant levels of VEGF under normal culture conditions. Inhibition of proteasome activity increased the secretion of VEGF by 2-fold. In contrast to the increase in HIF activity, NF-κB activation was reduced by proteasome inhibition. In addition, the expression and secretion of monocyte chemoattractant protein-1 (MCP-1) by RPE were substantially attenuated by the inhibition of proteasome activity. These data demonstrate that the UPP plays an important role in modulating the activities of HIF and NF-κB in the RPE. Consequences of an impairment of the UPP include accumulation of HIF-1α and diminished NF-κB activation, which lead to enhanced expression and secretion of pro-angiogenic factors and attenuated expression of MCP-1. Taken together, these data predict that the impairment of the UPP could lead to the development of AMD-related phenotypes.
Keywords: age-related macular degeneration, angiogenesis, signal transduction, retinal pigment epithelium, ubiquitin, proteasome, hypoxia-inducible factor, vascular endothelial growth factor, monocyte chemoattractant protein-1
1. Introduction
The ubiquitin-proteasome pathway (UPP) is the major non-lysosomal proteolytic pathway within cells (Glickman and Ciechanover, 2002; Ciechanover, 2003). In this pathway, proteins destined for degradation must be conjugated with a ubiquitin chain in order to be recognized and degraded by a large protease complex called the proteasome. Ubiquitin conjugation, or ubiquitination, is a highly ordered process in which a ubiquitin-activating enzyme (E1) first activates and transfers ubiquitin to a ubiquitin-conjugating enzyme (E2), which then acts in concert with one of a large family of ubiquitin protein ligases (E3) to transfer ubiquitin to a lysine residue on the target substrate (Pickart, 2001; Glickman and Ciechanover, 2002). In most cases, additional ubiquitins are conjugated to the initial ubiquitin moiety to form polyubiquitin chains. A chain of at least four ubiquitin moieties is required for substrate recognition by the 26S proteasome complex (Beal et al., 1996; Pickart, 1997; Lam et al., 2002). The proteasome complex consists of a 20S proteolytic core and typically two regulatory 19S caps. The 19S cap cleaves ubiquitin moieties from the substrate, unfolds the polypeptide and feeds it through the narrow channel of the proteolytic chamber of the 20S core (DeMartino and Slaughter, 1999).
The UPP is an important protein quality control system (Goldberg, 2003), which selectively degrades mutant, misfolded, or damaged proteins (Ward et al., 1995; Shang et al., 2001; Marques et al., 2004). Timely removal of abnormal or damaged proteins by the UPP is essential for the cells to with-stand and recover from various environmental stresses (Dudek et al., 2005; Shang et al., 2005). However, the UPP itself is also a target of such stresses. All of the three classes of ubiquitination enzymes (E1, E2 and E3) have a cysteine in their active sites and therefore the activities of these enzymes are subject to redox regulation (Jahngen-Hodge et al., 1997; Obin et al., 1998). S-Nitrosylation could also inactivate these enzymes (Yao et al., 2004). Reactive oxygen species and reactive lipid peroxidation products, such as 4-hydroxynonenal, also impair the function of the proteasome (Conconi et al., 1998; Okada et al., 1999; Caballero et al., 2003; Ishii et al., 2005). Since the UPP plays critical roles in almost every aspect of cellular function, abnormal regulation or inactivation of this pathway has been implicated in many age-related diseases, such as Alzheimer’s disease (Hope et al., 2003), Parkinson’s disease (Dawson and Dawson, 2003), and cataract (Jahngen-Hodge et al., 1992; Shang et al., 1997; Shang et al., 2001; Dudek et al., 2005).
Like most other types of cells, RPE have an active UPP. In a cell-free system, RPE supernatant is capable of degrading a variety of substrates for the UPP, such as histone 2A, oxidized RNase, transducin, and β-lactoglobulin (Obin et al., 1994, 1995). In contrast with many other types of cells, RPE have limited levels of free ubiquitin. Thus, addition of exogenous ubiquitin to RPE supernatant generated significantly higher levels of ubiquitin conjugates and also enhanced the proteasome-dependent proteolysis (Obin et al., 1994, 1995; Shang et al., 2005). The limited levels of endogenous ubiquitin may render the UPP in RPE more vulnerable to a variety of environmental stresses.
Age-related macular degeneration (AMD) is a complex and multifactorial disease and the leading cause of blindness in industrialized countries. The mechanism of this disease remains elusive and current therapeutic options are limited. An increasing body of literature indicates that development of AMD is related to dysfunction of RPE (Boulton et al., 2004; Zarbin, 2004; Zhou et al., 2005). One essential function of the postmitotic RPE is phagocytosis and degradation of shed photoreceptor outer segments. The completely degraded material is subsequently released at the basal side, where it is normally cleared by the choriocapillaris. Another function of RPE is to nurture photoreceptors and choriocapillaris by secreting various growth factors and cytokines, such as pigment epithelium-derived factor (PEDF), vascular endothelial growth factor (VEGF) and monocyte chemoattractant protein-1 (MCP-1) (Elner et al., 1997; Schlingemann, 2004).
Choroidal neovascularization (CNV) represents the most common cause of severe vision loss in AMD (Ambati et al., 2003a; Ishida et al., 2003). Maintenance of the physiological architecture of the choriocapillaris requires a well-regulated balance between various factors derived from the RPE, such as PEDF, VEGF, angiopoietin-1 (Ang-1) and angiopoietin-2 (Ang-2). VEGF, which can be induced by hypoxia, induces the formation of new blood vessels (Ishida et al., 2003), while PEDF serves as an inhibitory factor. In addition to VEGF and PEDF, recent results indicate that Ang-1 and Ang-2 and their receptor Tie-2 play a role in the formation of CNV (Hangai et al., 2001; Jain, 2003). Ang-1 promotes vascular integrity and maturation, inhibits apoptosis, and reduces vascular permeability. Ang-2 is an antagonist of Ang-1 and promotes VEGF-induced proliferation of endothelial cells. The receptor Tie-2, along with Ang-1 and Ang-2, was shown to be present in choroidal neovascular tissue (Hangai et al., 2001). The expression and secretion of VEGF, PEDF, Ang-1 and Ang-2 are under the control of the hypoxia-inducible factor (HIF), either directly or indirectly (Pugh and Ratcliffe, 2003). HIF is an αβ-heterodimer, first recognized as a DNA-binding factor that mediates hypoxia-induced activity of the erythropoietin 3′ enhancer (Semenza and Wang, 1992; Wang et al., 1995). In addition, the HIF system is a key regulator of a broad range of hypoxia-related processes, including angiogenesis (Wenger, 2002). Both HIF-1α and HIF-1β subunits exist as a series of isoforms encoded by distinct genetic loci. HIF-1β subunits are constitutive nuclear proteins, whereas HIF-1α subunits are induced by hypoxia. The abundance of HIF-1α subunits is mainly regulated by the rate of degradation. Oxygen-dependent hydroxylation at two proline residues (Pro402 and Pro564) of human HIF-1α mediates its interactions with a ubiquitin ligase (E3), von Hippel-Lindau (VHL), which targets HIF-1α for proteasomal degradation (Ivan et al., 2001; Jaakkola et al., 2001), and contributes to the extremely rapid proteolysis of HIF-1α in cells under normoxia conditions (Masson et al., 2001).
