

Abstract
The coordination chemistry of the 2-His-1-carboxylate facial triad mimics 3,3-bis(1-methylimidazol-2-yl)propionate (MIm2Pr) and 3,3-bis(1-ethyl-4-isopropylimidazol-2-yl) propionate (iPrEtIm2Pr) towards ZnCl2 was studied both in solution and in the solid state. Different coordination modes were found depending both on the stoichiometry and on the ligand that was employed. In the 2:1 ligand-to-metal complex [Zn(MIm2Pr)2], the ligand coordinates in a tridentate, tripodal N,N,O fashion similar to the 2-His-1-carboxylate facial triad. However, the 1:1 ligand-to-metal complexes [Zn(MIm2Pr)Cl(H2O)] and [Zn(iPrEtIm2Pr)Cl] were crystallographically characterized and found to be polymeric in nature. A new, bridging coordination mode of the ligands was observed in both structures comprising N,N-bidentate coordination of the ligand to one zinc atom and O-monodentate coordination to a zinc second atom. A rather unique transformation of pyruvate into oxalate was found with [Zn(MIm2Pr)Cl], which resulted in the isolation of the new, oxalato bridged zinc coordination polymer [Zn2(MIm2Pr)2(ox)]·6H2O, the structure of which was established by X-ray crystal structure determination.
Keywords: Zinc; N,N,O ligands; Pyruvic acid; Oxalic acid
Introduction
The 2-His-1-carboxylate facial triad is a structural motif of increasing prominence among structurally characterized metalloenzymes. Next to the rapidly growing subgroup of non-heme iron enzymes [1, 2], several mononuclear zinc enzymes also feature this triad at their active site [3]. The proteases thermolysin, carboxypeptidase, and neutral protease, for example, catalyze the hydrolysis of peptide bonds. The zinc ion in these enzymes is coordinated by three endogenous residues, i.e., one glutamate and two histidine residues (Fig. 1). The fourth coordination site of the pseudo-tetrahedral zinc center is occupied by a catalytically important water or hydroxide ligand. This 2-His-1-carboxylate facial triad is a variation of the most commonly observed structural motif for the active sites of zinc-containing enzymes. Typically, the tetrahedral zinc centers in these enzymes are bound by a combination of histidine (N), glutamate or aspartate (O) and/or cysteine (S) residues. The resulting NxOySz donor set determines its biological function [3]. Accordingly, it is important to understand how a particular combination of donor groups modulates the reactivity of the Zn(II) metal center.
Fig. 1.

Active site of a zinc-containing enzyme that features the 2-His-1-carboxylate facial triad: neutral protease from Bacillus cereus (Protein Data Bank accession code 1NPC.pdb) [4]
The study of small synthetic analogues has focused on establishing a basis for such a structure–function relationship for these mononuclear zinc-containing enzymes [3, 5]. Initially, pioneering studies by the groups of Vahrenkamp, Parkin, Kitajima, and others employed all-N donor ligands such as the trispyrazolylborates [6–8]. In order to obtain close structural models for the 2-His-1-carboxylate facial triad containing zinc enzymes, one would preferentially like to construct small analogues based on a tripodal N,N,O ligand framework that incorporates the biologically relevant donor groups. Of the complexes with a mixed N/O donor set reported to date [9–22], only a few make use of such tridentate, tripodal N,N,Ocarboxylato ligands. These tripodal ligands should be particularly well suited for the structural modeling of tetrahedral metal centers [23]. Dowling and Parkin [24] and Ghosh and Parkin [25] reported the first example of an N,N,O model complex with a carboxylato donor group, which resulted from the insertion of carbon dioxide into a borohydride bond. More recently, the groups of Burzlaff [9, 12] and Carrano [11, 18, 19] have reported studies on the zinc coordination chemistry of the bispyrazolylacetate ligand family. Beck et al. [9], for instance, reported that a 2:1 ligand–zinc complex was obtained with bis(3,5-dimethylpyrazol-2-yl)acetate (bdmpza), a ligand closely related to 3,3-bis(1-methylimidazol-2-yl)propionate (MIm2Pr). These 2:1 complexes were obtained regardless of the ratio of reactants, i.e., even with stoichiometric amounts of ligand and zinc(II). Very recently, Friese et al. [22] constructed mononuclear zinc models via a new approach, i.e., by the use of sterically hindered carboxylato and N-donor ligand building blocks.
We have been studying the copper and iron coordination chemistry of the new ligand family of the substituted 3,3-bis(1-alkylimidazol-2-yl)propionates as accurate structural mimics of the 2-His-1-carboxylate facial triad [26–28]. These ligands incorporate the biologically relevant donor groups, i.e., two imidazoles and a carboxylate group, into a monoanionic, tripodal framework. Their general biomimetic potential has been illustrated by the structurally characterized copper [27, 28] and iron [26] complexes of these ligands. Here, our studies are expanded to the structural modeling of the zinc-containing enzymes and describe the zinc coordination chemistry of the ligands MIm2Pr and 3,3-bis(1-ethyl-4-isopropylimidazol-2-yl)propionate (iPrEtIm2Pr) (Fig. 2). Furthermore, the attempted synthesis of zinc complexes with 3,3-bis(1-methylimidazol-2-yl)acetate (MIm2Ac), a direct analogue of the bispyrazolylacetates, is described. During the course of these studies, we found that zinc complexes of ligand MIm2Pr catalyze the unexpected and unprecedented conversion of pyruvate to oxalate in aqueous solution.
Fig. 2.

Ligands 3,3-bis(1-methylimidazol-2-yl)propionate (MIm2Pr), 3,3-bis(1-ethyl-4-isopropylimidazol-2-yl)propionate (iPrEtIm2Pr), and 3,3-bis(1-methylimidazol-2-yl)acetate (MIm2Ac) and the synthesis of the zinc complexes [Zn(MIm2Pr)2], [Zn(MIm2Pr)Cl], and [Zn(iPrEtIm2Pr)Cl]
Materials and methods
Air-sensitive organic reactions were carried out under an atmosphere of dry, oxygen-free N2 using standard Schlenk techniques. Tetrahydrofuran (THF) and diethyl ether were dried over sodium benzophenone ketyl and distilled under N2 prior to use. Methanol was dried over magnesium methoxide and distilled under N2 prior to use. 1H and 13C NMR spectra were recorded using a Varian AS400, a Varian Inova 300, or a Varian Mercury 200 spectrometer, operating at 25 °C. IR spectra were recorded with a PerkinElmer Spectrum One Fourier transform IR instrument. Elemental microanalyses were carried out by the Microanalytisches Laboratorium Dornis & Kolbe, Mulheim a.d. Ruhr, Germany. ESI–MS spectra were recorded using a Micromass liquid chromatography time-of-flight mass spectrometer at the Biomolecular Mass Spectrometry Group, Utrecht University. Bis(1-methylimidazol-2-yl)methane (MIm2CH2) [29,] 3,3-bis(1-methylimidazol-2-yl)propionic acid (HMIm2Pr), potassium and tetrabutylammonium 3,3-bis(1-methylimidazol-2-yl)propionate (K[MIm2Pr] and [Bu4N][MIm2Pr]) [27], potassium 3,3-bis(1-ethyl-4-isopropylimidazol-2-yl)propionate (K[iPrEtIm2Pr]) [26], and zinc bis(trimethylsilyl)amide [30] were prepared according to published procedures. All other chemicals were obtained commercially and used as received.
Benzyl bis(1-methylimidazol-2-yl)acetate
To a cooled solution of MIm2CH2 (0.52 g, 3.0 mmol) in dry THF (50 mL) at −78 °C was added dropwise a solution of n-butyl lithium in hexane (2.0 mL, 3.2 mmol). The reaction mixture was stirred for 1 h at −78 °C, after which benzylchloroformate (1.19 mL, 3.5 mmol, 50 wt% solution in toluene) was added dropwise. During the addition, a white precipitate formed. The solution was allowed to warm to room temperature overnight. The reaction mixture was quenched with H2O (20 mL) and all volatiles were removed in vacuo. The aqueous layer was extracted with diethyl ether (4 × 20 mL) and the combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude oil was purified by column chromatography (SiO2, ethyl acetate–methanol 2:1). Benzyl bis(1-methylimidazol-2-yl)acetate (BnMIm2Ac) was isolated as a yellow oil in 62% yield (0.58 g, 1.9 mmol). 1H NMR (300 MHz, CD3CN, 25 °C): δ = 3.44 (s, 6H, NCH3), 5.23 (s, 2H, CH2), 5.62 (s, 1H, CH), 6.85 (s, 2H, Him), 6.96 (s, 2H, Him), 7.35 (s, 5H, HPh) ppm. Anal. for C17H18N4O2 (310.35): calc. C 65.79, H 5.85, N 18.05; found C 65.88, H 5.74, N 17.92. IR (solid): ν = 3,122.9, 2,911.5, 1,734.5, 1,547.4, 1,510.2, 1,454.4, 1,373.6, 1,263.4, 1,220.8, 1,150.4, 978.2, 958.3, 907.1, 847.8, 785.7, 776.5, 740.1, 696.7, 683.5, 665.3 cm−1. ESI–MS: m/z = 409.0 ([M–Cl]+, calc. 409.0).
[Zn(MIm2Pr)2]
To a hot, colorless solution of HMIm2Pr (282 mg, 1.20 mmol) in dry acetonitrile (100 mL) was added a solution of Zn[N(SiMe3)2]2 (232 mg, 0.60 mmol) in dry diethyl ether (20 mL) via a cannula. The resulting colorless, clear solution was stirred for 60 h at elevated temperature, during which gradually a white precipitate formed. The reaction mixture was concentrated in vacuo to give the crude product as a white powder. Recrystallization from an acetone–water mixture (4:1 v/v) gave [Zn(MIm2Pr)2] as a colorless crystalline solid (230 mg, 72%). 1H NMR (300 MHz, D2O, 25 °C): δ = 2.69 (d, 2H, J = 6.3 Hz, CHCH2), 3.74 (s, 6H, NCH3), 4.93 (t, 1H, J = 6.0 Hz, CHCH2), 6.69 (s, 2H, Him), 7.00 (s, 2H, Him) ppm. 13C{1H} NMR (50 MHz, D2O, 25 °C): δ = 30.2, 33.4, 43.6, 122.7, 125.7, 146.8, 177.2 ppm. Anal. for C22H26ZnN8O4 (531.90): calc. C 49.68, H 4.93, N 21.07; found C 49.76, H 5.06, N 20.97. IR (solid): ν = 3,120.2, 2,951.7, 2,907.1, 2,815.2, 1,580.5, 1,508.1, 1,427.1, 1,391.0, 1,306.4, 1,288.0, 1,229.3, 1,168.8, 1,143.3, 1,044.8, 956.9, 906.2, 769.3, 751.1 cm−1. ESI–MS: m/z = 531.18 ([M + H]+, calc. 531.14), 553.18 ([M+Na]+, calc. 553.13), 569.16 ([M + K]+, calc. 569.10).
