

Abstract
Type 2 diabetes is associated with excessive food intake and a sedentary lifestyle. Local inflammation of white adipose tissue induces cytokine-mediated insulin resistance of adipocytes. This results in enhanced lipolysis within these cells. The fatty acids that are released into the cytosol can be removed by mitochondrial β-oxidation. The flux through this pathway is normally limited by the rate of ADP supply, which in turn is determined by the metabolic activity of the adipocyte. It is expected that the latter does not adapt to an increased rate of lipolysis. We propose that elevated fatty acid concentrations in the cytosol of adipocytes induce mitochondrial uncoupling and thereby allow mitochondria to remove much larger amounts of fatty acids. By this, release of fatty acids out of adipocytes into the circulation is prevented. When the rate of fatty acid release into the cytosol exceeds the β-oxidation capacity, cytosolic fatty acid concentrations increase and induce mitochondrial toxicity. This results in a decrease in β-oxidation capacity and the entry of fatty acids into the circulation. Unless these released fatty acids are removed by mitochondrial oxidation in active muscles, these fatty acids result in ectopic triacylglycerol deposits, induction of insulin resistance, beta cell damage and diabetes. Thiazolidinediones improve mitochondrial function within adipocytes and may in this way alleviate the burden imposed by the excessive fat accumulation associated with the metabolic syndrome. Thus, the number and activity of mitochondria within adipocytes contribute to the threshold at which fatty acids are released into the circulation, leading to insulin resistance and type 2 diabetes.
Keywords: Adipocytes, Fatty acids, HAART, Mitochondria, Thiazolidinedione, Type 2 diabetes mellitus
Introduction
Type 2 diabetes mellitus is generally associated with an enhanced energy intake and too little physical exercise. Also genetic factors determine the susceptibility to develop this disease. Whole-body insulin resistance and a state of low grade inflammation are early marker of the disease process [1]. Subsequently, hyperglycaemia develops due to an accelerated decline in beta cell function [2–4]. Type 2 diabetes mellitus is often accompanied by other co-morbidities such as hypertension and dyslipidaemia. Together, they constitute the metabolic syndrome [2].
Here we present the concept that removal of fatty acids within white adipocytes by fatty acid-induced uncoupled mitochondrial β-oxidation protects the organism against fatty acid leakage out of adipocytes, thereby preventing fatty acid-induced insulin resistance in liver and muscle and lipotoxicity in pancreatic beta cells. A consequence of this concept is that mitochondrial dysfunction in adipocytes, either inherited or acquired, makes the organism more prone to develop insulin resistance and type 2 diabetes.
Mitochondria and fatty acids
One of the main functions of mitochondria, in addition to producing ATP, is to remove fatty acids by β-oxidation. In this way, mitochondria are able to remove NEFA and to protect the organism against fatty acid-induced insulin resistance and pancreatic beta cell lipotoxicity. β-Oxidation can take place in all cell types relevant for glucose homeostasis, including muscle, liver and adipocytes. During this process, fatty acids are oxidised in the mitochondrial matrix by NAD+ and flavin adenine dinucleotide (FAD), yielding acetyl coenzyme A, which is further degraded to CO2 by the citric acid cycle. The resulting NADH and FADH2 need to be recycled into NAD+ and FAD by the respiratory chain so that additional fatty acid molecules can be oxidised. This requires a supply of ADP, which is converted into ATP by respiratory chain activity. The rate of conversion of ATP back into ADP, which is determined by the metabolic activity of the cell, determines the rate at which fatty acids can be removed by mitochondrial activity. If the capacity of the cell to oxidise fatty acids is to be enhanced, ATP needs to be reconverted into ADP at an increased rate. This occurs during exercise in contracting muscle. The resulting enhanced rate of ADP generation provides a major additional sink for the oxidation of fatty acids within the body. Only when mitochondria are in the uncoupled state can regeneration of NAD+ and FAD occur without the conversion of ATP into ADP. The released energy is then converted into heat. Uncoupling is a physiological adaptation process, normally regulated by specific proteins [5]. It is remarkable that, like uncoupling proteins, fatty acids are capable of inducing mitochondrial uncoupling, especially when their concentrations exceed the binding capacity of fatty acid binding proteins [6–8]. This implies that when unbound fatty acids are present in the cytosol of cells, mitochondria become less efficient at ATP production and generate more heat during the oxidation of NADH and FADH2 derived from food.