In addition to HIF, other transcription factors, such as NF-κB, are also under the control of the UPP. NF-κB proteins are present in the cytoplasm in association with inhibitory proteins called inhibitors of κB (I-κBs). Upon activation by a variety of inducers, the I-κB proteins are phosphorylated, ubiquitinated, and subsequently degraded by the proteasome (Maniatis, 1999; Tanaka et al., 2001). Degradation of I-κB allows the translocation of NF-κB into the nucleus and binding to its cognate DNA binding sites to regulate the transcription of a large number of genes, including those involved in angiogenesis, such as cytokines and stress response proteins.
Given the fact that several transcription factors, such as HIF-1α and NF-κB, are under the control of the UPP (Alkalay et al., 1995; Cho et al., 2001), we hypothesized that impairment of the UPP in RPE could result in altered gene expression that promotes the development of AMD-like phenotypes. To test this hypothesis, we inhibited proteasome activity in cultured RPE and assessed the effects on the expression and secretion of growth factors and chemokines that may be involved in the development of AMD. We demonstrate that inhibition of the UPP in RPE results in the accumulation of HIF-1α and an increase in the expression of VEGF and Ang-2. In addition, inhibition of the UPP in RPE also decreases the expression of MCP-1. Because overproduction of VEGF and deficiency in MCP-1 are both associated with development of AMD (Ambati et al., 2003b), these data support our hypothesis that impairment of the UPP in RPE could contribute to the development of AMD.
2. Materials and methods
2.1. Cell culture and treatments
The retinal pigment epithelial cell line ARPE-19 (Dunn et al., 1996) was obtained from ATCC. The cells were routinely maintained at 37 °C under 5% CO2 atmosphere and cultured in Dulbecco’s modified Eagle medium (DMEM; Gibco/BRL, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA) and containing 100 U/ml penicillin G and 100 μg/ml streptomycin. Most of the experiments were performed using subconfluent (∼90% confluence) RPE (Figs. 1-5). To determine if these findings pertain to differentiated RPE, the cells were cultured under confluent conditions for 3 weeks and then treated with proteasome inhibitors (Fig. 6). Before treatments, cell monolayers were rinsed once with PBS and fresh medium was added. For proteasome inhibition studies, MG132 (Calbiochem, La Jolla, CA, USA) and epoxomicin (Boston Biochem, Cambridge, MA, USA) were prepared in DMSO at 10 mM and diluted to10 μM (MG 132) and 5 μM (epoxomicin) in the culture medium immediately before use. Cells were incubated with proteasome inhibitors for different periods of time as indicated in the figure legends. For hypoxia studies, ARPE-19 cells were cultured in 21% or 1% oxygen for 16 h, or first in 1% for 16 h and then returned to 21% for 5 min. The hypoxic cell culture was performed in an INVIVO2 200 hypoxia working station (Biotrace, Bridgend, UK). To test the effect of HIF-1α levels on VEGF expression, sub-confluent RPE were transfected with empty pcDNA3 vector or pcDNA3-HIF-1α using Superfect™ transfect reagent (Qiagen, Valencia, CA, USA). Cellular RNA was extracted 48 h after transfection for determining VEGF mRNA levels.
Fig. 1.
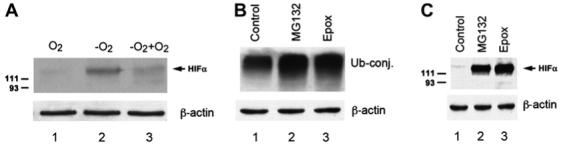
Hypoxia or inhibition of proteasome stabilizes HIF-1α in cultured RPE. (A) ARPE-19 cells were cultured in 21% oxygen (lane 1) or 1% oxygen for 16 h (lane 2), or first in 1% oxygen for 16 h and then returned to 21% oxygen for 5 min (lane 3). (B, C) ARPE-19 cells were cultured under normoxic conditions (21% oxygen) with or without proteasome inhibitors (MG132 or epoxomicin) for 8 h. Levels of endogenous ubiquitin conjugates (B) and HIF-1α (A, C) were determined by Western blotting analysis using a polyclonal anti-ubiquitin antibody or a monoclonal anti-HIF-1α antibody. (A) and (C) represent the data of five independent experiments with similar results whereas (B) was repeated twice with similar results.
Fig. 5.
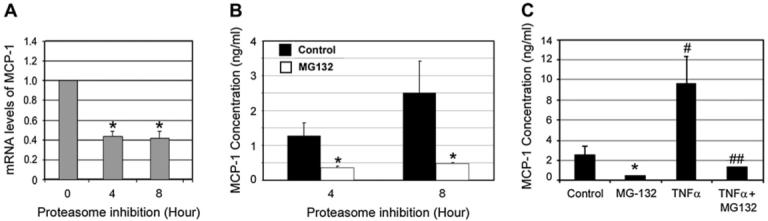
Proteasome inhibition down-regulates MCP-1 in RPE. ARPE-19 cells were cultured in the presence of MG132 for 0, 4, and 8 h. Levels of mRNA for MCP-1 were assessed by real-time RT-PCR analysis (A). GAPDH mRNA was used as a control to normalize the total mRNA levels. Levels of MCP-1 in the medium were detected by ELISA after the ARPE-19 cells were cultured in the presence or absence of MG 132 for 4 and 8 h (B) or after stimulation with or without TNFα in the absence or presence of MG132 for 8 h (C). The data presented is the result of three different experiments. *P < 0.01 in comparison with cells not treated with proteasome inhibitor in the absence of TNFα. #P < 0.01 in comparison between TNFα-treated and untreated cells in the absence of proteasome inhibitor. ##P < 0.01 in comparison between MG132-treated and untreated cells in the presence of TNFα.
Fig. 6.
