

Abstract
Lipopolysaccharides (LPS), play a key role in the pathogenesis of septic shock, a major cause of mortality in the critically ill patient. The only therapeutic option aimed at limiting downstream systemic inflammatory processes by targeting lipopolysaccharide is Toraymyxin™, an extracorporeal hemoperfusion device using solid phase-immobilized polymyxin B (PMB). While PMB is known to effectively sequester LPS, its severe systemic toxicity proscribes its parenteral use, and hemoperfusion may not be feasible in patients in shock. In our continuing efforts to develop small-molecule mimics which display the LPS-sequestering properties, but not the toxicity of PMB, a series of mono- and bis-substituted dialkylpolyamines were synthesized and evaluated. We show that EVK-203, analkylpolyamine compound specifically binds to, and neutralizes, the activity of LPS, and affords complete protection in a murine model of endotoxic shock. EVK-203 is without any apparent toxicity when administered to mice at multiples of therapeutic doses for several days. The specific endotoxinsequestering property along with a very favorable therapeutic index renders this compound an ideal candidate for preclinical development.
Keywords: Endotoxin, Lipopolysaccharide, Sepsis, Septic Shock, Alkylpolyamine, Lipopolyamine
Introduction
Endotoxins, or lipopolysaccharides (LPS), the predominant structural component of the outer membrane of Gram-negative bacteria,1 play a pivotal role in septic shock, a syndrome of systemic toxicity which occurs frequently as a sequel to serious systemic Gram-negative infections.2;3 The activation by LPS of the innate immune response, mediated via toll-like receptor-4,4 leads to an uncontrolled production of numerous inflammatory mediators, including tumor necrosis factor-α (TNF-α), interleukin-1 β (IL-1β), and interleukin-6 (IL-6),5 precipitating a systemic inflammatory response. This culminates in the frequently fatal syndrome of multiple system organ failure.6 Despite continuing advances in antimicrobial chemotherapy, the incidence of sepsis has risen almost three-fold from 1979 through 2000,7 emphasizing an urgent, unmet need to develop therapeutic options specifically targeting the pathophysiology of sepsis.
The toxicity of LPS resides in its structurally highly conserved glycolipid component called Lipid A,8 which is composed of a hydrophilic, bis-phosphorylated diglucosamine backbone, and a hydrophobic domain of 6 (E. coli) or 7 (Salmonella) acyl chains in amide and ester linkages (Fig. 1). Polymyxin B (PMB) is a membrane-active peptide antibiotic known to sequester LPS and abrogate its toxicity. The pronounced oto- and nephrotoxicity of PMB precludes its systemic use and has led to the development of an extracorporeal hemoperfusion cartridge based on PMB covalently immobilized on a polystyrene based fiber (Toraymyxin™, Toray Industries Inc., Tokyo),9;10 which was approved for clinical use in Japan in late 2000. The favorable clinical outcome associated with the use of Toraymyxin in small pilot study11 is suggestive of the therapeutic potential of sequestering circulating LPS, and warrants further validation in large randomized and controlled clinical trials.
Figure 1.
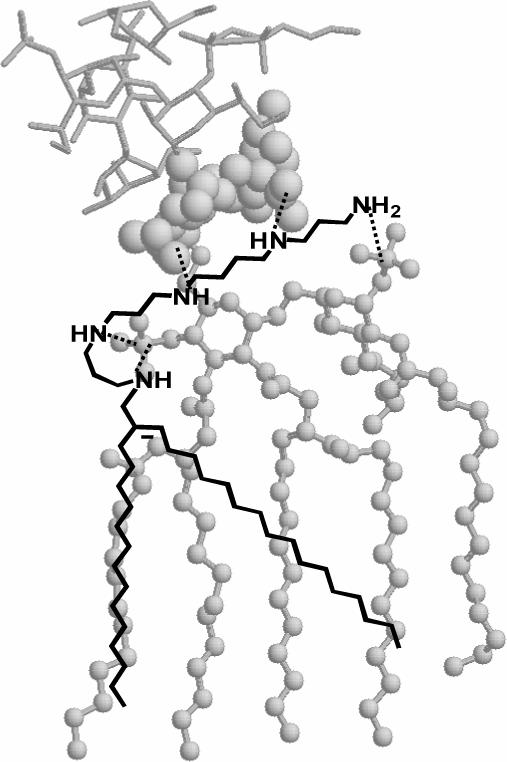
Molecular-modeling derived geometry of the complex between LPS and EVK-203. The atomic coordinates of LPS were derived from its crystal structure (PDB code: 1FCP).82 The lipid A moiety of LPS is depicted in ball-and-stick, the KDO sugars in spacefill, and the inner core glycolipid region as sticks. The mono-homologated spermine backbone is predicted to form salt-bridges (dotted lines) with both phosphate groups on lipid A, as well as participate in additional ionic H-bonds with the inner core KDO sugars as determined by docking studies.
A major goal in our laboratory has been to develop small-molecule analogues of PMB that would sequester LPS with a potency comparable to that of PMB and, importantly, be nontoxic and safe, so that it can be used parenterally for the prophylaxis or therapy of Gram-negative sepsis. Based on an NMR-derived model of PMB-lipid A complex,12 we had identified the pharmacophore necessary for optimal recognition and neutralization of lipid A (reviewed in Ref. 13). The pharmacophore is based on two simple heuristics: (a) an optimal distance of ∼14 Å is necessary between protonatable functions in bis-cationic molecules for simultaneous ionic interactions with the glycosidic phosphates on lipid A, the bis-cationic scaffold being the principal determinant of binding affinity;14;15 (b) binding is necessary, but not sufficient for LPS-neutralizing activity, and an additional, appropriately positioned hydrophobic group is obligatory for the interaction of the polyamine backbone with lipid A to manifest in neutralization of endotoxicity.16;17