Mitochondrial uncoupling in adipocytes by fatty acids may protect the organism against fatty acid-induced insulin resistance and lipotoxicity
White adipocytes contain large amounts of mitochondria in their tiny cytosolic compartment [9, 10]. There is no obvious reason why these cells need such a large capacity to produce ATP. We propose that, when uncoupled by fatty acids, these mitochondria prevent the release of fatty acids out of the adipocytes when the antilipolytic action of insulin is attenuated.
Evidence is accumulating that inflammation of adipose tissue is an early step in the pathogenesis of type 2 diabetes. The combination of excessive food intake with a sedentary lifestyle results in the formation of large adipocytes overloaded with triacylglycerols. Through as yet unknown mechanisms the adipose tissue becomes inflamed and infiltrated by leucocytes. This leads to the release of inflammatory cytokines such as TNF-α [11–14], a potent inducer of insulin resistance in adipocytes which also induces lipolysis. As a result, fatty acids are released out of the large triacylglycerol pool [15]. We propose that, initially, a part of these fatty acids are removed inside adipocytes by partially uncoupled mitochondrial β-oxidation. In doing so, these mitochondria create a threshold for fatty acid release into the circulation, which would otherwise trigger the development of whole-body insulin resistance and pancreatic beta cell lipotoxicity, the latter expected to occur when the body cannot utilise these released fatty acids as fuel, for example, in response to high food intake and little physical exercise. Our proposed mechanism is outlined in Fig. 1. When low amounts of fatty acids are released out of the triacylglycerol pool in insulin-resistant adipocytes, these fatty acids can be removed by coupled β-oxidation. The rate of fatty acid removal by this pathway is determined by the rate of ADP regeneration within adipocytes, which is likely to be a more or less constant factor, merely determined by cellular metabolism and not affected by variations in the lipolytic rate. We propose that, when the rate of fatty acid release exceeds the rate of fatty acid removal by coupled β-oxidation, the increase in cytosolic concentrations of fatty acids not bound to binding proteins (unbound) induces a partial uncoupling of the mitochondria. Uncoupling by fatty acids has been shown in multiple in vitro experiments [5–8, 16]. Evidence of uncoupling in vivo is provided by a study in which exposure of heart muscle to fatty acids resulted in a decrease in ATP production and a concomitant increase in oxygen consumption [17]. When uncoupled, the mitochondria are able to remove much larger quantities of fatty acids through generation of heat. The degree of uncoupling increases in with further elevations in fatty acid concentration, thus generating more heat. This represents a dynamic adaptation of the efficiency of the mitochondrion, which is determined by the intracellular concentration of fatty acids. If, however, the rate of fatty acid release exceeds the maximum clearance rate, fatty acids may reach concentrations that are toxic to the mitochondrion [18]. At this point β-oxidation capacity collapses and fatty acids are released out of adipocytes and redistributed over other tissues in the body. These ectopic triacylglycerol deposits are associated with the development of insulin resistance in muscle and liver and lipotoxicity in pancreatic beta cells [19]. At this stage of the disease process physical exercise will, by virtue of the generation of large amounts of ADP in muscle tissue, protect against lipotoxicity and the development of insulin resistance. This is because the muscle switches to β-oxidation to regenerate ATP when fatty acids reach a concentration sufficient to make the muscle resistant to insulin-stimulated glucose uptake [20, 21].
Fig. 1.