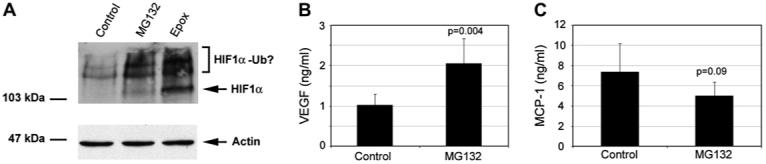
Proteasome inhibition in differentiated ARPE-19 stabilizes HIF-1α, increases VEGF secretion and decreases MCP-1 production. ARPE-19 cells were cultured under confluent conditions for 3 weeks and then treated with 10 μM MG132 or 5 μM epoxomicin for 8 h in fresh medium. Levels of HIF-1α were detected by Western blot (A). Levels of VEGF (B) and MCP-1 (C) in the medium were determined by ELISA. (A) represents the results of two independent experiments. (B) and (C) are given as mean ± S.D. of three different experiments.
2.2. Western blot analysis
Whole cell lysates were prepared for the use in Western blot analysis. After treatment, cells were rinsed once with ice-cold PBS and immediately collected in SDS loading buffer. Since SDS-gel loading buffer is not compatible with most of the commonly used protein quantification methods, we assessed the protein concentrations in the lysate by determining the densities of the protein bands on the gel after separation on SDS-PAGE and staining with Coomassie brilliant Blue R-250. Standard curves were established using 3 known concentrations of RPE supernatant. Cell lysates were then denatured at 100 °C for 3 min. Equal amounts of protein (20 μg) were resolved on 7.5-12% SDS-polyacrylamide gels and transferred to nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA, USA). Membranes were probed with rabbit polyclonal antibody against ubiquitin (produced in New Zealand White rabbits in this laboratory, 1:1000 dilution), or mouse monoclonal antibodies against HIF-1α (Novus Biologicals, Littleton, CO, USA) (1:500 dilution) and β-actin (Sigma, St. Louis, MO, USA) (1:10,000 dilution). After incubation with corresponding HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, USA), the specific bound antibody was visualized using Super Signal chemiluminescence detection kit (Pierce, Rockford, IL, USA).
2.3. Enzyme-linked immunosorbent assay (ELISA)
Levels of VEGF and MCP-1 secreted into the medium by RPE were determined by ELISA. The DuoSet ELISA kits were obtained from R&D Systems (Minneapolis, MN, USA). The medium was diluted 2 times for determining VEGF levels and 10 times for determining levels of MCP-1. All ELISAs were performed according to the manufacturer’s instructions.
2.4. Real-time RT-PCR
Total RNA was extracted from ARPE-19 cells using the RNeasy mini kit (Qiagen). Two micrograms of total RNA from each sample were used for reverse transcription using SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA, USA). The sequences of the primers used for real-time RT-PCR are summarized in Table 1. Real-time RT-PCR analysis was conducted on Stratagene Mx4000® multiplex quantitative PCR system using SYBR Green PCR master mix (Qiagen) according to the manufacturer’s instructions. The expression levels of the genes of interest were normalized using GAPDH as a housekeeping gene.
Table 1.
Primers for real-time RT-PCR
| Gene | Forward primer | Reverse primer | Tm (°C) | Size of product (bp) |
|---|---|---|---|---|
| VEGF | 5′-CAGAATCATCACGAAGTG | 5′-TCTGCATGGTGATGTTGGAC | 60 | 216 |
| Ang-1 | 5′-GTCGGAGATGGCCCAGATAC | 5′-GCTTTCTGGTCTGCTCTGCAG | 60 | 110 |
| Ang-2 | 5′-TTCCTCCTGCCAGAGATGGA | 5′-TGCACCGAGTCATCGTATTCG | 61 | 103 |
| MCP-1 | 5′AGATCTCAGTGCAGAGGCTCG | 5′-CACAGATCTCCTTGGCCACAA | 60 | 103 |
| GAPDH | 5′-TCACCATCTTCCAGGAGCGA | 5′-CTTCTCCATGGTGGTGAAGAC | 60 | 103 |
2.5. Preparation of nuclear extracts and DNA-binding assays
Following treatment of cell cultures, cells were rinsed with ice-cold PBS containing 1 mM EDTA and collected by scraping. After a brief centrifugation, the cell pellet was resuspended in ice-cold hypotonic lysis buffer (10 mM HEPES, pH 7.9, 0.1 mM EDTA, 0.1 mM EGTA, 10 mM KCl, 0.3% NP-40, 1 mM DTT, 1 mM AEBSF). After 15 min incubation on ice, the lysates were vortexed and centrifuged at 15,000 × g for 15 s to recover intact nuclei. The nuclear pellets were rinsed in the above buffer without NP-40 and then resuspended in ice-cold nuclear protein extraction buffer (20 mM HEPES, pH 7.9, 420 mM NaCl, 1 mM EDTA, 0.1 mM EGTA and a cocktail of protease inhibitors) and incubated on ice for an additional 30 min. The samples were centrifuged at 15,000 × g for 5 min. The resulting supernatants, the source of DNA-binding proteins, were mixed 1:1 with ice-cold 20 mM HEPES, pH 7.9, 20% glycerol, 100 mM KCl, 1 mM EDTA, 0.1 mM EGTA, 1% NP-40 and stored at -70 °C until use. Protein concentrations in the extracts were determined by the Coomassie Plus protein assay (Pierce), using bovine serum albumin as the standard.
Electrophoretic mobility shift assays were used to determine the DNA binding activity of NF-κB in the nuclear extracts. Briefly, equal amounts of nuclear extract (2 μg protein) were incubated for 20 min at RT with 10 fmol of 32P-labeled oligonucleotide (1 × 105 cpm) specific for NF-κB in 20 μl binding buffer (10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 1 mM MgCl2, 1 mM DTT, 1 mM EDTA, 4% glycerol, 1 μg poly(dI-dC), 1 μg BSA). The DNA-protein complexes formed were resolved on 5% nondenaturing polyacrylamide gels using 0.5× TBE (45 mM Tris-borate and 1 mM EDTA). Autoradiography was performed to visualize the shifted DNA-protein complexes. The NF-κB DNA-binding double-stranded oligonucleotide used was 5′-AGTTGAGGGGACTTTCCCAGGC-3′ (Promega, Madison, WI, USA). Specific DNA-binding complexes of NF-κB were identified as the band that disappeared when 50-fold excess of cold oligonucleotide competitor was added in binding assays. For supershift analysis, specific antibodies against NF-κB subunits were added for 30 min before binding reactions started and supershifts were resolved as indicated above. Antibodies used were RelA(p65) (sc-109) and NFκB1(p50) (sc-114) (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA).