Lipopolyamines of the conjugated spermine class, originally developed as DNA transfection reagents, were the first small molecules embodying the LPS-recognition pharmacophore shown to have significant LPS-sequestering activity;18 these compounds are of particular interest because they are active in vivo and afford protection in animal models of Gram-negative sepsis, are synthetically easily accessible, and, importantly, would likely be nontoxic, on account of their degradation to physiological constituents.18 Indeed, several members of this class of compounds have been approved for human use as alternatives to viral vectors.19;20
In our continuing efforts to refine our understanding of the structure-activity relationships in such molecules, we have synthesized and evaluated the LPS-binding and –neutralizing properties of several series of lipopolyamines, examining, with each increment, the length and placement of the hydrophobic (acyl or alkyl) functionality, the length and nature of the backbone, and the role of H-bond-donor and -acceptor functionalities on the scaffold. Studies on monoacylated homologated spermine derivatives established that an optimal chain length of 16 carbons for the acyl unit corresponded to optimal activity.21 Similar results were obtained in acylated lysine-spermine conjugates wherein the additional aminopropyl unit in homospermine was replaced by lysine.22 A careful examination of a 540-membered combinatorial library of bis-amide analogues with the internal secondary amines and polymethylene units of spermine replaced by amides and amino acid side chains23 led to the conclusion that the simple spermine scaffold offered analogues with the highest activity, reflecting the importance of H-bond donor -NH- groups on the scaffold which are predicted by molecular modeling studies24 to form salt bridges with the 3-Deoxy-D-manno-octulosonic acid (KDO) sugars 25 in the inner-core region of LPS (Fig. 1). Surprisingly, we observed that compounds with N-dialkyl branched scaffolds were associated with significant, unexpected acute toxicity in a murine model of septic shock.26 We further learned that the placement of the long-chain hydrophobic group was crucial, since analogues with centrally placed groups should significantly poorer activity, which we attribute to unfavorable steric interactions with the polyacyl domain of lipid A.27 Guided by experimental data from our earlier studies, as well as ongoingin silico modeling, our attention has converged on linear, unbranched spermine-type scaffolds with terminally placed hydrophobic functionalities. In the present study, we have examined a series of mono- and bis-alkyl polyamine compounds, and we report that a N1-alkyl-monohomologated spermine derivative (EVK-203) binds LPS and neutralizes its toxicity, affording complete protection in a murine model of LPS-induced lethality. Furthermore, EVK-203 is without any apparent nontoxicity, even at high doses, in a vertebrate animal model.
Results
Syntheses:
Syntheses of the various lipopolyamine derivatives, as desired for the present studies, were accomplished following the reaction schemes described below. Towards synthesizing the N-alkenyl substituted polyamine 5 (EVK-203), the 2,3-disubstituted acrolein derivative 2 was obtained via self- condensation of hexadecanal (1) as shown in Scheme 1. The Z-stereochemistry of the product was assigned based on the 1H NMR chemical shift of the olefinic proton (d 6.45), which is in good agreement with the literature reported values for similarly substituted Z-acrolein derivatives.28 Subsequent reductive amination involving the aldehyde 2 and the tetra-N-Boc-protected polyamine 321 resulted in the corresponding N-alkenyl adduct 4. Finally, removal of the Boc-protection under standard conditions provided the desired alkenyl polyamine 5.
Scheme 1.