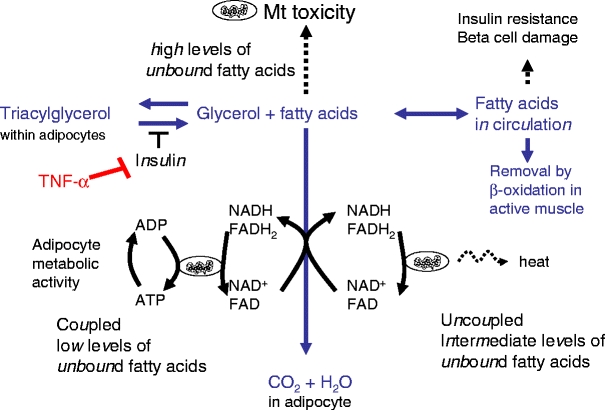
Consequences of fatty acid release from the adipocyte triacylglycerol pool. In insulin-sensitive adipocytes, fatty acid concentrations are kept low by insulin-induced antilipolytic action, re-esterification of fatty acids and mitochondrial β-oxidation. TNF-α induces insulin resistance and lipolysis. At low cytosolic concentrations of unbound fatty acids, the flux through mitochondrial β-oxidation is limited by the rate of ADP generation by cellular metabolism. At intermediate concentrations of unbound fatty acids, uncoupling of mitochondria is induced leading to continuous oxidation of fatty acids, independent of ADP supply. This process generates heat and keeps cytosolic fatty acid concentrations low. When the rate of fatty acid release from the triacylglycerol pool exceeds the rate of fatty acid removal, high cytosolic concentrations of unbound fatty acids develop, which induce mitochondrial damage [18]. This results in a decline in the capacity to remove fatty acids and the release of large amounts of fatty acids into the circulation. Unless these are removed by muscle activity they form ectopic triacylglycerol deposits and induce whole-body insulin resistance and beta cell damage. Dotted arrows indicate consequences; continuous arrows, fluxes
Discussion
In our model for the development of obesity-induced whole-body insulin resistance and beta cell damage leading to type 2 diabetes, we propose a key role for fatty acid removal by adipose tissue mitochondria. The disease process, as outlined in Fig. 2, is initiated by the development of cytokine-induced adipose tissue insulin resistance [11–15] and results in the release of fatty acids into the cytosol of the adipocytes. Further increases in cytosolic fatty acids induce mitochondrial dysfunction [18], decreasing the rate of β-oxidation, augmenting the fatty acid concentration, leading to their release into the circulation. These fatty acids are responsible for the development of insulin resistance in liver and muscle and lipotoxicity in pancreatic beta cells. Fatty acids do not appear to induce insulin resistance in adipocytes [22], which otherwise would create a positive feedback loop whereby the release of fatty acids induces additional insulin resistance and further promotes the release of fatty acids.
Fig. 2.
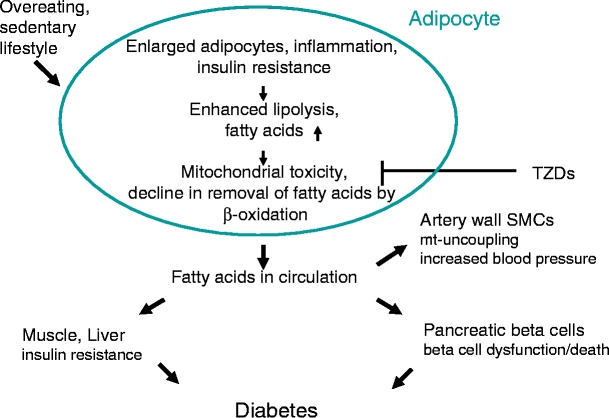
Proposed sequence of events leading to the development of hyperglycaemia during the metabolic syndrome. When adipocytes become overloaded with triacylglycerol, low-grade inflammation develops and inflammatory cytokines such as TNF-α induce insulin resistance in the adipocytes. This results in an elevated state of lipolysis. When fatty acids are inadequately removed within adipocytes because of mitochondrial dysfunction (for example, induced by fatty acids or HAART therapy, fatty acids appear in the circulation, where they induce insulin resistance of muscle and liver and malfunction of pancreatic beta cells. The elevated circulating fatty acid concentrations may also uncouple mitochondria in artery wall smooth muscle cells, thereby elevating the risk of hypertension [46]. Thiazolidinediones (TZDs) ameliorate the disease process in two ways: (1) by creating more mitochondria in adipose cells [23, 36, 37], thereby enhancing the capacity for oxidation of fatty acids; and (2) by enhanced re-esterification of fatty acids [39, 40]
Is there any evidence to support our model? Several studies have shown that in adipose tissue from obese animal models and humans, mitochondrial (mt) DNA copy number and expression of mitochondrial genes is decreased [23, 24].