[Zn(MIm2Pr)Cl]
To a hot, colorless solution of K[MIm2Pr] (255 mg, 0.93 mmol) in dry methanol (5 mL) was added a solution of ZnCl2 (127 mg, 0.93 mmol) in methanol (5 mL) via a cannula. Immediately upon addition a white precipitate formed and the suspension was stirred at elevated temperature for 1 h. The white precipitate was separated by centrifugation and washed with methanol (3 × 20 mL). [Zn(MIm2Pr)Cl] was obtained as a white powder in almost quantitative yield (305 mg, 98%). 1H NMR (300 MHz, D2O, 25 °C): δ = 2.80 (d, 3H, J = 3.3 Hz, CHCH2), 3.85 (s, 6H, NCH3), 4.99 (t, 1H, CH2CH), 6.99 (s, 2H, Him), 7.14 (s, 2H, Him) ppm. 13C{1H} NMR (50 MHz, D2O, 25 °C): δ = 29.8, 33.4, 42.6, 122.5, 125.5, 146.1, 177.3 ppm. Anal. for C11H13ClN4O2Zn (334.11): calc. C 39.55, H 3.92, N 16.77; found: C 39.46, H 4.05, N 16.85. IR (solid): ν = 3,138.0, 3,117.2, 1,614.8, 1,508.2, 1389.8, 1,299.8, 1,289.5, 1,151.7, 1,142.5, 1,089.7, 976.2, 949.7, 796.3, 765.3, 736.1 cm−1. ESI–MS: m/z = 296.94 ([M–Cl]+, calc. 297.03), 332.92 ([M + H]+, calc. 333.01), 413.97 ([3M–Zn–3Cl + H]2+, calc. 414.09), 531.02 ([2M–Zn–2Cl + H]+, calc. 531.15), 628.97 ([2M–Cl]+, calc. 629.03), 827.10 ([3M–Zn–3Cl]+, calc. 827.17).
[Zn(MIm2Pr)Cl(H2O)]
Colorless crystals of [Zn(MIm2Pr)Cl(H2O)] suitable for X-ray diffraction were obtained upon standing of a concentrated solution of [Zn(MIm2Pr)Cl] in H2O for several weeks. The coordination polymer obtained is insoluble in all common organic solvents and water of neutral pH. Anal. for C11H15ClN4O3Zn (352.12): calc. C 37.52, H 4.29, 15.91; found C 37.44, H 4.27, N 15.97. IR (solid): ν = 3,408.6, 3,113.5, 3,134.7, 1,594.5, 1,504.5, 1,403.1, 1,306.7, 1,255.5, 1,155.5, 1,136.3, 1,087.4, 983.7, 959.1, 934.9, 849.7, 764.6, 743.1, 717.6 cm−1.
[Zn(iPrEtIm2Pr)Cl]
To a colorless solution of ZnCl2 (30 mg, 0.22 mmol) in dry methanol (10 mL) was added a solution of K[iPrEtIm2Pr] (85 mg, 0.22 mmol) in dry methanol (15 mL) via a cannula. The clear solution was stirred overnight at room temperature and evaporated in vacuo. The product was redissolved in dichloromethane and insoluble KCl was separated off by centrifugation. The solution was filtered over Celite and concentrated in vacuo to give [Zn(iPrEtIm2Pr)Cl] as a white powder (97 mg, 99%). Colorless crystals suitable for X-ray diffraction were obtained from a methanolic solution of [Zn(iPrEtIm2Pr)Cl] upon standing. 1H NMR (300 MHz, D2O, 25 °C): δ = 1.22 (d, 6H, J = 7.2 Hz, CH3CHCH3), 1.25 (d, 6H, J = 6.8 Hz, CH3CHCH3), 1.44 (t, 6H, J = 7.2 Hz, CH3CH2), 2.88 (d, 2H, J = 6.0 Hz, CHCH2), 3.25 (m, 2H, CH3CHCH3), 4.21 (ABX3, 2H, J = 7.2, CHHCH3), 4.32 (ABX3, 2H, J = 7.2, CHHCH3), 5.02 (t, 1H, J = 5.7 Hz, CHCH2), 6.99 (s, 2H, Him) ppm. 13C{1H} NMR (100 MHz, CD3OD, 25 °C): δ = 16.6, 22.1, 24.1, 28.0, 31.1, 42.9, 116.0, 146.2, 149.9, 175.7 ppm. Anal. for C19H29ClN4O2Zn (446.32): calc. C 51.13, H 6.55, N 12.55; found: C 51.05, H 6.43, N 12.37. IR (solid): ν = 3,101.4, 2,967.9, 2,873.7, 1,626.6, 1,572.8, 1,494.8, 1,448.9, 1,464.1, 1,381.1, 1,324.8, 1,257.2, 1,175.8, 1,153.0, 1,027.6, 976.1, 801.2, 757.7, 660.5 cm−1. ESI–MS: m/z = 409.13 ([2M–2Cl]2+, calc. 409.16), 445.11 ([M + H]+, calc. 445.13), 483.04 ([M + K]+, calc. 483.09), 631.21 ([3M–2Cl]2+, calc. 631.22), 853.29 ([2M–Cl]+, calc. 853.29), 889.20 ([2M + H]+, calc. 889.26), 1297.02 ([3M + H]+, calc. 1297.42).
[Zn(BnMIm2Ac)Cl2]
To a hot solution of BnMIm2Ac (259 mg, 0.84 mmol) in dry methanol (5 mL) was added a solution of anhydrous ZnCl2 (115 mg, 0.84 mmol) in dry MeOH (5 mL) via a cannula. The reaction mixture was stirred at elevated temperature for 1 h and gradually the product formed as a white precipitate. The product [Zn(BnMIm2Ac)Cl2] was separated by centrifugation and was obtained as a white powder in quantitative yield. Single crystals suitable for X-ray analysis were obtained by slow evaporation of a solution of [Zn(BnMIm2Ac)Cl2] in acetone. 1H NMR (300 MHz, CD3CN, 25 °C): δ = 3.79 (2, 6H, CH3), 5.12 (s, 2H, CH2), 5.60 (s, 1H, CH), 7.13 (d, 2H, J = 0.75 Hz, Him), 7.22 (d, 2H, J = 0.75 Hz, Him), 7.25 (m, 2H, HPh), 7.33 (m, 3H, HPh) ppm. 13C{1H} NMR (100 MHz, CD3OD, 25 °C): δ = 34.9, 40.6, 69.7, 124.5, 126.5, 129.1, 129.5, 129.7, 136.1, 142.3, 166.2 ppm. Anal. for C17H18Cl2N4O2Zn (446.67): calc. C 45.71, H 4.06, N 12.54; found C 45.64, H 3.95, N 12.61. IR (solid): ν = 3,122.9, 2,911.5, 1,734.5, 1,547.4, 1,510.2, 1,454.4, 1,373.6, 1,263.4, 1,220.8, 1,150.4, 978.2, 958.3, 907.1, 847.8, 785.7, 776.5, 740.1, 696.7, 683.5, 665.3 cm−1. ESI–MS: m/z = 409.0 ([M–Cl]+, calc. 409.0).
[Zn2(MIm2Pr)2(ox)] (direct synthesis)
To a solution of [Zn(MIm2Pr)Cl] (50 mg, 0.15 mmol) in water (5 mL) was added a solution of potassium oxalate hydrate (14 mg, 0.075 mmol) in water (3 mL). The solution was stirred overnight at room temperature, during which gradually a white precipitate formed. The precipitate was separated by centrifugation and washed three times with water (3 × 20 mL). The product [Zn2(MIm2Pr)2(ox)] was dried in vacuo and obtained as a white powder in 82% yield (42 mg, 0.061 mmol). Anal. for C24H26N8O8Zn2·6H2O (685.33): calc. C 36.33, H 4.83, N 14.12; found: C 36.45, H 4.50, N 14.25. IR (solid): ν = 3,476.8, 3,135.4, 1,671.3, 1,651.6, 1,600.2, 1,544.9, 1,504.8, 1,423.6, 1,382.7, 1,320.1, 1,287.2, 1,158.1, 1,137.9, 985.1, 769.4, 745.1, 710.4 cm−1.
[Zn2(MIm2Pr)2(ox)]·6H2O, reaction of [Zn(MIm2Pr)Cl] with sodium pyruvate
For NMR investigation of reaction mixtures, stock solutions of [Zn(-MIm2Pr)Cl] (20 mg, 0.06 mmol) in H2O (1 mL) and sodium pyruvate (13.2 mg, 0.06 mmol) in H2O (2 mL) were prepared. NMR tubes were filled with 0.1 mL of each stock solution and kept either at room temperature or heated to 50 °C. The NMR samples were analyzed at different times after mixing the reagents. Before acquisition of the NMR spectrum, 0.4 mL D2O was added to the reaction mixture. Crystals usually deposited within 2 or 3 days in the NMR tube.
In the case of the catalytic runs, 0.1 mL of a solution containing 20 equiv of sodium pyruvate was added to 0.1 mL of the [Zn(MIm2Pr)Cl] stock solution and dimethyl sulfoxide (5 equiv, 11 μL) was added as an internal standard. The reaction mixture was kept at 50 °C and the NMR spectrum was recorded with addition of 0.4 mL D2O after 24 h.
On a preparative scale, to a solution of ZnCl2 (37 mg, 0.27 mmol) and K[MIm2Pr] (75 mg, 0.27 mmol) in H2O (10 mL) was added sodium pyruvate (30 mg, 0.27 mmol). The solution was heated to 50 °C and gradually an off-white precipitate was formed. After 1 week, the product was separated by centrifugation (29 mg, 31%). The IR spectrum of the product was identical to that of independently synthesized [Zn2(MIm2Pr)2(ox)].