2.6. Statistical analyses
Statistical analysis was performed using Student’s t-test assuming equal variances for all data points.
3. Results
3.1. Levels of HIF-1α in RPE are regulated in an oxygen-and proteasome dependent manner
HIF-1 is an αβ-heterodimer composed of HIF-1α and HIF-1β. HIF-1β subunit is a constitutive nuclear protein and therefore the HIF-1 activity is controlled by levels of HIF-1α. It has been demonstrated in other types of cells that the abundance of HIF-1α subunit is mainly regulated by oxygen-dependent hydroxylation and subsequent UPP-mediated degradation. However, the molecular regulation of HIF activity in RPE is poorly understood. To determine if HIF-1α levels in RPE cells are regulated in an oxygen dependent manner, confluent monolayers of ARPE-19 were cultured under normoxic (21% oxygen) or hypoxic conditions (1% oxygen) for 16 h and the levels of HIF-1α were determined by Western blotting. As in many other types of cells, HIF-1α was barely detectable in ARPE-19 cells when they were cultured under normoxic conditions (Fig. 1A and C, lane 1). In contrast, when RPE were cultured under hypoxic conditions (1% oxygen), HIF-1α was readily detected (Fig. 1A, lane 2). When hypoxia-treated ARPE-19 cells were returned to normoxic conditions, levels of HIF-1α decreased rapidly (Fig. 1A, lane 3). The half-life of HIF-1α was ∼5 min under normoxic conditions.
To further determine if levels of HIF-1α in RPE were regulated by UPP-mediated degradation, we assessed the effects of proteasome inhibition on HIF-1α levels in ARPE-19 cells cultured under normoxic conditions (21% oxygen). Upon treatment with 10 μM MG132 or 5 μM epoxomicin, two commonly used inhibitors of the proteasome, the levels of endogenous ubiquitin-protein conjugates increased ∼2-fold (Fig. 1B, compare lanes 2 and 3 with lane 1), indicating that proteasome activity is inhibited. Peptidase activity assays showed that the chymotrypsin-like activity of the proteasome was inhibited by 90% under these conditions. HIF-1α levels in RPE increased in a time-dependent manner upon inhibition of the proteasome by MG132 or epoxomicin and reached the maximum by 8 h. Fig. 1C shows HIF-1α levels in RPE upon 8 h of proteasome inhibition (compare lanes 2 and 3 with lane 1). High molecular weight species that react with anti-HIF-1α antibody were also detected upon inhibition of proteasome (similar to those shown in Fig. 6A). The high molecular weight species that react with HIF-1α are probably the ubiquitin-HIF-1α conjugates, but their identities remain to be further verified. The data show that levels of HIF-1α in RPE are controlled by UPP-mediated degradation and that impairment of the UPP in RPE can result in the accumulation of HIF-1α, even under normoxic conditions.
3.2. Proteasome inhibition in RPE results in enhanced expression and secretion of VEGF and other pro-angiogenic factors
To assess the physiological consequences of proteasome inhibition and elevated levels of HIF-1α in RPE, we examined the expression and secretion of VEGF, Ang-1 and Ang-2. The UPP plays important roles in many aspects of cellular functions. To avoid cytotoxicity associated with prolonged UPP inhibition, we determined the time course of VEGF expression and secretion upon proteasome inhibition (0, 2, 4, 8, 16 and 24 h). We found that the expression of VEGF started to increase after inhibiting the proteasome for 4 h and this increase lasted ∼16 h. If the proteasome was inhibited for longer than 16 h, the expression of VEGF, along with many other genes we have tested, decreased. The cross board decline in gene expression associated with prolonged proteasome inhibition might be caused by cytotoxicity of the inhibitors. Therefore, we only presented the data obtained upon 8 h of proteasome inhibition. As shown in Fig. 2A, mRNA levels for VEGF increased 3- or 6-fold upon hypoxia treatment or inhibition of the proteasome, respectively. Consistent with previous work (Zhou et al., 2005), we found that RPE cells secrete significant amounts of VEGF even under normal conditions. The concentration of VEGF-A165 in RPE-conditioned medium was 1.75 ng/ml under normal culture conditions (Fig. 2B). Under hypoxic conditions, the VEGF concentration in the medium increased to 2.8 ng/ml. If proteasome activity was inhibited, levels of VEGF in the medium increased to 3.5 ng/ml (Fig. 2B). Since both hypoxia and proteasome inhibition stabilize HIF-1α in RPE cells and both enhance the expression and secretion of VEGF, these data suggest that stabilization of HIF-1α and enhanced expression of VEGF are functionally related. To further test this hypothesis, we assessed the effect of HIF-1α levels on VEGF expression by overexpression of HIF-1α in RPE. As compared to RPE transfected with empty pcDNA3 vector, cells transfected with pcDNA3 vector encoding HIF-1α expressed higher levels of VEGF (∼2.5-fold) (Fig. 2C). Because only ∼40% of the cells were transfected under these conditions, the effect of HIF-1α on VEGF expression was underestimated. Taken together, these data indicate that accumulation of HIF-1α in RPE accounts, at least in part, for the enhancement in expression and production of VEGF upon proteasome inhibition. However, we cannot rule out the possibility that enhanced expression and secretion of VEGF upon proteasome inhibition is caused by other mechanisms, in addition to accumulation of HIF-1α.
Fig. 2.

Inhibition of proteasome in RPE increases expression and secretion of VEGF. ARPE-19 cells were cultured under normoxic conditions (21% oxygen) with or without MG132 for 8 h, or cultured under hypoxic conditions (1% oxygen) for 16 h. (C) ARPE-19 were transfected with empty pcDNA3 vector or pcDNA3-HIF-1α for 48 h. VEGF mRNA levels were determined by real-time RT-PCR assay (A and C). GAPDH mRNA was used as a control to normalize the total mRNA levels. VEGF protein levels in the medium were determined by ELISA (B). Results are given as mean ± S.D. of three different experiments. *P < 0.05, and **P < 0.01.
In addition to VEGF, other factors, such as Ang-1 and Ang-2, also play roles in the formation and remodeling of new vessels. As shown in Fig. 3, the expression of these angiogenic factors also transiently increased following proteasome inhibition. Upon inhibition of the proteasome for 4 h, Ang-1 and Ang-2 mRNA levels increased 2.5-fold and 6.8-fold, respectively (Fig. 3B and C). Unlike the prolonged over-expression of VEGF, the expression of Ang-1 and Ang-2 returned to basal levels when the proteasome was inhibited for longer than 8 h (Fig. 3B and C). It is known that expression of Ang-1 and Ang-2 is under the control of the HIF system, therefore the transient increase in expression of Ang-1 and Ang-2 by proteasome inhibition is probably mediated via the stabilization of HIF-1α in RPE cells. However, the down-regulation of these genes upon prolonged inhibition of the proteasome suggests that the expression of Ang-1 and Ang-2 may also be controlled by other unidentified mechanisms.