In the diabetic state, mitochondrial β-oxidation of fatty acids was found to be attenuated. In addition, mitochondria showed an abnormal morphology [24]. These data support the concept of mitochondrial dysfunction in adipose tissue in states of obesity and type 2 diabetes.
Furthermore, intake of high-fat food by healthy persons rapidly increases the basal metabolic rate. This increase is blunted in obese individuals, whereas obese individuals exhibit an increased thermogenesis. These observations are suggestive for a rapid effect of ingested fatty acids on thermogenesis by mitochondrial uncoupling. The data also suggest that, in obese individuals, mitochondria are already uncoupled or damaged and less responsive to further fatty acid-induced uncoupling [25].
In vitro studies have shown that fatty acid-induced uncoupling of the mitochondrial respiratory chain depends on the chemical nature of these fatty acids. Unsaturated fatty acids, such as oleic acid, are better uncouplers than saturated fatty acids [8]. This could imply that the inclusion of saturated fatty acids in the diet makes mitochondria less prone to uncoupling, resulting in the earlier release of fatty acids, i.e. at a lower fatty acid level.
A further implication of our model is that a decline in mitochondrial function in adipocytes reduces the ability of these cells to store triacylglycerol. Inadvertently released fatty acids out of the triacylglycerol pool enter the circulation and are redistributed in other tissues. This occurs, for example, in individuals starting on highly active antiretroviral therapy (HAART) [26–29]. The nucleoside analogues included in HAART inhibit mtDNA polymerase and induce a ∼30–50% reduction in mtDNA content in adipocytes and other tissues. HAART is associated with the redistribution of peripheral fat to the central and other compartments and with an elevated risk of developing the metabolic syndrome and type 2 diabetes. We see this clinical phenotype as a result of release of fatty acids by peripheral adipocytes, which in turn is due to a decrease in mitochondrial capacity to remove fatty acids. The fatty acids, after entering the circulation, induce insulin resistance and lipotoxicity to pancreatic beta cells and become stored as ectopic triacylglycerol deposits.
Another example of a mitochondrial dysfunction is represented by patients carrying a 3243A>G mutation in mtDNA. This mutation results in an attenuated mitochondrial function. Most of these patients develop the maternally inherited diabetes and deafness (MIDD) syndrome. Remarkably, these patients usually have a BMI of <25 kg/m2. Furthermore, they exhibit ectopic triacylglycerol deposits in multiple tissues [30–33]. This clinical picture suggests the attenuated storage of triacylglycerol in adipose tissue. Around mid-life, these patients show a decrease in insulin secretion, which may result from lipotoxicity to pancreatic beta cells.
Congenital lipodystrophy is another clinical example showing that an inadequate storage of triacylglycerol in adipocytes contributes to ectopic triacylglycerol deposits and the development of severe whole-body insulin resistance and diabetes [34].
In contrast, improved mitochondrial function specifically in adipocytes is seen in individuals taking thiazolidinediones. These drugs are widely used to ameliorate whole-body insulin resistance in patients suffering from type 2 diabetes mellitus and the metabolic syndrome. These drugs bind to peroxisome proliferator-activated receptor-γ receptors which are highly expressed in adipocytes and in cells from the immune system, but low in liver and muscle [35]. These drugs increase mitochondrial copy number and mitochondrial gene expression specifically in adipose tissue [23, 36, 37]. Clinically, these drugs improve storage of triacylglycerol in peripheral adipocytes and ameliorate fatty acid-related insulin resistance. Patients taking these drugs tend to store more triacylglycerol in their adipocytes, leading to weight gain [38]. Together, these clinical data suggest that correct mitochondrial function is needed for adequate storage of fatty acids as triacylglycerol in adipocytes. By this means the organism is protected against the fatty acid-induced development of insulin resistance and lipotoxicity to the pancreas.