X-ray crystal structure determinations of [Zn(MIm2Pr)Cl(H2O)], [Zn(iPrEtIm2Pr)Cl], ZnCl2(BnMIm2Ac)], [ZnCl2(MIm2CH2)], and [Zn2(MIm2Pr)2(ox)]·6H2O
Reflections were measured with a Nonius Kappa CCD diffractometer with a rotating anode (graphite monochromator, λ = 0.71073 Å) at −123 °C. The structures were refined with SHELXL-97 [31] against F2 of all reflections. Non-hydrogen atoms were refined with anisotropic displacement parameters. Geometry calculations and checking for higher symmetry were performed with the PLATON program [32]. Further details are given in Table 1.
Table 1.
Crystallographic data for compounds [Zn(MIm2Pr)Cl(H2O)], [Zn(iPrEtIm2Pr)Cl], ZnCl2(BnMIm2Ac)], [ZnCl2(MIm2CH2)], and [Zn2(MIm2Pr)2(ox)]·6H2O
| Compound | [Zn(MIm2Pr)Cl(H2O)] | [Zn(iPrEtIm2Pr)Cl] | [ZnCl2(BnMIm2Ac)] | [ZnCl2(MIm2CH2)] | [Zn2(MIm2Pr)2(ox)]·6H2O |
|---|---|---|---|---|---|
| Formula | C11H15ClN4O3Zn | C19H29ClN4O2Zn | C17H18Cl2N4O2Zn | C9H12Cl2N4Zn | C24H26N8O8Zn2·6H2O |
| FW | 352.09 | 446.28 | 446.62 | 312.50 | 793.36 |
| Crystal size (mm3) | 0.30 × 0.12 × 0.09 | 0.18 × 0.03 × 0.03 | 0.60 × 0.03 × 0.03 | 0.30 × 0.15 × 0.06 | 0.24 × 0.18 × 0.09 |
| Crystal color | Colorless | Colorless | Colorless | Colorless | Colorless |
| Crystal system | Orthorhombic | Monoclinic | Monoclinic | Monoclinic | Triclinic |
| Space group | P212121 (no. 19) | Cc (no. 9) | C2/c (no. 15) | P21/c (no. 14) |  |
| a (Å) | 8.7159(1) | 10.1766(2) | 35.5761(4) | 7.3547(1) | 9.9634(7) |
| b (Å) | 10.1124(1) | 23.8561(6) | 7.4531(1) | 14.9538(2) | 12.0617(11) |
| c (Å) | 15.1177(2) | 9.1995(2) | 14.6106(2) | 12.3489(2) | 14.8889(9) |
| α (°) | – | – | – | – | 113.021(3) |
| β (°) | – | 107.0078(14) | 101.8858(5) | 117.5968(6) | 90.365(3) |
| γ (°) | – | – | – | – | 102.513(3) |
| V (Å) | 1332.45(3) | 2135.72(8) | 3790.97(8) | 1203.62(3) | 1599.4(2) |
| Z | 4 | 4 | 8 | 4 | 2 |
| Dcalc. (g cm−3) | 1.755 | 1.388 | 1.565 | 1.724 | 1.647 |
| μ (mm−1) | 2.056 | 1.296 | 1.597 | 2.461 | 1.580 |
| Absorption correction | Multiscan | Multiscan | Multiscan | Multiscan | Multiscan |
| Absorption correction range | 0.71–0.84 | 0.86–0.96 | 0.85–0.96 | 0.74–0.86 | 0.64–0.87 |
| sin(θ/λ)max (Å−1) | 0.65 | 0.65 | 0.60 | 0.65 | 0.65 |
| Reflections (measured/unique) | 21,618/3,063 | 11,758/4,644 | 28,566/3,424 | 27,341/2,759 | 35,709/7,342 |
| Parameters/restraints | 241/0 | 251/2 | 237/0 | 193/0 | 438/0 |
| R1/wR2 [I > 2σ(I)] | 0.0182/0.0447 | 0.0312/0.0694 | 0.0309/0.0704 | 0.0306/0.0793 | 0.0389/0.0867 |
| R1/wR2 (all reflections) | 0.0190/0.0452 | 0.0384/0.0723 | 0.0512/0.0786 | 0.0389/0.0852 | 0.0540/0.0947 |
| S | 1.062 | 1.041 | 1.081 | 1.089 | 1.083 |
| Flack parameter [37] | −0.006(7) | 0.305(9) | – | – | – |
| Residual density (e Å−3) | −0.36/0.19 | −0.27/0.41 | −0.31/0.43 | −0.54/1.23 | −0.79/0.51 |
MIm2Pr 3,3-bis(1-methylimidazol-2-yl)propionate, iPrEtIm2Pr 3,3-bis(1-ethyl-4-isopropylimidazol-2-yl)propionate, and BnMIm2Ac benzyl bis(1-methylimidazol-2-yl)acetate, 22, bis(1-methylimidazol-2-yl)methane
For Zn(MIm2Pr)Cl(H2O)] the structure was solved with SHELXS-97 [33] using direct methods. All hydrogen atoms were located in the difference Fourier map and refined freely with isotropic displacement parameters.
For [Zn(iPrEtIm2Pr)Cl] the structure was solved with DIRDIF-99 [34] using automated Patterson methods. Refinement was performed as an inversion twin. All hydrogen atoms were located in the difference Fourier map and refined with a riding model.
For ZnCl2(BnMIm2Ac)] the structure was solved with DIRDIF-99 [34] using automated Patterson methods. All hydrogen atoms were located in the difference Fourier map and refined with a riding model.
For [ZnCl2(MIm2CH2)] the structure was solved with SIR-97 [35] using Direct methods. All hydrogen atoms were located in the difference Fourier map and refined freely with isotropic displacement parameters.
For [Zn2(MIm2Pr)2(ox)]·6H2O the crystal was nonmerohedrally twinned with a twofold rotation about the crystallographic c-axis as a twin operation. This twin law was taken into account during the intensity integration using the program EvalCCD [36]. The structure was solved on the nonoverlapping reflections with DIRDIF-99 [34] using automated Patterson methods. All hydrogen atoms were located in the difference Fourier map and refined with a riding model. The twin fraction refined to 0.127(3).
Crystallographic data (without structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre (CCDC) as supplementary publication nos. CCDC-637452 for Zn(MIm2Pr)Cl(H2O)], CCDC-637453 for [Zn(iPrEtIm2Pr)Cl], CCDC-637454 for ZnCl2(BnMIm2Ac)], CCDC-637455 for [ZnCl2(MIm2CH2)], and CCDC-637456 for [Zn2(MIm2Pr)2(ox)]·6H2O. Copies of the data can be obtained free of charge from the CCDC (12 Union Road, Cambridge CB2 1EZ, UK; Tel.: +44-1223-336408; Fax: +44-1223-336003; e-mail: deposit@ccdc.cam.ac.uk; Web site http://www.ccdc.cam.ac.uk).
Results
As part of our efforts to study the coordination chemistry of the substituted 3,3-bis(1-alkylimidazol-2-yl)propionate ligand family, the complexes of MIm2Pr and iPrEtIm2Pr with ZnCl2 were synthesized and structurally characterized (Fig. 2). MIm2Pr is the parent ligand of the family and iPrEtIm2Pr offers the most steric hindrance of the ligands studied so far. The coordination chemistry of MIm2Pr and iPrEtIm2Pr with zinc was found to be stoichiometry-dependent.
Zinc complexes of MIm2Pr
The formation of different species upon addition of either 0.5 or 1.0 equiv of ZnCl2 to a solution of the potassium salt of MIm2Pr in D2O is evidenced by the respective 1H NMR spectra in D2O (Fig. 3).
Fig. 3.
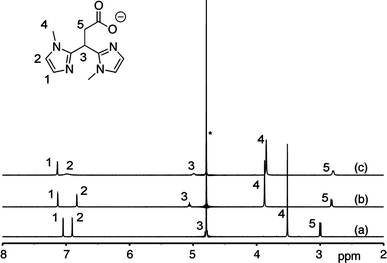
400 MHz 1H NMR spectra of the titration of K[MIm2Pr] with ZnCl2 in D2O at room temperature: a K[MIm2Pr]; b K[MIm2Pr] and ZnCl2 2:1; c K[MIm2Pr] and ZnCl2 1:1. Asterisks denote the residual solvent peak
All four resonances associated with ligand MIm2Pr (spectrum a) shifted upon addition of 0.5 equiv of ZnCl2 (spectrum b). The observation of two sharp signals of equal intensity for the imidazole protons shows that all four imidazole groups are equivalent. This is indicative of symmetric binding of both ligands to the zinc ion. The resonances of the methyl groups and the methine proton at the bridging carbon shift downfield relative to the free ligand, whereas the propionate CH2 protons shift upfield. Note that the methine proton resonance of the free ligand is hidden under the residual solvent signal in spectrum a. The lack of any other resonances suggests the clean formation of a neutral, 2:1 [Zn(MIm2Pr)2] complex. Upon addition of another 0.5 equiv of ZnCl2 several resonances shift and signal broadening is observed (spectrum c). Especially, one of the two imidazole signals is severely broadened. Again, no free ligand is observed in the spectrum. These observations indicate a dynamic system with an overall 1:1 ligand–zinc stoichiometry.
The isolation and purification of the complex formed with 2 equiv of MIm2Pr with respect to ZnCl2 proved troublesome. The separation of the inorganic salts formed as a by-product in the synthesis by means of repetitive washing in all cases resulted in partial loss of coordinated ligand (as observed by NMR), regardless of the A[MIm2Pr] salt (A is K+, [Bu4N]+) originally employed. We therefore attempted the synthesis of this complex by the reaction of 2 equiv of the acid HMIm2Pr with Zn[N(SiMe3)2]2. Zinc bis(trimethylsilyl)amide can be synthesized in sizeable quantities [30] and was found to be a convenient reagent in this case. Indeed, the reaction of Zn[N(SiMe3)2]2 with 2 equiv of HMIm2Pr in acetonitrile resulted in the formation of a white precipitate, which was recrystallized from an acetonitrile–water solution and was identified as the 2:1 complex [Zn(MIm2Pr)2].