Fig. 3.
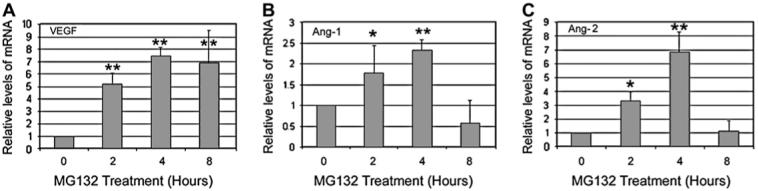
Time course of proteasome inhibition-induced expression of pro- angiogenic factors in RPE. ARPE-19 cells were cultured in the presence of MG132 for 0, 2, 4, and 8 h. Total RNA was extracted from these cells and levels of VEGF (A), Ang-1 (B) and Ang-2 (C) were determined by real-time RT-PCR analysis. GAPDH mRNA was used as a control to normalize the total mRNA levels. Results are given as mean ± S.D. of three different experiments. *P < 0.05, **P < 0.01.
3.3. Proteasome inhibition decreases NF-κB activation in RPE cells
We next determined whether proteasome inhibition affects NF-κB activation. As in many other types of cells, the activation of NF-κB in RPE is a transient event. Addition of fresh medium to the cells is sufficient to trigger the activation of NF-κB. This activation is likely induced by growth factors and cytokines in the medium. It is known that various cytokines can activate this pathway. The physical stress associated with medium change could be also a factor for NF-κB activation, since NF-κB is a known stress-responsive transcription factor. The pilot experiments showed that activation of NF-κB was detectable as early as 15 min upon stimulation and reached peak levels by 30-60 min. Thereafter the signal declined and was barely detectable by 4 h. Therefore, we chose 30 and 60 min time points to study the effect of proteasome inhibition on NF-κB activation in these experiments. ARPE-19 cells were cultured in the presence or absence of MG132 for 30 and 60 min, nuclear extracts were prepared and electrophoretic mobility shift assays were performed for NF-κB. As shown in Fig. 4A, NF-κB activity in control cells increased in a time-dependent manner, peaking at 1 h after changing the medium. When proteasome activity was inhibited by MG132, the levels of NF-κB in the nuclei were less than 50% of those found in control cells at each time point (Fig. 4A, compare lanes 3 and 4 with lanes 1 and 2, respectively). Upon stimulation with TNFα, a classic activator of the NF-κB pathway, NF-κB DNA-binding activity increased significantly (Fig. 4B, lanes 1 and 2). Treatment with proteasome inhibitor substantially attenuated the TNFα-induced activation of NF-κB (Fig. 4B, compare lanes 3 and 4 with lanes 1 and 2). The supershift assay demonstrated that the NF-κB dimmer in ARPE-19 cells was mainly the p65/p50 heterodimer (Fig. 4C). These data indicate that NF-κB activity in the RPE is also regulated by the UPP.
Fig. 4.

Proteasome inhibition decreases NF-κB activation. ARPE-19 cells were cultured in the presence or absence of MG132 for 30 and 60 min, nuclear extracts were prepared, and electrophoretic mobility shift assays were performed for NF-κB binding (A). The specific DNA-binding complexes of NF-κB were identified as the band that disappeared in the presence of 50-fold excess of cold oligonucleotide competitor in the binding assay (A, lane 5). The effects of TNFα and proteasome inhibition on NF-κB activity are shown in (B). Supershift analysis using specific antibodies to RelA(p65) and NF-κB1(p50) (C). Arrows indicate NF-κB-specific DNA-binding complexes and the corresponding supershifted DNA complexes identifying RelA(p65) and NFkB1(p50) as protein subunits of NF-κB. The figure represents the data of three independent experiments with similar results.
3.4. Inhibition of the UPP in RPE reduces the expression of MCP-1
It is known that the expression of MCP-1 is under the control of NF-κB (Uetama et al., 2003). Given the role of the UPP in regulating NF-κB activity, we assessed the effects of proteasome inhibition on the expression of MCP-1. ARPE-19 cells were cultured in the presence of MG132 for 4 or 8 h. MCP-1 mRNA levels were assessed by real-time RTePCR analysis and protein levels were determined by ELISA. As shown in Fig. 5A, levels of MCP-1 mRNA decreased significantly after proteasome inhibition for 4 h or longer. Under normal conditions, levels of MCP-1 in the medium increased in a time-dependent manner (Fig. 5B), indicating that the RPE cells continuously secrete MCP-1. Upon proteasome inhibition, the concentration of MCP-1 secreted by RPE decreased 70-80% as compared to untreated controls (Fig. 5B). To confirm that the decreased expression of MCP-1 upon inhibition of the proteasome is caused by inhibition of NF-κB activation, we tested the effect of TNFα on expression of MCP-1. As predicted, levels of MCP-1 increased dramatically upon incubation of ARPE-19 with TNFα (Fig. 5C). Even in the presence of TNFα, proteasome inhibition reduced the production of MCP-1 (Fig. 5C). These data suggest that the decreased MCP-1 production by ARPE-19 cells upon inhibition of the proteasome is related to down-regulation of NF-κB activity.
3.5. Proteasome inhibition in differentiated ARPE-19 is associated with accumulation of HIF-1α, increased production of VEGF and decreased production of MCP-1
It is known that ARPE-19 differentiates and acquires many RPE-like properties with prolonged time in culture at confluence. To determine if the data obtained using undifferentiated ARPE-19 cells pertains to differentiated RPE, ARPE-19 cells were cultured at confluence for 3 weeks and then we assessed the effects of proteasome inhibition on levels of HIF-1α, production of VEGF and MCP-1. Fig. 6 shows that, as in undifferentiated ARPE-19, proteasome inhibition also resulted in accumulation of HIF-1α (Fig. 6A), increased production of VEGF (Fig. 6B), and decreased production of MCP-1 (Fig. 6C). However, the responses to proteasome inhibition in differentiated RPE were less dramatic than those found in undifferentiated RPE. The attenuated responses of differentiated RPE to proteasome inhibitors may partially be related to the decreased cell permeability of these inhibitors, since the RPE were surrounded by large quantity of extracellular matrix under confluent conditions.