As thiazolidinediones also induce the expression of genes involved in the neogenesis of glycerol, these drugs also enhance the re-esterification of fatty acids [36, 39, 40]. This also contributes to the removal of fatty acids from the circulation and protects adipocyte mitochondria against fatty acid-induced damage. Because part of the glycerol in adipocytes is synthesised through pathways that are dependent upon mitochondrial function [36], this suggests that changes in mitochondrial function also may affect the rate of glycerol production and thereby the rate of fatty acid esterification.
To be able to perform, uncoupled β-oxidation mitochondria require the presence of sufficient amounts of oxygen. It has been suggested that a state of hypoxia occurs in expanding adipose tissue during the development of obesity and that this state may contribute to the development of the metabolic syndrome [41, 42]. Our model predicts that hypoxia in adipocytes would lead to a reduced rate of fatty acid removal inside these cells and to more fatty acid release into the circulation.
The sensitivity of an individual to the development of type 2 diabetes as a result of lifestyle is determined by genetic factors. Multiple genetic variants that modulate the risk of an individual for the development of diabetes have been identified, including genetic variants related to mitochondrial function. A high penetrance mutation in mtDNA has been found to be associated with the MIDD syndrome. This mutation predominantly affects the activity of complex I of the respiratory chain [43]. In the same biochemical pathway a mutation in the LARS2 gene, which encodes mitochondrial leucyl-tRNA synthetase 2, also modulates the risk for diabetes [44]. Furthermore, genetically determined changes in expression levels of components of the mitochondrial respiratory chain have been found to be associated with an increased risk of developing type 2 diabetes mellitus [45]. These genetic factors are likely to result in a decreased capacity of the mitochondria to remove fatty acids through uncoupled β-oxidation. Thereby, a genetic predisposition can lower the threshold for fatty acid release by adipocytes into the circulation and as a result enhance the risk for type 2 diabetes.
Our model of the pathogenesis of type 2 diabetes mellitus requires experimental verification of several points, and it certainly does not exclude the involvement of additional factors such as the coregulation of whole-body insulin action and insulin secretion through adipokines and the involvement of uncoupling proteins in setting the threshold for fatty acid-induced uncoupling of mitochondria. However, we see the way fatty acids interact with mitochondria in the cytosol of adipocytes as a major initiating event in the disease process leading to the metabolic syndrome and type 2 diabetes mellitus.
Acknowledgments
Duality of interest The authors declare that there is no duality of interest associated with this manuscript.
Abbreviations
- FAD
flavin adenine dinucleotide
- HAART
highly active antiretroviral therapy
- MIDD
maternally inherited diabetes and deafness
- mt
mitochondrial
References
- 1.Kahn SE (2003) The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes mellitus. Diabetologia 46:3–19 [DOI] [PubMed]
- 2.Reaven GM (2003) The insulin resistance syndrome. Curr Atheroscler Rep 5:364–371 [DOI] [PubMed]
- 3.Ferrannini E, Vichi S, Beck-Nielsen H, Laakso M, Paolisso G, Smith U (1996) Insulin action and age. European group for the study of insulin resistance (EGIR). Diabetes 45:947–953 [DOI] [PubMed]
- 4.Fritsche A, Madaus A, Stefan N et al (2002) Relationships among age, proinsulin conversion, and beta-cell function in nondiabetic humans. Diabetes 51(Suppl 1):S234–S239 [DOI] [PubMed]