Unfortunately, all attempts at obtaining single crystals of [Zn(MIm2Pr)2] failed. On the basis of ESI–MS, IR, and the previously described NMR analysis, we propose that [Zn(MIm2Pr)2] (Fig. 4) is isostructural to the crystallographically characterized analogous complex [CuII(MIm2Pr)2] [27]. The position of the symmetric and asymmetric carbonyl stretching vibrations proved to be indicative of the binding mode of MIm2Pr (Bruijnincx et al., submitted) [27]. The νas(C=O) and νs(C=O) are found at 1,581 and 1,391 cm−1, respectively, indicative of a monodentate binding mode of the carboxylate [Δ(νas − νs) = 190 cm−1, identical to Δionic] [38]. Furthermore, the clean ESI–MS spectrum of [Zn(MIm2Pr)2] confirmed the structure of a mononuclear, neutral 2:1 species with [M + X]+ (X is H, Na, K) as the prominent ions.
Fig. 4.
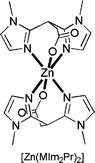
The 2:1 complex [Zn(MIm2Pr)2]
The 1:1 complex [Zn(MIm2Pr)Cl)] was synthesized by the reaction of equimolar amounts of ZnCl2 and K[MIm2Pr] in dry methanol. This led to the precipitation and isolation of [Zn(MIm2Pr)Cl] in almost quantitative yield. The broadened signals in the 1H NMR spectrum of [Zn(MIm2Pr)Cl)] in D2O prompted us to study this complex by variable-temperature NMR (Fig. 5). As complex [Zn(MIm2Pr)Cl)] is insoluble in organic solvents, the variable-temperature NMR studies were limited to aqueous solutions, which restricted the temperature range that could be studied. Spectra were recorded in the range from 2 to 70 °C.
Fig. 5.

1H NMR resonances of the imidazole rings of [Zn(MIm2Pr)Cl] at various temperatures in D2O
Figure 5 shows that at elevated temperatures two sharp imidazole resonances are observed. In the spectrum recorded at 70 °C, the (small) coupling between the two imidazole protons H1 and H2 can also be discerned. The imidazole signal at higher field broadens upon cooling and ultimately splits into two separate (broad) resonances. This implies that at higher temperature the imidazole H1 protons (Fig. 3) become magnetically equivalent, whereas at lower temperature exchange slows down with the result that decoalesence into two broad resonances occurs. This behavior can be tentatively explained by the possibility of two different spatial orientations of the imidazole rings with respect to the carboxylate group of a second ligand upon coordination to zinc (vide infra), which would result in nonequivalent imidazole H1 protons at lower temperature. At higher temperatures these orientations average on the NMR timescale and only one signal is observed.
The asymmetric and symmetric carbonyl stretch vibrations of isolated [Zn(MIm2Pr)Cl] were observed at 1,615 and 1,390 cm−1 [Δ(νas − νs) = 225 cm−1], respectively. The asymmetric stretch thus shifted to higher wavenumbers compared with that observed for [Zn(MIm2Pr)Cl] and a different, but still monodentate coordination mode of the carboxylate must therefore be present in [Zn(MIm2Pr)Cl]. The ESI–MS spectrum of [Zn(MIm2Pr)Cl] showed the presence of several different species, with ions corresponding to, e.g., [MIm2Pr + Zn]+, [2(MIm2Pr) + Zn + H]+, [2(MIm2Pr) + 2Zn + Cl]+, [3(MIm2Pr) +2Zn + H]2+ and [3(MIm2Pr) + 2Zn]+. On the basis of the NMR, IR, and ESI–MS data, the structure of [Zn(MIm2Pr)Cl] can be described as an oligomeric/polymeric species both in the solid state and in solution. This was confirmed by the single-crystal structure determination of [Zn(MIm2Pr)Cl(H2O)].
Crystal structure of [Zn(MIm2Pr)Cl(H2O)]
Colorless single crystals of complex [Zn(MIm2Pr)Cl(H2O)] suitable for X-ray diffraction were obtained from a solution of [Zn(MIm2Pr)Cl] in H2O upon standing. The molecular structure of [Zn(MIm2Pr)Cl(H2O)] is depicted in Fig. 6, with selected bond lengths and angles presented in Table 2.
Fig. 6.

Molecular structure of [Zn(MIm2Pr)Cl(H2O)] in the crystal, showing three molecules of the carboxylato-bridged coordination polymer, forming a one-dimensional chain in the crystallographic c-direction. C–H hydrogen atoms have been omitted for clarity. Displacement ellipsoids are drawn at the 50% probability level. Symmetry operation a 3/2 − x, 1 − y, z − 1/2
Table 2.
Selected bond lengths (angstroms) and bond angles (degrees) for [Zn(MIm2Pr)Cl(H2O)]
| Bond | Bond length | Angle | Bond angle | Angle | Bond angle |
|---|---|---|---|---|---|
| Zn1–N2 | 2.1203 (15) | N4–Zn1–N2 | 88.05 (6) | N2–Zn1–O1a | 92.41 (5) |
| Zn1–N4 | 2.0517 (14) | N2–Zn1–O3 | 173.58 (5) | O1a–Zn1–O3 | 82.24 (5) |
| Zn1–O1a | 1.9975 (11) | O3–Zn1–N4 | 90.38 (5) | N4–Zn1–Cl1 | 119.50 (4) |
| Zn1–Cl1 | 2.2844 (4) | O3–Zn1–Cl1 | 90.23 (4) | Cl1–Zn1–O1a | 130.40 (4) |
| Zn1–O3 | 2.1860 (13) | Cl1–Zn1–N2 | 95.97 (4) | O1a–Zn1–N4 | 109.55 (5) |
Symmetry operation a 3/2 − x, 1 − y, z − 1/2
The single-crystal structure determination reveals that the structure of the metal complex is that of a coordination polymer. The zinc atom is coordinated by two 1-methylimidazole groups of one MIm2Pr ligand, while the carboxylato group of this ligand coordinates to a neighboring zinc atom. In this way an infinite one-dimensional chain parallel to the crystallographic c-axis is formed. The chlorido anion and a coordinated water molecule complete the coordination sphere around each zinc center and render it five-coordinate. Thus, a new, bridging coordination mode of MIm2Pr is observed in the structure of [Zn(MIm2Pr)Cl(H2O)] comprising N,N-bidentate coordination to one metal center and O-monodentate coorrdination to a second one. This coordination mode complements the alternative N,N,O-tridentate facial capping mode of the ligand, which is commonly observed for divalent transition metals [26, 27, 39]. The coordination geometry around the five-coordinate Zn atom is distorted trigonal bipyrimidal (τ = 0.72) [40]. The equatorial positions are occupied by N4 and the monoanionic carboxylato O1a and chlorido Cl1 ligands with O1a–Zn1–N4, Cl1–Zn1–O1a, and N4–Zn1–Cl1 angles of 109.55(5), 130.40(4), and 109.50(4)°, respectively (Σangles = 360°). The axial positions are occupied by O3 of the water molecule and imidazole N2 [O3–Zn1–N2 173.58(5)°]. The Zn1–N2 and Zn1–N4 bond lengths are different [2.1203(15) and 2.0517(14) Å], consistent with their respective axial and equatorial positions. The Zn1–O1a distance of 1.9975(11) Å is slightly longer than the Zn–Ocarboxylato bond length observed in [Zn(bdatBu2,Me2)Cl]2 (1.942 Å) [12] [bpatBu2,Me2 is (3,5-di-tert-butylpyrazol-1-yl)(3′,5′-dimethylpyrazol-1-yl)acetate], in which a carboxylato group is similarly coordinated to the metal center. The zinc-aquo distance of 2.1860(13) Å in [Zn(MIm2Pr)Cl(H2O)] is rather long compared with most reported Zn–OH2 distances (mean Zn–OH2 distance for five-coordinate zinc complexes is 2.046 Å; Cambridge Structural Database version 5.27) [41]. In the related coordination polymer [Zn(bpatBu,Me)(H2O)](ClO4) [11] [bpatBu,Me is bis(5-tert-butyl-3-methylpyrazol-2-yl)acetate], where the water molecule occupies a position in the basal plane of the trigonal bipyramid, the zinc–water distance, for example, amounts to 1.961 Å. [Zn(MIm2Pr)Cl(H2O)] crystallizes in the noncentrosymmetric space group P212121 and the determination of the Flack parameter indicates that the crystal is enantiomerically pure.
The one-dimensional linear chains in Zn(MIm2Pr)Cl(H2O)] are interconnected via hydrogen-bonding interactions into a two-dimensional network (Fig. 7, Table 3). The water molecule binds via its two hydrogen atoms by hydrogen bonding to a carbonyl O and a chlorido anion as hydrogen acceptors of a [Zn(MIm2Pr)Cl(H2O)] molecule in a neighboring strand.
Fig. 7.
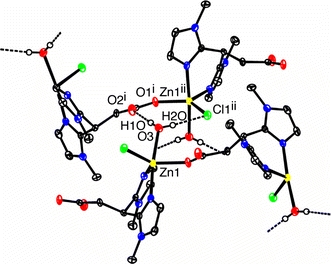
Hydrogen-bonding network in [Zn(MIm2Pr)Cl(H2O)]. Hydrogen bonds connect the infinite one-dimensional chains into a two-dimensional network. C–H hydrogen atoms have been omitted for clarity. Symmetry operations i 1 − x, 1/2 + y, 1/2 − z; ii −1/2 + x, 3/2 − y, −z
Table 3.
Selected hydrogen bond lengths (angstroms) and angles (degrees) for [Zn(MIm2Pr)Cl(H2O)]
| Donor–H···acceptor | D–H | H···A | D···A | D–H···A |
|---|---|---|---|---|
| O3–H1O···O2i | 0.78 (3) | 1.95 (3) | 2.7307 (19) | 176 (3) |
| O3–H2O···Cl1ii | 0.84 (3) | 2.31 (3) | 3.1472 (15) | 171 (3) |
Symmetry operations i 1 − x, 1/2 + y, 1/2 − z; ii −1/2 + x, 3/2 − y, −z
Zinc complexes of iPrEtIm2Pr
In order to assess the influence of the presence of steric bulk in the ligand framework, we also studied the zinc coordination chemistry of ligand iPrEtIm2Pr. The ethyl and isopropyl substituents of this ligand provide significant steric hindrance and, moreover, increase the solubility of its complexes in organic solvents. The 1:1 complex [Zn(iPrEtIm2Pr)Cl] was synthesized by the reaction of equimolar amounts of ZnCl2 and K[iPrEtIm2Pr] in methanol. The complex was isolated in almost quantitative yield. The IR spectrum of [Zn(iPrEtIm2Pr)Cl] showed the asymmetric and symmetric carbonyl stretch vibrations at 1,627 and 1,381 cm−1 [Δ(νas − νs) = 246 cm−1], respectively. The structure of [Zn(iPrEtIm2Pr)Cl] was determined by X-ray crystal structure determination.