4. Discussion
Pro-angiogenic factors produced by RPE, such as VEGF, play a central role in CNV development (Campochiaro et al., 1999; Witmer et al., 2003). Although oxidative stress appears to play a role in triggering expression of these growth factors (Beatty et al., 2000; Zhou et al., 2005), the molecular mechanisms underlying this molecular change remain unknown. Since the UPP is involved in regulating several signal transduction pathways and its activity can be compromised upon oxidative stress, we hypothesize that oxidative damage to the UPP in RPE is a potential factor for stress-induced expression of proangiogenic factors. Our current study tested this hypothesis by inhibiting the UPP in RPE and assessing its effects on signal transduction and on the expression of VEGF, Ang-1 and Ang-2. Consistent with our hypothesis, we found that inhibiting proteasome activity stabilized HIF-1α (Figs. 1 and 6) and diminished the activation of NF-κB (Fig. 4) in RPE. Consequently, the expression and secretion of VEGF by RPE increased at least 2-fold (Figs. 2 and 6). The expression of Ang-1 and Ang-2 also transiently increased upon inhibition of the proteasome (Fig. 3). We also found that the expression and secretion of MCP-1 by RPE decreased (60-80%) when the UPP function was impaired (Fig. 6).
It is well known that the HIF system is a key transcriptional regulator of pro-angiogenic factors, such as VEGF (Pugh and Ratcliffe, 2003) and that HIF-1α is a substrate for the UPP. In other types of cells, the levels of HIF-1α are mainly regulated by oxygen-dependent and UPP-mediated degradation (Salceda and Caro, 1997; Semenza, 2003; Hagg and Wennstrom, 2005). The current work demonstrates that HIF-1α in RPE is also regulated in an UPP-dependent manner. Consistent with this finding, a recent study showed that hypoxia could markedly increase the expression of HIF-1α in RPE cells (Wang et al., 2005). These data suggest that if the UPP function is compromised, RPE cells behave as if they were under hypoxic conditions, expressing and secreting high levels of pro-angiogenic factors (Blaauwgeers et al., 1999; Mousa et al., 1999; Schlingemann, 2004; Vinores et al., 2006).
Regulation of pro-angiogenic factors is controlled by various transcription factors. The stabilization of HIF-1α is likely one of the mechanisms of the increased VEGF expression and secretion by RPE upon proteasome inhibition. However, it is not clear how proteasome inhibition enhanced the expression of Ang-1 and Ang-2. Although expression of Ang-2 is upregulated under hypoxia conditions (Mandriota et al., 2000), the molecular structure of hypoxia-responsive element has not been identified in the promoter region of Ang-2. Ang-1 and Ang-2 are a pair of closely related pro-angiogenic factors and they share a common receptor, Tie-2. Unlike VEGF, Ang-1 does not promote proliferation of endothelial cells, but it inhibits apoptosis of endothelial cells and promotes migration of endothelial cells and the maturation of newly formed vessels. Ang-2, by antagonizing the function of Ang-1, promotes VEGF-induced endothelial proliferation and sprouting (Hangai et al., 2001). Because Ang-1 and Ang-2 compete for the same receptor (Tie-2), the net effect may be determined by the Ang-1/Ang-2 ratio. Although both Ang-1 and Ang-2 were transiently up-regulated upon proteasome inhibition, the extent of the up-regulation is different. Whereas expression of Ang-1 increased by 2.5-fold, expression of Ang-2 increased up-to 7-fold upon proteasome inhibition, resulting in a dramatic decrease in the Ang-1/Ang-2 ratio. The parallel regulation in the expression of Ang-1 and Ang-2 upon proteasome inhibition suggests that the cells have a self-control mechanism that maintains the ratios of Ang-1/Ang-2 in a certain range. Data from the present study indicate that impairment of the UPP function in RPE could trigger the expression and secretion of growth factors that favor the proliferation and migration of endothelial cells and the angiogenesis process.
A recent study showed that deficiency in MCP-1 or its cognate receptor Ccr-2 causes AMD-like features in mice, including accumulation of RPE-lipofuscin, drusen, atrophy and CNV (Ambati et al., 2003b). Since MCP-1 expression is controlled by NF-κB (Ueda et al., 1994; Barnes and Karin, 1997; Martin et al., 1997; Marumo et al., 1999) and NF-κB activation is regulated by the UPP, we expected that inhibition of proteasome would down-regulate MCP-1 expression. Consistent with our prediction, we found that proteasome inhibition reduced NF-κB activation as demonstrated by decreasing binding of NF-κB to DNA (Fig. 4), and decreased MCP-1 secretion by RPE (Fig. 5). Consistent with the role of NF-κB in regulating MCP-1 expression, activation of NF-κB by TNFα increased the expression of MCP-1 (Fig. 5C). This result is consistent with a previous report that showed that MCP-1 gene expression was blocked by proteasome inhibitors in endothelial cells (Parry et al., 1998).
Taken together, these data suggest that the UPP plays an important role in regulating gene expression in RPE. It is plausible that impairment of the UPP by oxidative stress or other insults may trigger the development of CNV and other features of AMD. However, further studies are needed to elucidate if impairment of the UPP in RPE is related to AMD in animals or humans.
Acknowledgments
This work is supported by NIH grants EY 11717 (to F.S.) and EY 13250 (to A.T.), a grant from the Portuguese Foundation for Science and Technology POCI/SAU-OBS/57772/2004 (to P.P.), and US.D.A CRIS 1950-51000-060-01A. A.F. is a recipient of a Fellowship from the Portuguese Foundation for Science and Technology (SFRH/BD/19039/2004). The authors thank Dr. E. Whitcomb for her critical reading of the manuscript.