- 5.Di Paola M, Lorusso M (2006) Interaction of free fatty acids with mitochondria: coupling, uncoupling and permeability transition. Biochim Biophys Acta 1757:1330–1337 [DOI] [PubMed]
- 6.Skulachev VP (1999) Anion carriers in fatty acid-mediated physiological uncoupling. J Bioenerg Biomembr 31:431–445 [DOI] [PubMed]
- 7.Vaartjes WJ, van den Bergh SG (1978) The oxidation of long-chain unsaturated fatty acids by isolated rat liver mitochondria as a function of substrate concentration. Biochim Biophys Acta 503:437–449 [DOI] [PubMed]
- 8.Borst TP, Loos JA, Christ EJ, Slater EC (1962) Uncoupling activity of long-chain fatty acids. Biochim Biophys Acta 62:509–518 [DOI] [PubMed]
- 9.Loncar D, Afzelius BA, Cannon B (1988) Epididymal white adipose tissue after cold stress in rats. II. Mitochondrial changes. J Ultrastruct Mol Struct Res 101:199–209 [DOI] [PubMed]
- 10.Chen CH, Lin EC, Cheng WT, Sun HS, Mersmann HJ, Ding ST (2006) Abundantly expressed genes in pig adipose tissue: an expressed sequence tag approach. J Anim Sci 84:2673–2683 [DOI] [PubMed]
- 11.Greenberg AS, Obin MS (2006) Obesity and the role of adipose tissue in inflammation and metabolism. Am J Clin Nutr 83:461S–465S [DOI] [PubMed]
- 12.Xu H, Barnes GT, Yang Q et al (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–1830 [DOI] [PMC free article] [PubMed]
- 13.Rajala MW, Scherer PE (2003) The adipocyte—at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 144:3765–3773 [DOI] [PubMed]
- 14.Hotamisligil GS, Spiegelman BM (1994) Tumor necrosis factor alpha: a key component of the obesity-diabetes link. Diabetes 43:1271–1278 [DOI] [PubMed]
- 15.Ryden M, Dicker A, van Harmelen V et al (2002) Mapping of early signaling events in tumor necrosis factor-α-mediated lipolysis in human fat cells. J Biol Chem 277:1085–1091 [DOI] [PubMed]
- 16.Chan SH, Higgins E Jr (1979) Uncoupling activity of endogenous free fatty acids in rat liver mitochondria. Can J Biochem 56:111–116 [DOI] [PubMed]
- 17.Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED (2005) Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 112:2686–2695 [DOI] [PubMed]
- 18.Weinberg JM (2006) Lipotoxicity. Kidney Int 70:1560–1566 [DOI] [PubMed]
- 19.Raz I, Eldor R, Cernea S, Shafrir E (2005) Diabetes: insulin resistance and derangements in lipid metabolism. Cure through intervention in fat transport and storage. Diabetes Metab Res Rev 21:3–14 [DOI] [PubMed]
- 20.Roden M (2005) Muscle triglycerides and mitochondrial function: possible mechanisms for the development of type 2 diabetes mellitus. Int J Obes (Lond) 29(Suppl 2):S111–S115 [DOI] [PubMed]
- 21.Goodpaster BH, Brown NF (2005) Skeletal muscle lipid and its association with insulin resistance: what is the role for exercise? Exerc Sport Sci Rev 33:150–154 [DOI] [PubMed]
- 22.Lundgren M, Eriksson JW (2004) No in vitro effects of fatty acids on glucose uptake, lipolysis or insulin signaling in rat adipocytes. Horm Metab Res 36:203–209 [DOI] [PubMed]
- 23.Bogacka I, Xie H, Bray GA, Smith SR (2005) Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes 54:1392–1399 [DOI] [PubMed]
- 24.Choo HJ, Kim JH, Kwon OB et al (2006) Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 49:784–791 [DOI] [PubMed]
- 25.Blaak EE, Hul G, Verdich C et al (2007) Impaired fat-induced thermogenesis in obese subjects: the NUGENOB study. Obesity (Silver Spring) 15:653–663 [DOI] [PubMed]
- 26.Brinkman K, Smeitink JA, Romijn JA, Reiss P (1999) Mitochondrial toxicity induced by nucleoside-analogue reverse-transcriptase inhibitors is a key factor in the pathogenesis of antiretroviral-therapy-related lipodystrophy. Lancet 354:1112–1115 [DOI] [PubMed]