Crystal structure of [Zn(iPrEtIm2Pr)Cl]
Colorless single crystals of [Zn(iPrEtIm2Pr)Cl] suitable for X-ray diffraction were obtained from a solution of [Zn(iPrEtIm2Pr)Cl] in methanol. The molecular structure of [Zn(iPrEtIm2Pr)Cl] is depicted in Fig. 8, with selected bond lengths and angles presented in Table 4. The crystal structure shows that whereas like [Zn(MIm2Pr)Cl(H2O)] [Zn(iPrEtIm2Pr)Cl] also crystallizes as a coordination polymer, the molecular structure of [Zn(iPrEtIm2Pr)Cl] is noticeably different. Unlike in the structure of [Zn(MIm2Pr)Cl(H2O)], no water molecule is found to coordinate to zinc in [Zn(iPrEtIm2Pr)Cl]. Instead of the five-coordinate structure in [Zn(MIm2Pr)Cl(H2O)], the zinc atom in [Zn(iPrEtIm2Pr)Cl] is four-coordinate. It has a pseudotetrahedral coordination geometry with two imidazole groups of one iPrEtIm2Pr ligand, a chlorido, and a carboxylato group of a neighboring ligand coordinated to it. The bridging mode of iPrEtIm2Pr observed in [Zn(iPrEtIm2Pr)Cl] is the same as found in [Zn(MIm2Pr)Cl(H2O)]. The deviation from ideal tetrahedral geometry is reflected in the observed bond angles [91.90(9)–119.66(6)°]. The smallest angle (N2–Zn1–N4) is caused by the inherent geometrical restrictions imposed by the ligand and the largest angle (Cl1–Zn1–O1a) probably reflects the steric influence of the isopropyl groups. The Zn–N, Zn–O, and Zn–Cl bond lengths in [Zn(iPrEtIm2Pr)Cl] are shorter than the corresponding distances in [Zn(MIm2Pr)Cl(H2O)], which reflects the stronger Lewis acidity of zinc(II) in a four-coordinate versus a five-coordinate geometry. The Zn1–O1a bond length of 1.9628(19) Å is comparable to values reported for other tetrahedral N,N,O zinc complexes with a monodentate carboxylate [9, 11, 12, 18]. The different coordination geometry at zinc results in a more folded zigzag polymer chain in [Zn(iPrEtIm2Pr)Cl], compared with the more stretched zigzag chain observed for [Zn(MIm2Pr)Cl(H2O)].
Fig. 8.
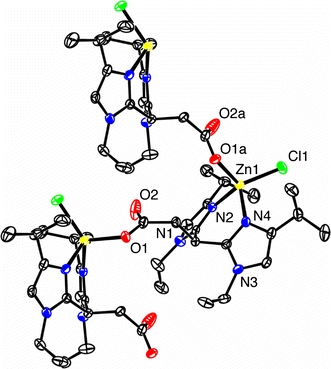
Molecular structure of [Zn(iPrEtIm2Pr)Cl] in the crystal, showing three molecules of the carboxylato-bridged coordination polymer. C–H hydrogen atoms have been omitted for clarity. Displacement ellipsoids are drawn at the 50% probability level. Symmetry operation a x, 1 − y, z − 1/2
Table 4.
Selected bond lengths (angstroms) and bond angles (degrees) for [Zn(iPrEtIm2Pr)Cl]
| Bond | Bond length | Angle | Bond angle | Angle | Bond angle |
|---|---|---|---|---|---|
| Zn1–N2 | 2.038 (2) | Cl1–Zn1–N2 | 116.85 (7) | N2–Zn1–O1a | 108.85 (8) |
| Zn1–N4 | 2.045 (2) | Cl1–Zn1–N4 | 112.12 (7) | N4–Zn1–O1a | 103.15 (9) |
| Zn1–O1a | 1.9628 (19) | Cl1–Zn1–O1a | 119.66 (6) | ||
| Zn1–Cl1 | 2.2146 (7) | N2–Zn1–N4 | 91.90 (9) |
Symmetry operation a x, 1 − y, z − 1/2
The structure of [Zn(iPrEtIm2Pr)Cl] and more generally the coordination chemistry of iPrEtIm2Pr with Zn in methanol solution were studied by 1H NMR spectroscopy and ESI–MS. The 1H NMR spectra of a titration experiment comprising the addition of ZnCl2 to the potassium salt of iPrEtIm2Pr in methanol solution are shown in Fig. 9.
Fig. 9.
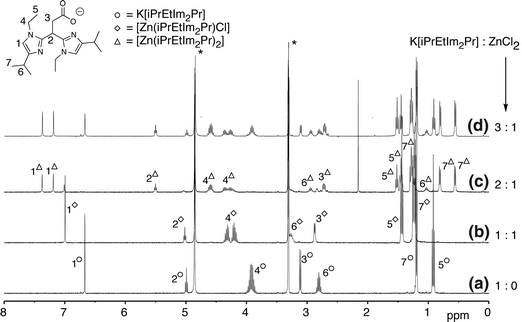
400 MHz 1H NMR spectra of the titration of K[iPrEtIm2Pr] with ZnCl2 in CD3OD at room temperature: a K[iPrEtIm2Pr] and ZnCl2 1:0; b K[iPrEtIm2Pr] and ZnCl2 1:1; c K[iPrEtIm2Pr] and ZnCl2 2:1; d K[iPrEtIm2Pr] and ZnCl2 3:1. Asterisks denote the residual solvent peak and H2O
Comparison of the 1H NMR spectra of K[iPrEtIm2Pr] (Fig. 9, spectrum a) with that of an equimolar K[iPrEtIm2Pr]–ZnCl2 mixture shows significant shifts pointing to the formation of a [Zn(iPrEtIm2Pr)Cl] complex (spectrum b). In the 1H NMR spectrum of [Zn(iPrEtIm2Pr)Cl] one sharp resonance is observed for the imidazole protons at 6.99 ppm, which is shifted to lower field compared with that of the free ligand. The two imidazole rings are equivalent on the NMR timescale, suggesting that they are symmetrically bound to zinc. Importantly, the CH2 resonance of the ethyl groups is shifted and split into two signals of equal intensity (4.20 and 4.32 ppm). Similarly, the methyl groups of the isopropyl substituent now appear as two doublets of equal intensity. These splittings confirm that the CH2(Et) and CH3(iPr) groupings contain diastereotopic protons and methyl groups, respectively. No splitting or change in multiplicity of the resonances associated with the propionate backbone is observed, which is indicative of mirror symmetry of the complex in solution. Although at first sight the NMR data would allow an assignment of the solution structure for [Zn(iPrEtIm2Pr)Cl] as a mononuclear zinc complex with a facially capped ligand, a bridged oligomeric species (similar as the one discussed before for [Zn(MIm2Pr)Cl]) is preferred. ESI–MS data support the latter description since trimeric, dimeric, and monomeric species are observed. Prominent ions corresponding to [Zn(iPrEtIm2Pr)Cl + H]+, [Zn2(iPrEtIm2Pr)2Cl2 + H]+, and [Zn3(iPrEtIm2Pr)3Cl3 + H]+ are, for instance, observed. The structure of [Zn(iPrEtIm2Pr)Cl] in solution can therefore best be described as a coordination polymer similar to [Zn(MIm2Pr)Cl].
Upon addition of 2 equiv of K[iPrEtIm2Pr] to ZnCl2 in CD3OD (Fig. 9, spectrum c), a second species is formed next to the 1:1 complex [Zn(iPrEtIm2Pr)Cl]. The fact that two species are observed, indicates that an equilibrium is obtained, i.e., the clean formation of a symmetrical 2:1 iPrEtIm2Pr to zinc complex, as previously observed for MIm2Pr, does not occur. Further inspection of the 1H NMR spectrum indicated that all resonances of the 1-ethyl-4-isopropylimidazole groups of the new species are split into two signals of equal intensity, e.g., the imidazole ring protons at 7.37 and 7.19 ppm. This suggests that the two imidazole groups are inequivalent and experience a drastically different magnetic environment. For instance, the two resonances of the CH protons of the two isopropyl groups are found at 2.96 and 1.03 ppm. Furthermore, the methine proton at the bridging carbon is still a single triplet, whereas the CH2 group of the propionate backbone has become diastereotopic. Variable-temperature NMR studies over a temperature range from −40 to 60 °C did not show significant spectral changes. The ESI–MS spectrum of a 2:1 K[iPrEtIm2Pr]–ZnCl2 solution showed prominent ions corresponding to the [Zn(iPrEtIm2Pr)2 + H]+ and [Zn(iPrEtIm2Pr)2 + 2H]2+ cations.
On the basis of these data, the structure of the new species is proposed to be a 2:1 ligand-to-zinc complex [Zn(iPrEtIm2Pr)2] in which each ligand is coordinated via its carboxylate group and one imidazole group, resulting in a four-coordinated zinc(II) ion (Fig. 10). The coordination of two of the three donor atoms in a 2:1 ligand-to-metal complex has also been observed with bulky N,N,S(thiolate) ligands in the complexes [Zn(L2S)2] [L2SH is (3-tert-butyl-5-methyl-2-thiophenyl)bis(3,5-dimethyl pyrazolyl) methane] and [Zn(L2)2] [L2 is 1-methoxy-2-methyl-1,1-bis(1-methyl-4,5-diphenyl-1H-imidazol-2-yl)propane-2-thiol] [42, 43]. The addition of a third equivalent of K[iPrEtIm2Pr] (spectrum d) results in the disappearance of the signals associated with [Zn(iPrEtIm2Pr)Cl] and both free ligand K[iPrEtIm2Pr] and [Zn(iPrEtIm2Pr)2] are detected. No attempts to isolate [Zn(iPrEtIm2Pr)2] were undertaken.