References
- Alkalay I, Yaron A, Hatzubai A, Orian A, Ciechanover A, Ben-Neriah Y. Stimulation-dependent IκB phosphorylation marks NF-κB inhibitor for degradation via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA. 1995;92:10599–10603. doi: 10.1073/pnas.92.23.10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambati J, Ambati BK, Yoo SH, Ianchulev S, Adamis AP. Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv. Ophthalmol. 2003a;48:257–293. doi: 10.1016/s0039-6257(03)00030-4. [DOI] [PubMed] [Google Scholar]
- Ambati J, Anand A, Fernandez S, Sakurai E, Lynn BC, Kuziel WA, Rollins BJ, Ambati BK. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat. Med. 2003b;9:1390–1397. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Beal R, Deveraux Q, Xia G, Rechsteiner M, Pickart C. Surface hydrophobic residues of multiubiquitin chains essential for proteolytic targeting. Proc. Natl. Acad. Sci. USA. 1996;93:861–866. doi: 10.1073/pnas.93.2.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000;45:115–134. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- Blaauwgeers HG, Holtkamp GM, Rutten H, Witmer AN, Koolwijk P, Partanen TA, Alitalo K, Kroon ME, Kijlstra A, van Hinsbergh VW, Schlingemann RO. Polarized vascular endothelial growth factor secretion by human retinal pigment epithelium and localization of vascular endothelial growth factor receptors on the inner choriocapillaris. Evidence for a trophic paracrine relation. Am. J. Pathol. 1999;155:421–428. doi: 10.1016/S0002-9440(10)65138-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton M, Roanowska M, Wess T. Ageing of the retinal pigment epithelium: implications for transplantation. Graefes Arch. Clin. Exp. Ophthalmol. 2004;242:76–84. doi: 10.1007/s00417-003-0812-8. [DOI] [PubMed] [Google Scholar]
- Caballero M, Liton PB, Epstein DL, Gonzalez P. Proteasome inhibition by chronic oxidative stress in human trabecular meshwork cells. Biochem. Biophys. Res. Commun. 2003;308:346–352. doi: 10.1016/s0006-291x(03)01385-8. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Soloway P, Ryan SJ, Miller JW. The pathogenesis of choroidal neovascularization in patients with age-related macular degeneration. Mol. Vis. 1999;5:34. [PubMed] [Google Scholar]
- Cho S, Choi YJ, Kim JM, Jeong ST, Kim JH, Kim SH, Ryu SE. Binding and regulation of HIF-1alpha by a subunit of the proteasome complex, PSMA7. FEBS Lett. 2001;498:62–66. doi: 10.1016/s0014-5793(01)02499-1. [DOI] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin proteolytic system and pathogenesis of human diseases: a novel platform for mechanism-based drug targeting. Biochem. Soc. Trans. 2003;31:474–481. doi: 10.1042/bst0310474. [DOI] [PubMed] [Google Scholar]
- Conconi M, Petropoulos I, Emod I, Turlin E, Biville F, Friguet B. Protection from oxidative inactivation of the 20S proteasome by heat-shock protein 90. Biochem. J. 1998;333(Pt 2):407–415. doi: 10.1042/bj3330407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- DeMartino GN, Slaughter CA. The proteasome, a novel protease regulated by multiple mechanisms. J. Biol. Chem. 1999;274:22123–22126. doi: 10.1074/jbc.274.32.22123. [DOI] [PubMed] [Google Scholar]
- Dudek EJ, Shang F, Valverde P, Liu Q, Hobbs M, Taylor A. Selectivity of the ubiquitin pathway for oxidatively modified proteins: relevance to protein precipitation diseases. FASEB J. 2005;19:1707–1709. doi: 10.1096/fj.05-4049fje. [DOI] [PubMed] [Google Scholar]
- Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp. Eye Res. 1996;62:155–169. doi: 10.1006/exer.1996.0020. [DOI] [PubMed] [Google Scholar]
- Elner VM, Burnstine MA, Strieter RM, Kunkel SL, Elner SG. Cell-associated human retinal pigment epithelium interleukin-8 and monocyte chemotactic protein-1: immunochemical and in-situ hybridization analyses. Exp. Eye Res. 1997;65:781–789. doi: 10.1006/exer.1997.0380. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Hagg M, Wennstrom S. Activation of hypoxia-induced transcription in normoxia. Exp. Cell Res. 2005;306:180–191. doi: 10.1016/j.yexcr.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Hangai M, Murata T, Miyawaki N, Spee C, Lim JI, He S, Hinton DR, Ryan SJ. Angiopoietin-1 upregulation by vascular endothelial growth factor in human retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 2001;42:1617–1625. [PubMed] [Google Scholar]
- Hope AD, de Silva R, Fischer DF, Hol EM, van Leeuwen FW, Lees AJ. Alzheimer’s associated variant ubiquitin causes inhibition of the 26S proteasome and chaperone expression. J. Neurochem. 2003;86:394–404. doi: 10.1046/j.1471-4159.2003.01844.x. [DOI] [PubMed] [Google Scholar]
- Ishida S, Usui T, Yamashiro K, Kaji Y, Amano S, Ogura Y, Hida T, Oguchi Y, Ambati J, Miller JW, Gragoudas ES, Ng YS, D’Amore PA, Shima DT, Adamis AP. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J. Exp. Med. 2003;198:483–489. doi: 10.1084/jem.20022027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T, Sakurai T, Usami H, Uchida K. Oxidative modification of proteasome: identification of an oxidation-sensitive subunit in 26s proteasome. Biochemistry. 2005;44:13893–13901. doi: 10.1021/bi051336u. [DOI] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Jahngen-Hodge J, Cyr D, Laxman E, Taylor A. Ubiquitin and ubiquitin conjugates in human lens. Exp. Eye Res. 1992;55:897–902. doi: 10.1016/0014-4835(92)90016-l. [DOI] [PubMed] [Google Scholar]
- Jahngen-Hodge J, Obin M, Gong X, Shang F, Nowell T, Gong J, Abasi H, Blumberg J, Taylor A. Regulation of ubiquitin conjugating enzymes by glutathione following oxidative stress. J. Biol. Chem. 1997;272:28218–28226. doi: 10.1074/jbc.272.45.28218. [DOI] [PubMed] [Google Scholar]
- Jain RK. Molecular regulation of vessel maturation. Nat. Med. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- Lam YA, Lawson TG, Velayutham M, Zweier JL, Pickart CM. A proteasomal ATPase subunit recognizes the polyubiquitin degradation signal. Nature. 2002;416:763–767. doi: 10.1038/416763a. [DOI] [PubMed] [Google Scholar]