- 27.Sutinen J, Hakkinen AM, Westerbacka J et al (2002) Increased fat accumulation in the liver in HIV-infected patients with antiretroviral therapy-associated lipodystrophy. AIDS 16:2183–2193 [DOI] [PubMed]
- 28.Shain CS, Cleary JD (2002) Buffalo hump in an HIV-infected patient. Ann Pharmacother 36:1807 [DOI] [PubMed]
- 29.Buffet M, Schwarzinger M, Amellal B et al (2005) Mitochondrial DNA depletion in adipose tissue of HIV-infected patients with peripheral lipoatrophy. J Clin Virol 33:60–64 [DOI] [PubMed]
- 30.Maassen JA, ’T Hart LM, Van Essen E et al (2004) Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes 53(Suppl 1):S103–S109 [DOI] [PubMed]
- 31.Suzuki Y, Nishimaki K, Taniyama M et al (2004) Lipoma and opthalmoplegia in mitochondrial diabetes associated with small heteroplasmy level of 3243 tRNALeu(UUR) mutation. Diabetes Res Clin Pract 63:225–229 [DOI] [PubMed]
- 32.Guillausseau PJ, Massin P, Dubois-LaForgue D et al (2001) Maternally inherited diabetes and deafness: a multicenter study. Ann Intern Med 134:721–728 [DOI] [PubMed]
- 33.Suzuki S, Oka Y, Kadowaki T et al (2003) Clinical features of diabetes mellitus with the mitochondrial DNA 3243 (A–G) mutation in Japanese: maternal inheritance and mitochondria-related complications. Diabetes Res Clin Pract 59:207–217 [DOI] [PubMed]
- 34.Capeau J, Magre J, Lascols O et al (2005) Diseases of adipose tissue: genetic and acquired lipodystrophies. Biochem Soc Trans 33:1073–1077 [DOI] [PubMed]
- 35.Knouff C, Auwerx J (2004) Peroxisome proliferator-activated receptor-γ calls for activation in moderation: lessons from genetics and pharmacology. Endocr Rev 25:899–918 [DOI] [PubMed]
- 36.Wilson-Fritch L, Nicoloro S, Chouinard M et al (2004) Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest 114:1281–1289 [DOI] [PMC free article] [PubMed]
- 37.Wilson-Fritch L, Burkart A, Bell G et al (2003) Mitochondrial biogenesis and remodeling during adipogenesis and in response to the insulin sensitizer rosiglitazone. Mol Cell Biol 23:1085–1094 [DOI] [PMC free article] [PubMed]
- 38.Larsen TM, Toubro S, Astrup A (2003) PPARγ agonists in the treatment of type II diabetes: is increased fatness commensurate with long-term efficacy? Int J Obes Relat Metab Disord 27:147–161 [DOI] [PubMed]
- 39.Franckhauser S, Munoz S, Pujol A et al (2002) Increased fatty acid re-esterification by PEPCK overexpression in adipose tissue leads to obesity without insulin resistance. Diabetes 51:624–630 [DOI] [PubMed]
- 40.Cadoudal T, Blouin JM, Collinet M et al (2007) Acute and selective regulation of glyceroneogenesis and cytosolic phosphoenolpyruvate carboxykinase in adipose tissue by thiazolidinediones in type 2 diabetes. Diabetologia 50:666–675 [DOI] [PubMed]
- 41.Hosogai N, Fukuhara A, Oshima K et al (2007) Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56:901–911 [DOI] [PubMed]
- 42.Trayhurn P, Wood IS (2004) Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr 92:347–355 [DOI] [PubMed]
- 43.van den Ouweland JM, Lemkes HH, Ruitenbeek W et al (1992) Mutation in mitochondrial tRNALeu(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat Genet 1:368–371 [DOI] [PubMed]
- 44.’t Hart LM, Hansen T, Rietveld I et al (2005) Evidence that the mitochondrial leucyl tRNA synthetase (LARS2) gene represents a novel type 2 diabetes susceptibility gene. Diabetes 54:1892–1895 [DOI] [PubMed]
- 45.Mootha VK, Lindgren CM, Eriksson KF et al (2003) PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34:267–273 [DOI] [PubMed]
- 46.Bernal-Mizrachi C, Gates AC, Weng S et al (2005) Vascular respiratory uncoupling increases blood pressure and atherosclerosis. Nature 435:502–506 [DOI] [PubMed]