Reagents: a. AlCl3, py, reflux. b. MgSO4, THF–MeOH, rt, followed by, NaBH4, MeOH. c. CF3CO2H (excess), rt.
The tetra-Boc-N-alkenyl polyamine intermediate 4 also provided an easy access to the corresponding double bond reduced analog 7 (Scheme 2). Thus, catalytic hydrogenation of 4 led to the uneventful formation of the saturated alkyl polyamine 6, which on subsequent Boc-deprotection yielded the branched N-alkyl polyamine 7 in good overall yield.
Scheme 2.

Reagents: a. 10% Pd(OH)2 – C, H2, MeOH, room temp. b. CF3CO2H (excess), room temp.
Starting from the tetra-Boc-protected polyamine 3 and following a concise sequence of reactions, the N-alkylated lipopolyamines 9, 11, and 13 (Scheme 3) were synthesized in an efficient manner. Thus, reductive alkylation of the amine 3 with commercially available 16-hentriacontanone yielded the corresponding adduct 8 (Scheme 3). Removal of the Boc-protection culminated in the C-1 branched alkyl polyamine 9 in good yield. Similarly, a one-pot reductive amination involving hexadecanal and the amine 3 resulted in the N-hexadecyl adduct 10, Boc-deprotection of which yielded the corresponding alkyl lipopolyamine derivative 11. On the other hand, N-alkylation of the secondary amine 10 with hexadecyl iodide to form the corresponding N,N-dialkylated product 12, and subsequent Boc-deprotection culminated in the dialkyl lipopolyamine 13. The activity profile of 11 (along with pharmacokinetic and toxicological studies are being reported elsewhere.
Scheme 3.

Reagents: a. (H31C15)2C=O, AcOH (cat.), C6H6, reflux, followed by, NaBH4, MeOH,rt. b. CF3CO2H (excess), room temp. c. H31C15CHO, NaBH4, MeOH, AcOH, rt. d. H31C15CH2I, C6H6, reflux.
Following a reaction strategy as described for the N-alkenyl polyamine 5, synthesis of the bis-homologated N1,N6-lipolyamine 16 was achieved starting from the earlier reported tetra-Boc-protected hexamine derivative 142 (Scheme 4). Accordingly, treatment of the amine 14 with an excess of hexadecanal, followed by reduction of the resulting terminal imines yielded the expected N1,N6-alkenyl substituted adduct 15 in moderate yield. Standard deprotection of Boc-groups provided the desired bis-alkenyl lipopolyamine 16. Similarly, reductive alkylation of the free diamine 14 with an excess of 16-hentriacontanone, forming the corresponding adduct 17 (Scheme 5), followed by removal of the Boc-protection completed the synthesis of the branched bis-alkyl polyamine derivative 18.
Scheme 4.

Reagents: a. Aldehyde 2 (excess), MgSO4, THF–MeOH, rt, then, NaBH4, MeOH. b. CF3CO2H, rt.
Scheme 5.

Reagents: a. (H31C15)2C=O (excess), AcOH (cat.), C6H6, reflux, then, NaBH4, MeOH, rt. b. CF3CO2H, rt.
Primary in vitro screen of mono- and bis-substituted alkylpolyamines, and selection of EVK-203 as a lead compound:
The de-repression and subsequent translocation of NF-κB to the nucleus is a pivotal transcriptional activation event that occurs in response to noxious stimuli. NF-κB induction is upstream of a plethora of signaling cascades which include the production of cytokines and other proinflammatory molecules. The use of HEK-Blue™ (Invivogen, San Diego, CA) cells (human embryonic kidney 293 cells stably transfected with a secreted alkaline phosphatase reporter gene under the control of NF-κB/AP-1 promoters along with the LPS receptor [Tlr-4] and co-receptors [CD-14 and MD-2]) provides a robust and easily quantifiable readout and is now our primary in vitro screen. As shown in Fig. 2, a clear segregation in IC50 values in the NF-κB inhibition assay was apparent, with the mono-substituted compounds 5 (EVK-203), 7, 9, and 13 being about an order of magnitude more potent that the bis-substituted 16 and 18 compounds. Of the mono-substituted compounds, EVK-203 (5) was found to be least hemolytic and cytotoxic as determined by erythrocyte lysis and XTT assays,21 respectively (Fig. 3), and was therefore chosen for further evaluation as described below.
Figure 2.