- Mandriota SJ, Pyke C, Di Sanza C, Quinodoz P, Pittet B, Pepper MS. Hypoxia-inducible angiopoietin-2 expression is mimicked by iodonium compounds and occurs in the rat brain and skin in response to systemic hypoxia and tissue ischemia. Am. J. Pathol. 2000;156:2077–2089. doi: 10.1016/S0002-9440(10)65079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T. A ubiquitin ligase complex essential for the NF-kappaB, Wnt/Wingless, and Hedgehog signaling pathways. Genes Dev. 1999;13:505–510. doi: 10.1101/gad.13.5.505. [DOI] [PubMed] [Google Scholar]
- Marques C, Pereira P, Taylor A, Liang JN, Reddy VN, Szweda LI, Shang F. Ubiquitin-dependent lysosomal degradation of the HNE-modified proteins in lens epithelial cells. FASEB J. 2004;18:1424–1426. doi: 10.1096/fj.04-1743fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin T, Cardarelli PM, Parry GC, Felts KA, Cobb RR. Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-kappa B and AP-1. Eur. J. Immunol. 1997;27:1091–1097. doi: 10.1002/eji.1830270508. [DOI] [PubMed] [Google Scholar]
- Marumo T, Schini-Kerth VB, Busse R. Vascular endothelial growth factor activates nuclear factor-kappaB and induces monocyte chemoattractant protein-1 in bovine retinal endothelial cells. Diabetes. 1999;48:1131–1137. doi: 10.2337/diabetes.48.5.1131. [DOI] [PubMed] [Google Scholar]
- Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001;20:5197–5206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousa SA, Lorelli W, Campochiaro PA. Role of hypoxia and extracellular matrix-integrin binding in the modulation of angiogenic growth factors secretion by retinal pigmented epithelial cells. J. Cell Biochem. 1999;74:135–143. [PubMed] [Google Scholar]
- Obin M, Nowell T, Taylor A. The photoreceptor G-protein transducin (Gt) is a substrate for ubiquitin-dependent proteolysis. Biochem. Biophys. Res. Commun. 1994;200:1169–1176. doi: 10.1006/bbrc.1994.1574. [DOI] [PubMed] [Google Scholar]
- Obin M, Nowell T, Taylor A. A comparison of ubiquitin-dependent proteolysis of rod outer segment proteins in reticulocyte lysate and a retinal pigment epithelial cell line. Curr. Eye Res. 1995;14:751–760. doi: 10.3109/02713689508995796. [DOI] [PubMed] [Google Scholar]
- Obin M, Shang F, Gong X, Handelman G, Blumberg J, Taylor A. Redox regulation of ubiquitin-conjugating enzymes: mechanistic insights using the thiol-specific oxidant diamide. FASEB J. 1998;12:561–569. doi: 10.1096/fasebj.12.7.561. [DOI] [PubMed] [Google Scholar]
- Okada K, Wangpoengtrakul C, Osawa T, Toyokuni S, Tanaka K, Uchida K. 4-Hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress. Identification of proteasomes as target molecules. J. Biol. Chem. 1999;274:23787–23793. doi: 10.1074/jbc.274.34.23787. [DOI] [PubMed] [Google Scholar]
- Parry GC, Martin T, Felts KA, Cobb RR. IL-1beta-induced monocyte chemoattractant protein-1 gene expression in endothelial cells is blocked by proteasome inhibitors. Arterioscler. Thromb. Vasc. Biol. 1998;18:934–940. doi: 10.1161/01.atv.18.6.934. [DOI] [PubMed] [Google Scholar]
- Pickart CM. Targeting of substrates to the 26S proteasome. FASEB J. 1997;11:1055–1066. doi: 10.1096/fasebj.11.13.9367341. [DOI] [PubMed] [Google Scholar]
- Pickart CM. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- Schlingemann RO. Role of growth factors and the wound healing response in age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 2004;242:91–101. doi: 10.1007/s00417-003-0828-0. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang F, Gong X, Palmer HJ, Nowell TR, Taylor A. Age-related decline in ubiquitin conjugation in response to oxidative stress in the lens. Exp. Eye Res. 1997;64:21–30. doi: 10.1006/exer.1996.0176. [DOI] [PubMed] [Google Scholar]
- Shang F, Nowell TR, Jr., Taylor A. Removal of oxidatively damaged proteins from lens cells by the ubiquitin-proteasome pathway. Exp. Eye Res. 2001;73:229–238. doi: 10.1006/exer.2001.1029. [DOI] [PubMed] [Google Scholar]
- Shang F, Deng G, Liu Q, Guo W, Haas AL, Crosas B, Finley D, Taylor A. Lys6-modified ubiquitin inhibits ubiquitin-dependent protein degradation. J. Biol. Chem. 2005;280:20365–20374. doi: 10.1074/jbc.M414356200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Kawakami T, Tateishi K, Yashiroda H, Chiba T. Control of IkappaBalpha proteolysis by the ubiquitin-proteasome pathway. Biochimie. 2001;83:351–356. doi: 10.1016/s0300-9084(01)01237-8. [DOI] [PubMed] [Google Scholar]
- Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T, Matsunaga K, Fukushima J, Kawamoto S, Ishigatsubo Y, Okubo T. NF-kappa B and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J. Immunol. 1994;153:2052–2063. [PubMed] [Google Scholar]
- Uetama T, Ohno-Matsui K, Nakahama K, Morita I, Mochizuki M. Phenotypic change regulates monocyte chemoattractant protein-1 (MCP-1) gene expression in human retinal pigment epithelial cells. J. Cell Physiol. 2003;197:77–85. doi: 10.1002/jcp.10342. [DOI] [PubMed] [Google Scholar]
- Vinores SA, Xiao WH, Aslam S, Shen J, Oshima Y, Nambu H, Liu H, Carmeliet P, Campochiaro PA. Implication of the hypoxia response element of the VEGF promoter in mouse models of retinal and choroidal neovascularization, but not retinal vascular development. J. Cell Physiol. 2006;206:749–758. doi: 10.1002/jcp.20525. [DOI] [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Li H, Yan H, Xiao JG. Genistein inhibited hypoxia-inducible factor-1alpha expression induced by hypoxia and cobalt chloride in human retinal pigment epithelium cells. Methods Find. Exp. Clin. Pharmacol. 2005;27:179–184. doi: 10.1358/mf.2005.27.3.890875. [DOI] [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002;16:1151–1162. doi: 10.1096/fj.01-0944rev. [DOI] [PubMed] [Google Scholar]
- Witmer AN, Vrensen GF, Van Noorden CJ, Schlingemann RO. Vascular endothelial growth factors and angiogenesis in eye disease. Prog. Retin. Eye Res. 2003;22:1–29. doi: 10.1016/s1350-9462(02)00043-5. [DOI] [PubMed] [Google Scholar]
- Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E, Uehara T, Lipton SA. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch. Ophthalmol. 2004;122:598–614. doi: 10.1001/archopht.122.4.598. [DOI] [PubMed] [Google Scholar]
- Zhou J, Cai B, Jang YP, Pachydaki S, Schmidt AM, Sparrow JR. Mechanisms for the induction of HNE-MDA- and AGE-adducts, RAGE and VEGF in retinal pigment epithelial cells. Exp. Eye Res. 2005;80:567–580. doi: 10.1016/j.exer.2004.11.009. [DOI] [PubMed] [Google Scholar]