Fig. 10.

Proposed structure for [Zn(iPrEtIm2Pr)2]. The structure was calculated by molecular mechanics geometry optimization (Spartan SGI [44] MMFF94)
Attempted synthesis of a zinc bis(1-methylimidazol-2-yl)acetate complex
Recently, the zinc coordination chemistry of bis(pyrazolyl)acetates (bpa) has been actively explored to mimic the 2-His-1-carboxylate facial triad found in several different zinc enzymes [9, 11, 12, 18, 19]. The backbone of bpa is more rigid than the backbone of the ligands MIm2Pr and iPrEtIm2Pr used in the present study. In particular, the bridging N,N-bidentate-O-monodentate coordination mode reported here is less favorable for bpa, for which a strong predisposition to an N,N,O facial capping mode is expected. The synthesis of zinc complexes of MIm2Ac would therefore be desirable, since it would allow a direct comparison of the respective pyrazole and imidazole donor groups (Fig. 11).
Fig. 11.

Ligands benzyl bis(1-methylimidazol-2-yl)acetate (BnMIm2Ac), MIm2Ac, and bis(pyrazolyl)acetate (bpa) and the synthesis of complexes [ZnCl2(BnMIm2Ac)] and [ZnCl2(MIm2CH2)]
We attempted the synthesis of ligand MIm2Ac, but found that the common synthetic procedure, i.e., lithiation of MIm2CH2 followed by reaction with carbon dioxide [45], did not result in product formation in acceptable yields. Instead, it was found that the ligand or an ester precursor of the ligand decarboxylates rather easily, as was also observed by Peters et al. [39]. The benzyl ester of bis(1-methylimidazol-2-yl)acetic acid (BnMIm2Ac), however, was sufficiently stable and could be isolated and purified after reaction of lithio bis(1-methylimidazol-2-yl)methane with benzylchloroformate. Since hydrolysis and hydrogenolysis of the ester moiety resulted in decomposition of the ligand, the 1:1 zinc dichloride complex with BnMIm2Ac was synthesized in the hope that autocatalytic ester hydrolysis by the zinc complex would yield the desired zinc bis(1-methylimidazol-2-yl)acetate complex. A similar approach, involving the autocatalytic saponification of a zinc bis(picolyl)glycine ethyl ester complex, has been reported by Abufarag and Vahrenkamp [46].
The reaction of equimolar amounts of ZnCl2 and BnMIm2Ac gave the complex [ZnCl2(BnMIm2Ac)] as a white powder in quantitative yield. The structure of [ZnCl2(BnMIm2Ac)] was determined by X-ray diffraction studies and showed the BnMIm2Ac ligand bound to the zinc center in an N,N-bidentate way (Fig. 12, top). However, stirring of [ZnCl2(BnMIm2Ac)] in H2O at room temperature in an attempt to obtain [Zn(MIm2Ac)Cl] resulted in decomposition of the ligand and formation of [ZnCl2(MIm2CH2)], which was isolated as one of the decomposition products. [ZnCl2(MIm2CH2)] was also characterized crystallographically (Fig. 12, bottom). On the basis of these findings, it was concluded that the MIm2Ac monoanion is too unstable for practical studies and the pursuit of this ligand was abandoned.
Fig. 12.

Molecular structures of [ZnCl2(BnMIm2Ac)] (top) and [ZnCl2(MIm2CH2)] (bottom) in the crystal. Displacement ellipsoids are drawn at the 50% probability level. All hydrogen atoms have been omitted for clarity. Selected bond lengths (angstroms) as follows: [ZnCl2(BnMIm2Ac)], Zn1–N2 2.011(2), Zn1–N4 1.994(2), Zn1–Cl1 2.2242(7), Zn1–Cl2 2.2554(7); [ZnCl2(MIm2CH2)], Zn1–N2 2.0247(17), Zn1–N4 2.0222(17), Zn1–Cl1 2.2110(6), Zn1–Cl2 2.2504(6)
Reaction of [Zn(MIm2Pr)Cl] with sodium pyruvate: oxalate formation
In an attempt to prevent the formation of coordination polymers of MIm2Pr and to obtain mononuclear complexes with the desired N,N,O facial capping mode of the ligand, several different monoanionic, bidendate coligands were used. In the course of this study, we also investigated the reaction of [Zn(MIm2Pr)Cl] with sodium pyruvate. The addition of 1 equiv of sodium pyruvate to a solution of [Zn(MIm2Pr)Cl] in D2O resulted in the appearance of a new singlet at 2.38 ppm in the 1H NMR spectrum (Fig. 13, top spectrum). The signal is not shifted with respect to the signal observed for free sodium pyruvate, which indicates that the interaction between [Zn(MIm2Pr)Cl] and the pyruvate anion is weak, at best. The ESI–MS spectrum of an aqueous solution of equimolar amounts of [Zn(MIm2Pr)Cl] and sodium pyruvate, however, did show some major ions corresponding to a mononuclear {[Zn(MIm2Pr)(pyruvate)] + Na}+ and a dinuclear {[Zn2(MIm2Pr)2(pyruvate)]}+ cation, in addition to the ions observed in the ESI–MS spectrum of a solution of [Zn(MIm2Pr)Cl] in water.
Fig. 13.

1H NMR spectra of a solution of [Zn(MIm2Pr)Cl] in D2O (asterisk) after addition of 1 equiv of sodium pyruvate and the same solution after 24 h
However, when the mixture of [Zn(MIm2Pr)Cl] and pyruvate in D2O was followed in time by NMR spectroscopy, the gradual decrease in intensity of the signal of the pyruvate methyl group was noticed until it ultimately disappeared (Fig. 13, bottom spectrum). After several days colorless crystals suitable for X-ray diffraction studies were formed in the NMR tube. To our surprise, the X-ray crystal structure showed the structure of the zinc complex [Zn2(MIm2Pr)2(ox)]·6H2O. [Zn2(MIm2Pr)2(ox)]·6H2O has the structure of a coordination polymer involving [Zn(MIm2Pr)]+ cations bridged by oxalato dianions.
To unequivocally prove the relevance of the structure obtained, zinc coordination polymer [Zn2(MIm2Pr)2(ox)] was also synthesized independently. Indeed, the addition of 0.5 equiv of K2C2O4·H2O to an aqueous solution of [Zn(MIm2Pr)Cl] yielded a white, insoluble powder. This powder was identified as [Zn2(MIm2Pr)2(ox)] by elemental analysis and IR spectroscopy. Typical features of the IR spectrum of [Zn2(MIm2Pr)2(ox)] include absorptions at 1,674, 1,638, 1,591, and 1,399 cm−1, which can be attributed to the oxalato and carboxylato groups in the polymer. Crystallization of [Zn2(MIm2Pr)2(ox)] proved difficult given its general insolubility. Single crystals could, however, be obtained via the gel crystallization method [47]. A gel formed from a solution of tetramethoxysilane and potassium oxalate hydrate in water was layered with an aqueous solution of K[MIm2Pr] and ZnCl2. Single crystals were obtained after several days and could be unequivocally identified as [Zn2(MIm2Pr)2(ox)] by X-ray crystal structure determination.
Crystal structure of [Zn2(MIm2Pr)2(ox)]·6H2O
The molecular structure of [Zn2(MIm2Pr)2(ox)]·6H2O is shown in Fig. 14, with selected bond lengths and angles presented in Table 5.1 The polymeric structure of [Zn2(MIm2Pr)2(ox)]·6H2O comprises two crystallographically inequivalent zinc atoms. Both zinc centers are five-coordinate and differ only in the slightly different bond lengths and angles. Each zinc atom is coordinated in an N,N-bidentate fashion by the 1-methylimidazole groups of one MIm2Pr ligand, two oxygen atoms of a bis O,O-bidentate bridging oxalato group, and a carboxylato O donor atom of a neighboring MIm2Pr ligand. The combination of the bridging oxalato and MIm2Pr ligands constructs a polymeric structure leading to the formation of two-dimensional sheets.
Fig. 14.
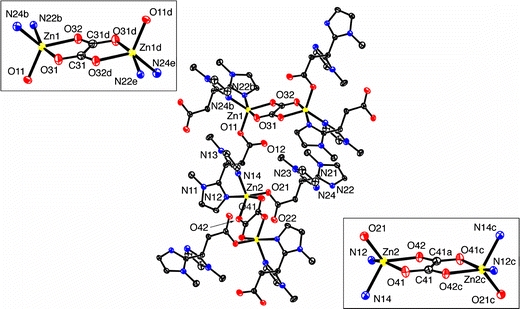
Molecular structure of [Zn2(MIm2Pr)2(ox)]·6H2O in the crystal. C–H hydrogen atoms and all solvent water molecules have been omitted for clarity. Displacement ellipsoids are drawn at the 50% probability level
Table 5.