Comparison of the inhibition profiles of LPS (100 ng/ml)-induced NF-κB induction in HEK-293 cells stably transfected with Tlr4, MD-2, CD14, and an NF-κB-secreted alkaline phosphatase reporter gene construct. The structures of the alkylpolyamines and the corresponding IC50 values are shown.
Figure 3.

(A) Hemolytic activity of the alkylpolyamines. A suspension of 1:1000 diluted, washed, human erythrocytes was used. Hemolysis was determined by automated video microscopy. (B) Cytotoxic/cytostatic activity measured by XTT assay.
Binding affinity and in vitro neutralization activity.
We first examined quantitatively the binding affinities of EVK-203 and PMB to LPS using an automated fluorescence displacement assay using BODIPY®-TR-cadaverine (BC)29. BC binds LPS resulting in quenching of its fluorescence. The addition of test compounds results in displacement of LPS-bound BC fluorescence, manifesting in emission intensity enhancements at λem=620 nm, from which ED50 values were computed using standard four-parameter logistic curve-fitting. This assay was performed in high-salt buffers to minimize nonspecific ion-pairing interactions 29;29;30. The ED50 values for PMB and EVK-203 were comparable, being 1.22 ± 0.16 μM, and 3.14 ± 0.12 μM, respectively (Fig. 4A).
Figure 4.