Selected bond lengths (angstroms) and angles (degrees) for [Zn2(MIm2Pr)2(ox)]·6H2O
| Bond | Bond length | Angle | Bond angle | Angle | Bond angle |
|---|---|---|---|---|---|
| Zn1–O31 | 2.258 (2) | O11–Zn1–O31 | 89.48 (8) | O21–Zn2–O41 | 90.33 (8) |
| Zn1–O32 | 2.018 (2) | O11–Zn1–O32 | 124.09 (9) | O21–Zn2–O42 | 125.19 (8) |
| Zn1–O11 | 1.9694 (19) | O11–Zn1–N22b | 108.91 (9) | O21–Zn2–N12 | 109.08 (9) |
| Zn1–N24b | 2.046 (2) | O11–Zn1–N24b | 107.61 (9) | O21–Zn2–N14 | 107.85 (9) |
| Zn1–N22b | 2.088 (2) | O31–Zn1–O32 | 77.16 (9) | O41–Zn2–O42 | 77.84 (8) |
| Zn2–O41 | 2.233 (2) | O31–Zn1–N22b | 161.43 (9) | O41–Zn2–N12 | 160.51 (8) |
| Zn2–O42 | 2.0179 (19) | O31–Zn1–N24b | 87.69 (9) | O41–Zn2–N14 | 87.24 (9) |
| Zn2–O21 | 1.9630 (19) | O32–Zn1–N22b | 89.94 (8) | O42–Zn2–N12 | 89.12 (8) |
| Zn2–N12 | 2.089 (2) | O32–Zn1–N24b | 125.38 (9) | O42–Zn2–N14 | 124.42 (9) |
| Zn2–N14 | 2.052 (2) | N22b–Zn1–N24b | 89.06 (9) | N12–Zn2–N14 | 88.34 (9) |
| C31–O31 | 1.248 (4) | C41–O41 | 1.250 (3) | ||
| C31d–O32 | 1.252 (4) | C41c–O42 | 1.254 (3) | ||
| C31–C31d | 1.544 (6) | C41–C41c | 1.537 (6) |
Symmetry operations b 1 + x, y, z; c −x, 1 − y, −z; d 1 − x, 1 − y, 1 − z
The coordination geometry around each zinc center is best described as a severely distorted trigonal bipyramid, with τ values of 0.60 and 0.59 for Zn1 and Zn2, respectively [40]. The equatorial positions are occupied by N24b/N14, and the anionic carboxylato O11/O21 and oxalato O32/O42 ligands for Zn1/Zn2, respectively. The axial positions are occupied by O31/O41 of the oxalato group and imidazole N22b/N12 for Zn1/Zn2 [O–Zn–N angles of 161.43(9)°/160.51(8)°]. The planar oxalato moieties are rather asymmetrically bound to the metal centers, with the Zn1–O31 and Zn1–O32 distances amounting to 2.258(2) and 2.018(2) Å, for instance. This asymmetry has also been observed in other bisbidentate bridged zinc–oxalato complexes with five-coordinated metal centers and is consistent with the occupation of one equatorial and one axial site of the trigonal bipyramid [10, 48]. The shortest Zn···Zn distances via the bridging oxalato group are 5.5667(7) Å for Zn1···Zn1d and 5.5332(7) Å for Zn2···Zn2c. The distances of the metal centers to the donor atoms of MIm2Pr follow the same trend as observed in [Zn(MIm2Pr)Cl(H2O)]. The crystal structure of [Zn2(MIm2Pr)2(ox)]·6H2O is further stabilized by 14 different hydrogen-bonding interactions, which involve the cocrystallized water molecules, the oxalato bridges, and the carboxylato groups (Fig. 15). The oxalato groups are, for instance, involved in two chelated, three-centered hydrogen bonds with two water molecules via four O–H···O hydrogen bonds.
Fig. 15.
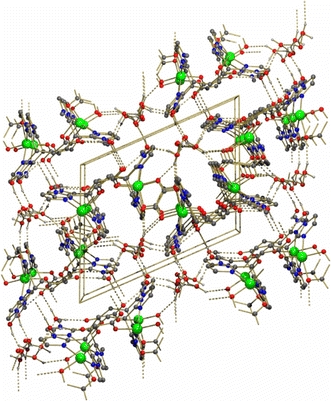
Hydrogen bonding network and crystal packing of [Zn2(MIm2Pr)2(ox)]·6H2O
Discussion
The aim of this investigation was to explore the biomimetic modeling potential of the MIm2Pr ligand family with respect to mononuclear zinc enzymes featuring the 2-His-1-carboxylate facial triad. Complex [Zn(MImPr)2] shows that the facial capping mode of MIm2Pr, as earlier observed with copper [27, 28] and iron (Bruijnincx et al., submitted) [26], is also accessible for zinc. The crystal structure of the 1:1 complex [Zn(MIm2Pr)Cl(H2O)], however, shows that MIm2Pr can also adopt other coordination modes. The formation of the oligomeric/polymeric structure of [Zn(MIm2Pr)Cl], both in solution and in the solid state, can be attributed to the flexibility of the ligand due to the CH2 spacer of the propionate backbone. The formation of polymeric [Zn(MIm2Pr)Cl(H2O)] is further aided by the insolubility of the resulting coordination polymer. Two strategies were explored to circumvent polymer formation, i.e., increasing the steric bulk of the ligand and elimination of the CH2 spacer. The latter approach demanded the synthesis of the bis(1-methylimidazol-2-yl)acetate analogue of MIm2Pr, which was found to be rather unstable and its zinc complexes could not be obtained. The effect of more steric bulk was studied with iPrEtIm2Pr, which contains the sterically more demanding isopropyl and ethyl substituents. Zinc complexes of iPrEtIm2Pr are polymeric in nature as well, albeit with a different coordination geometry at the zinc atom as observed in [Zn(MIm2Pr)Cl(H2O)]. The first coordination spheres of [Zn(MIm2Pr)Cl(H2O)] and [Zn(iPrEtIm2Pr)Cl] are compared in Fig. 16. The added steric bulk in iPrEtIm2Pr causes the zinc ion to adopt a pseudotetrahedral geometry in [Zn(iPrEtIm2Pr)Cl], instead of the distorted trigonal bipyramid observed for [Zn(MIm2Pr)Cl(H2O)]. The coordination around zinc in [Zn(iPrEtIm2Pr)Cl] resembles the first coordination sphere of the active sites of the zinc enzymes thermolysin, carboxypeptidase A, and neutral protease. The polymeric rather than monomeric nature of the complex, however, clearly limits its potential as a biological mimic.
Fig. 16.

Comparison of the first coordination spheres of the zinc atoms in the coordination polymers [Zn(MIm2Pr)Cl(H2O)] (top) and [Zn(iPrEtIm2Pr)Cl] (bottom)
The isolation of [Zn2(MIm2Pr)2(ox)] from the reaction of [Zn(MIm2Pr)Cl] with sodium pyruvate was rather unexpected and is, to the best of our knowledge, the first report of the nonoxidative formation of oxalate from pyruvate (Scheme 1).2 Interestingly, Takeste and Vahrenkamp [51] very recently reported a trispyrazolylborato–zinc complex with pyruvate, representing the first structurally characterized zinc complex of an α-keto acid. Although in their case the zinc–pyruvate complex obtained was stable, unexpected cleavage reactions with other α-keto acids, such as benzoylformic acid, and α-diketones were observed.
Scheme 1.
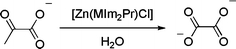
The observed transformation of pyruvate to oxalate mediated by [Zn(MIm2Pr)Cl]
A control experiment showed that an aqueous sodium pyruvate solution is stable under the reaction conditions. Apparently, [Zn(MIm2Pr)Cl] mediates the conversion of pyruvate to oxalate. The product [Zn2(MIm2Pr)2(ox)] invariably deposited as crystals in experiments done on the NMR scale. On a preparative scale (0.3 mmol), the reaction also takes place, yet the product was isolated as an insoluble white powder. The IR spectrum of this powder was identical to that obtained for independently synthesized [Zn2(MIm2Pr)2(ox)]. Most importantly, further experiments revealed that the reaction is catalytic in zinc. For example, addition of 20 equiv of sodium pyruvate to a solution of [Zn(MIm2Pr)Cl] in water under ambient conditions resulted in ten turnovers after 24 h. The disappearance of the pyruvate methyl signal was also observed in a solution containing ZnCl2 and sodium pyruvate without any further additives. Concomitantly, a precipitate was observed in the NMR tube. However, the composition of this precipitate, i.e., the formation of oxalate, could not be unambiguously established.
Since it is known that pyruvate keto–enol tautomerization is catalyzed by divalent metal ions such as Zn(II) [52, 53], the disappearance of the methyl signal could in principle also be attributed to consecutive H/D-exchange steps. This would yield the deuterated pyruvate-d3 anion, which is also invisible in 1H NMR spectroscopy. To be able to exclude this possibility, experiments were performed in H2O and samples were diluted with 2 equiv of D2O just before the measurement. The same observations were made as previously, i.e., gradual disappearance of the methyl signal and slow crystallization of the product in the NMR tube. The presence of appreciable amounts of enolate should also lead to the detection of the dimerization product of pyruvate [52, 53]. This pyruvate dimer, which results from an aldol condensation reaction and has characteristic signals at 1.38 (singlet) and 3.35 (AB pattern) ppm [52–54], was not observed under these conditions.
Phenyl pyruvic acid (and other α-ketoacids) is oxidized by molecular oxygen to give benzaldehyde and oxalate or the carbon oxides (CO and/or CO2) [55, 56]. This reaction is catalyzed by several metal ions, such as Mn(II), Fe(II), and Cu(II), but not by Zn(II) [57, 58], and has been attributed to the presence of the enol tautomer, which is capable of reacting with dioxygen [61]. In the absence of dioxygen, i.e., under an Ar atmosphere, pyruvate does react with [Zn(MIm2Pr)Cl] to form oxalate. The anaerobic reactivity cannot be attributed to any residual dioxygen traces, since phenyl pyruvate (a substrate more prone to the reaction with dioxygen) did not show any reactivity under these conditions.
The observed conversion of pyruvate to oxalate under anaerobic conditions clearly places it outside previously observed formation of oxalate from oxidative transformations of α-keto acids [55, 56, 59–63]. The observation is important, since pyruvate is a major metabolic junction and it is therefore of prime importance to understand its reactivity. At present no biological counterpart has been reported for the pyruvate–oxalate conversion catalyzed by [Zn(MIm2Pr)Cl]. The chemical transformation as reported here, however, takes place at room temperature and physiological pH and should therefore be considered as a possible alternative pathway in the reactivity of pyruvate. The mechanism of this conversion is currently not clear. No other products could be detected by NMR during the reaction, which precludes the formulation of a mass balance for the reaction and clearly hampers a mechanistic interpretation. In any case, the pyruvate to oxalate conversion requires the scission of a C–C bond. On the other hand, the reactivity of zinc enzymes and their model complexes is often of hydrolytic nature. This can be attributed to the generation of a nucleophilic hydroxide species and/or the activation of a coordinated substrate towards attack of such a nucleophile [3]. The formation of oxalate from pyruvate via a hydrolytic mechanism is, however, difficult to envision. Further investigations have to be performed to address the mechanistic questions concerning this transformation.
Conclusions
The difference in applied steric bulk by the ligands MIm2Pr and iPrEtIm2Pr is reflected in the coordination chemistry of the two ligands towards ZnCl2. Octahedral (MIm2Pr) versus tetrahedral (iPrEtIm2Pr) and trigonal pyramidal (MIm2Pr) versus tetrahedral (iPrEtIm2Pr) coordination geometries were obtained in the 1:1 and 2:1 ligand-to-metal complexes, respectively. The bridging binding mode of the two crystallographically characterized coordination polymers illustrates the intrinsic flexibility of the 3,3-bis(imidazol-2-yl)propionate ligand framework. The polymeric nature of the complexes could limit their biological relevance with respect to the 2-His-1-carboxylate facial triad in the case of zinc. On the other hand, attempts aimed at obtaining structurally more faithful mononuclear complexes led to the observation of the unprecedented Zn-mediated pyruvate to oxalate conversion. This new, nonoxidative transformation of an important metabolic junction is intriguing and warrants further investigation.