(A) Binding affinity of EVK-203 and PMB (reference) to LPS determined by BODIPY®-cadaverine displacement assay. (B) Inhibitory activity of EVK-203 and PMB on nitric oxide production (measured spectrophotometrically as nitrite using Griess assay) in murine J774 macrophage cells stimulated with 100 ng/ml LPS. Shown on the left are negative and positive (LPS alone and medium alone) controls.
Murine, but not human monocytes, produce measurable quantities of nitric oxide (NO), an important surrogate marker of immune activation by bacterial products31. In vitro NO inhibition assays have proven to be a reliable, rapid, and cost-effective primary biological screen for identifying anti-LPS compounds13, and is a key primary screen in the hierarchical screening strategy used in our laboratory. As shown in Fig. 4B, EVK-203 as well as PMB inhibited NO production in a dose-dependent manner in murine J774A.1 cells stimulated with 10 ng/ml LPS, the IC50 values for PMB and EVK-203 being 1.79 ± 3 μM and 2.22 ± 3.5 μM respectively.
Ex vivo neutralization activity in human blood.
It was important to verify that the endotoxinsequestering activity of EVK-203 would be manifested in the milieu of whole human blood, characterized not only by its high ionic strength (∼300 mOsmoles) which attenuates electrostatic interactions 15, but also by near-millimolar concentrations of albumin. Albumin has been shown to bind both LPS 32 as well as to a variety of lipopolyamines (manuscript in preparation). Also present in human serum are a number of high-affinity LPS-binding proteins such as soluble CD-1433 and LPS-binding protein34. Furthermore, given the amphipathic nature of both LPS and EVK-203, it is conceivable that substantial partitioning of both target and ligand into the lipoprotein constituents could occur as has been observed with E5564 (Eritoran™), a lipid A receptor antagonist currently undergoing clinical trials35. For these reasons, and prior to initiating in vivo evaluation, we compared the effects of EVK-203 and PMB in two ex vivo assays using whole human blood.
In the first, we sought to examine the specificity and potency of EVK-203 relative to PMB in inhibiting the phosphorylation of p38 mitogen activated protein kinase (p38MAPK), a key component of a phosphorylation cascade that is upstream of NF-κB. The p38 MAP kinases (α, β, γ, δ) are activated by dual phosphorylation on Thr and Tyr within the Thr-Gly-Tyr motif located in kinase subdomain VIII. Activation of p38 MAPK leads to the activation of multiple transcription factors (NF-κB, ATF-2, Elk-1, and CHOP) that result in the expression of many different genes, including those encoding proinflammatory cytokines36;37. Exposure of whole blood obtained from healthy human volunteers to 100 ng/ml LPS results in a marked elevation in T180/Y182 dual-phosphorylated p38MAPK as probed by flow cytometry using an Alexa Fluor® 647-labeled anti-phospho-p38 MAPK (T180/Y182) antibody (Fig. 5). The activation seems to occur chiefly in polymorphonuclear (PMN) cells since the positive population maps directly to the subset of cells adjudged as PMN based on forward and side-scatter characteristics. The addition of either PMB or EVK-203 concomitant with LPS exposure results in a dose-dependent attenuation of p38MAPK phosphorylation with the potencies of both compounds being very similar (PMB: 10 nM, EVK-203: 23 nM). Notably, p38MAPK phosphorylation induced by TNF-α is affected by neither compound, emphasizing again the specificity of action against LPS (Fig.5).
Figure 5.
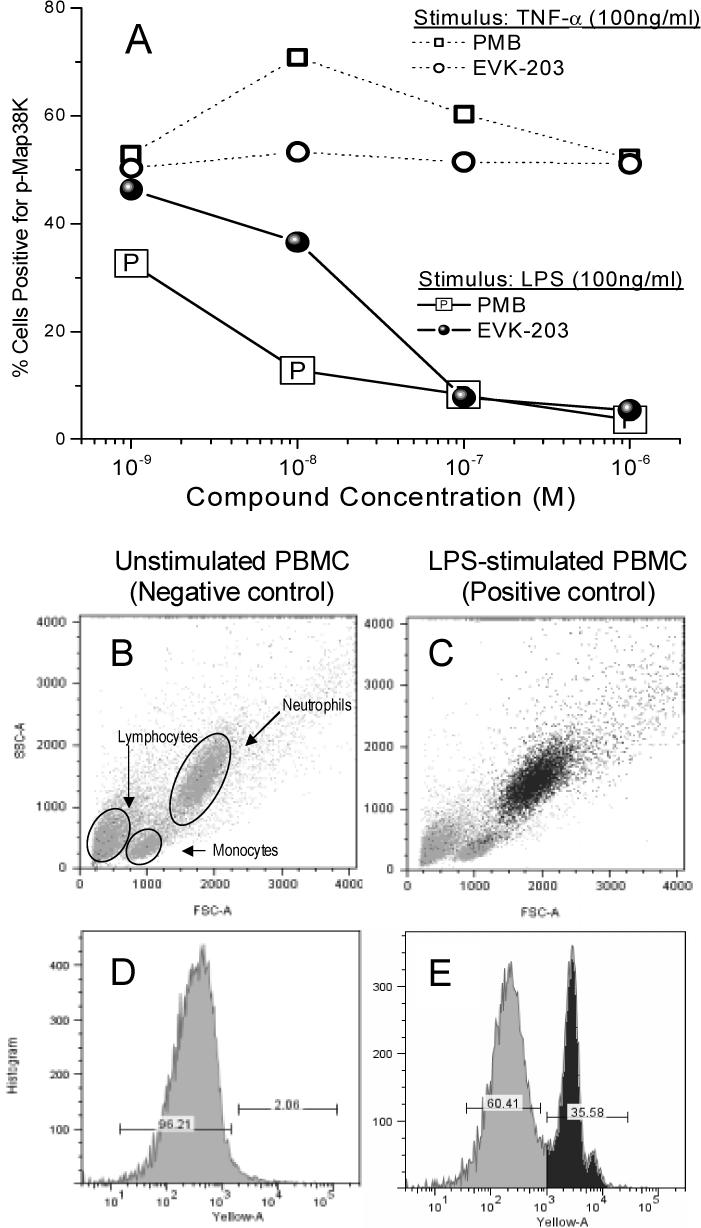
(A) Inhibition of phosphorylation of p38 MAP kinase in neutrophils in whole human blood (ex vivo), stimulated with either LPS or hTNF-α (100 ng/ml) for 15 minutes in the presence of graded concentrations of EVK-203 or PMB. Quantification of p38 MAPK was performed using flow cytometry. Panels B and D show forward scatter/side scatter profile and the gating for p38 MAPK-negative and -positive gates obtained on unstimulated cells (negative control), respectively. Panels C and E are corresponding results obtained from LPS stimulated cells (positive control). Back-gating on the p38 MAPK-positive cells (panel E: heavily-shaded peak) map to the polymorphonuclear population (panel C).
Having demonstrated that EVK-203 inhibited, in a specific manner, an early, upstream cell-signaling event which is consistent with the premise of the compound being an LPS sequestrant, we next evaluated the efficacy of the compound in inhibiting proinflammatory cytokine release using a multiplexed cytokine detection system21;22. As shown in Fig. 6, EVK-203 inhibits TNF-α, IL-6 and IL-8 production, with the IC50 of 3−4 μM (an LPS stimulus of 100 ng/ml was used), which is considerably lower than that of PMB (11−16 μM). As controls, we used phorbol myristate acetate (PMA; 100 ng/ml) plus ionomycin (1 μM), or PMA plus phytohemagglutinin (2 μg/ml) as non-LPS stimuli. Neither compound inhibited TNF-α, IL-6, or IL-8 appreciably up to concentrations of 40 μM, verifying that the inhibition observed was specific for LPS (data not shown).
Figure 6.