Footnotes
The possibility of the presence of an enolate dianion in the crystal structure of [Zn2(MIm2Pr)2(ox)]·6H2O could be unequivocally ruled out on the basis of crystallographic arguments.
Unexpected oxalate formation by Zn complexes is not completely unprecedented. The reaction of Zn(NO3)2 with pyridine-2,4,6-tricarboxylic acid in the presence of pyridine at room temperature resulted, for instance, in the formation of the zinc oxalato polymer [Zn(ox)(py)2]·H2O [49]. Another oxalato bridged zinc coordination polymer was obtained from the hydro(solvo)thermal reaction between Zn(NO3)2 and pyridine in ethanol or methanol [50]. It was proposed that the oxalate resulted from ethanol oxidation and oxidative coupling of methanol for the two different solvents, respectively. Clearly, different reaction mechanisms are operative in these cases.
Contributor Information
Anthony L. Spek, Email: a.l.spek@chem.uu.nl
Robertus J. M. Klein Gebbink, Phone: +31-30-2531889, FAX: +31-30-2523615, Email: r.j.m.kleingebbink@chem.uu.nl
References
- 1.Costas M, Mehn MP, Jensen MP, Que L Jr (2004) Chem Rev 104:939–986 [DOI] [PubMed]
- 2.Koehntop KD, Emerson JP, Que L Jr (2005) J Biol Inorg Chem 10:87–93 [DOI] [PubMed]
- 3.Parkin G (2004) Chem Rev 104:699–768 [DOI] [PubMed]
- 4.Stark W, Pauptit RA, Wilson KE, Jansonius JN (1992) Eur J Biochem 207:781–791 [DOI] [PubMed]
- 5.Vahrenkamp H (1999) Acc Chem Res 32:589–596 [DOI]
- 6.Alsfasser R, Trofimenko S, Looney A, Parkin G, Vahrenkamp H (1991) Inorg Chem 30:4098–4100 [DOI]
- 7.Kitajima N, Hikichi S, Tanaka M, Moro-oka Y (1993) J Am Chem Soc 115:5496–5508 [DOI]
- 8.Looney A, Han R, McNeill K, Parkin G (1993) J Am Chem Soc 115:4690–4697 [DOI]
- 9.Beck A, Weibert B, Burzlaff N (2001) Eur J Inorg Chem 521–527
- 10.Chiu Y-H, Canary JW (2003) Inorg Chem 42:5107–5116 [DOI] [PubMed]
- 11.Hammes BS, Kieber-Emmons MT, Letizia JA, Shirin Z, Carrano CJ, Zakharov LN, Rheingold AL (2003) Inorg Chim Acta 346:227–238 [DOI]
- 12.Hegelmann I, Beck A, Eichhorn C, Weibert B, Burzlaff N (2003) Eur J Inorg Chem 339–347
- 13.Maldonado Calva JA, Vahrenkamp H (2005) Inorg Chim Acta 358:4019–4026 [DOI]
- 14.Mareque Rivas JC, Torres Martín de Rosales R, Parsons S (2003) Dalton Trans 2156–2163
- 15.Niklas N, Walter O, Alsfasser R (2000) Eur J Inorg Chem 1723–1731
- 16.Niklas N, Zahl A, Alsfasser R (2003) Dalton Trans 778–786 [DOI] [PubMed]
- 17.Papish ET, Taylor MT, Jernigan FE III, Rodig MJ, Shawhan RR, Yap GPA, Jové FA (2006) Inorg Chem 45:2242–2250 [DOI] [PubMed]
- 18.Smith JN, Hoffman JT, Shirin Z, Carrano CJ (2005) Inorg Chem 44:2012–2017 [DOI] [PubMed]
- 19.Smith JN, Shirin Z, Carrano CJ (2003) J Am Chem Soc 125:868–869 [DOI] [PubMed]
- 20.Trösch A, Vahrenkamp H (1998) Eur J Inorg Chem 827–832
- 21.Trösch A, Vahrenkamp H (2001) Inorg Chem 40:2305–2311 [DOI] [PubMed]
- 22.Friese SJ, Kucera BE, Que L Jr, Tolman WB (2006) Inorg Chem 45:8003–8005 [DOI] [PubMed]
- 23.Parkin G (2000) Chem Commun 1971–1985
- 24.Dowling C, Parkin G (1996) Polyhedron 15:2463–2465 [DOI]
- 25.Ghosh P, Parkin G (1998) J Chem Soc Dalton Trans 2281–2283
- 26.Bruijnincx PCA, Lutz M, Spek AL, Hagen WR, Weckhuysen BM, van Koten G, Klein Gebbink RJM (2007) J Am Chem Soc 129:2275–2286 [DOI] [PubMed]
- 27.Bruijnincx PCA, Lutz M, Spek AL, van Faassen EL, Weckhuysen BM, van Koten G, Klein Gebbink RJM (2005) Eur J Inorg Chem 779–787
- 28.Kervinen K, Bruijnincx PCA, Beale AM, Mesu JG, van Koten G, Klein Gebbink RJM, Weckhuysen BM (2006) J Am Chem Soc 128:3208–3217 [DOI] [PubMed]
- 29.Braussaud N, Ruther T, Cavell KJ, Skelton BW, White AH (2001) Synthesis 626–632
- 30.Darensbourg DJ, Holtcamp MW, Struck GE, Zimmer MS, Niezgoda SA, Rainey P, Robertson JB, Draper JD, Reibenspies JH (1999) J Am Chem Soc 121:107–116 [DOI]
- 31.Sheldrick GM (1997) SHELXL-97. Program for crystal structure refinement. University of Göttingen, Germany
- 32.Spek AL (2003) J Appl Cryst 36:7–13 [DOI]
- 33.Sheldrick GM (1997) SHELXS-97. Program for crystal structure solution. University of Göttingen, Germany
- 34.Beurskens PT, Admiraal G, Beurskens G, Bosman WP, Garcia-Granda S, Gould RO, Smits JMM, Smykalla C (1999) The DIRDIF99 program system, The Netherlands, technical report of the Crystallography Laboratory, University of Nijmegen
- 35.Altomare A, Burla MC, Camalli M, Cascarano GL, Giacovazzo C, Guagliardi A, Moliterni AGG, Polidori G, Spagna R (1999) J Appl Crystallogr 32:115–119 [DOI]
- 36.Duisenberg AJM, Kroon-Batenburg LMJ, Schreurs AMM (2003) J Appl Crystallogr 36:220–229 [DOI]
- 37.Flack HD (1983) Acta Crystallogr Sect A 39:876–881 [DOI]
- 38.Robert V, Lemercier G (2006) J Am Chem Soc 128:1183–1187 [DOI] [PubMed]
- 39.Peters L, Hübner E, Burzlaff N (2005) J Organomet Chem 690:2009–2016 [DOI]
- 40.Addison WA, Rao TN, Reedijk J, van Rijn J, Verschoor GC (1984) J Chem Soc Dalton Trans 1349–1356
- 41.Allen FH (2002) Acta Crystallogr Sect B 58:380–388 [DOI] [PubMed]
- 42.Hammes BS Carrano CJ (1999) Inorg Chem 38:4593–4600 [DOI] [PubMed]
- 43.Karambelkar VV, diTargiani RC, Incarvito CD, Zakharov LN, Rheingold AL, Stern CL, Goldberg DP (2004) Polyhedron 23:471–480 [DOI]
- 44.Spartan SGI version 5.1.1. Wavefunction, Irvine
- 45.Otero A, Fernández-Baeza J, Antiñolo A, Tejeda J, Lara-Sánchez A (2004) Dalton Trans 1499–1510 [DOI] [PubMed]
- 46.Abufarag A, Vahrenkamp H (1995) Inorg Chem 34:2207–2216 [DOI]
- 47.Arend H, Connelly JJ (1982) J Cryst Growth 56:642–644 [DOI]
- 48.Curtis NF, McCormick IRN, Waters TN (1973) J Chem Soc Dalton Trans 1537–1548
- 49.Ghosh SK, Savitha G, Bharadwaj PK (2004) Inorg Chem 43:5495–5497 [DOI] [PubMed]
- 50.Evans OR, Lin W (2001) Cryst Growth Des 1:9–11 [DOI]
- 51.Takeste T, Vahrenkamp H (2006) Eur J Inorg Chem 5158–5164
- 52.Tallman DE, Leussing DL (1969) J Am Chem Soc 91:6253–6256 [DOI]
- 53.Tallman DE, Leussing DL (1969) J Am Chem Soc 91:6256–6262 [DOI]
- 54.Cheong M, Leussing DL (1989) J Am Chem Soc 111:2541–2549 [DOI]
- 55.Jefford CW, Knöpfel W, Cadby PA (1978) J Am Chem Soc 100:6432–6436 [DOI]
- 56.Jefford CW, Knöpfel W, Cadby PA (1978) Tetrahedron Lett 38:3585–3588 [DOI]
- 57.Nierop Groot MN, de Bont JAM (1998) Appl Environ Microbiol 64:3009–3013 [DOI] [PMC free article] [PubMed]
- 58.Nierop Groot MN, de Bont JAM (1999) Appl Environ Microbiol 65:5590–5593 [DOI] [PMC free article] [PubMed]
- 59.Smit BA, Engels WJM, Alewijn M, Lommerse GTCA, Kippersluijs EAH, Wouters JTM, Smit G (2004) J Agric Food Chem 52:1263–1268 [DOI] [PubMed]
- 60.Mahler HR, Roberts A (1950) J Am Chem Soc 72:5095–5098 [DOI]
- 61.Meyer K (1933) J Biol Chem 103:39–49
- 62.Niki E, Yamamoto Y, Saito T, Nagano K, Yokoi S, Kamiya Y (1983) Bull Chem Soc Jpn 56:223–228 [DOI]
- 63.Villablanca M, Cilento G (1987) Biochim Biophys Acta 926:224–230 [DOI] [PubMed]