(A-C) Dose-dependent inhibition of LPS-induced proinflammatory cytokine production in ex vivo whole human blood. Whole human blood was stimulated with LPS and graded concentrations of either EVK-203 or PMB. Cytokine levels were quantified using a multiplexed flow-cytometric bead array system (CBA).
Efficacy in murine models of septic shock.
A well-established murine model of lethal septic shock18;21;38 was employed to compare the potencies of EVK-203 with PMB. Cohorts of 10 CF-1 mice per group, sensitized to the lethal effects of LPS with D-galactosamine, were challenged with a supralethal dose of LPS (2X LD100 dose = 200 ng/animal) administered intraperitoneally (i.p.). This was preceded by a subcutaneous (s.c.) injection of graded doses of test compounds given one hour prior to LPS challenge. The difference in dose-dependent survival rates between animals receiving PMB and EVK-203 was statistically significant (p<0.005; Fisher one-tailed exact probability) with EVK-203 being more potent in preventing LPS-induced lethality (Fig. 7A).
Figure 7.

(A) Comparison of in vivo potency of PMB and EVK-203 in a D-galactosamine-primed murine model of lethal endotoxic shock. Dose-dependent increase in survival in mice challenged with a supralethal (200 ng/animal; 2X LD100) dose of LPS. The difference in dose-dependent survival rates between animals receiving PMB and EVK-203 was statistically significant (p<0.005; Fisher one-tailed exact probability). (B) Time-course (pharmacodynamics) of protection conferred by 8 mg/kg of EVK-203 administered at various times prior to, and following supralethal LPS challenge in a D-galactosamine-primed murine model of lethal endotoxic shock. LPS (200 ng/animal; 2X LD100) was administered at time = 0 h.
Pharmacokinetic experiments aimed at quantifying half-life of test-compounds have, on occasion, led to apparent incongruencies with pharmacodynamics data due to unusual partitioning and/or protein-binding behavior as has been observed with Eritoran™, for instance.39 We therefore elected to first characterize the pharmacodynamics of EVK-203. Mice were administered EVK-203 at a dose of 8 mg/kg s.c. at various time points before and after a supralethal LPS challenge. As depicted in Fig. 7B, EVK-203 is maximally effective when it is administered concurrent to LPS administration. The data further suggest that sufficiently high plasma concentrations persist even up to 6 h so as to provide partial protection against LPS-induced lethality (Fig. 7B). It is to be noted that EVK-203 is bereft of any protective effect if administered following LPS challenge, suggesting that once the innate immune system has responded to the presence of circulatory LPS, anti-endotoxin agents are unlikely to be of any benefit. This is crucial since this defines the subset of patients in whom a compound such as EVK-203 is likely to be of benefit.
We next sought to examine if the protection afforded by EVK-203 was attributable to attenuated LPS-induced cytokine production. Consistent with the premise of LPS sequestration by the alkylpolyamine compounds, we observed a dose dependent inhibition in peak levels of TNF-α, IL-6, and MCP-1 in vivo (Fig. 8). Furthermore, we confirmed that EVK-203 had no effect in preventing lethality induced by 100 ng/animal of recombinant murine TNF-α (data not shown).
Figure 8.

Time-course of cytokine profiles in plasma of mice receiving graded doses of EVK-203 and challenged with a lethal dose of LPS. Cohorts of 5 animals per group were administered graded doses of EVK-203 (or vehicle) subcutaneously at time = −1 h. Following lethal LPS challenge (200 ng/animal, administered intraperitoneally at t = 0 h), blood samples were obtained at t = 1, 2, and 3 h. Plasma samples were processed for cytokine analyses using the CBA assay.
Toxicity:
EVK-203 is a cationic amphipath and, as such would be expected to be surface active, with the possible consequence of nonspecific cytotoxicity. This was of concern when we started evaluating lipopolyamines18;21;22. As with those earlier compounds we found that our apprehension was unwarranted: the hemolytic activity of EVK-203 was virtually abrogated in the presence of physiological concentrations of albumin; in whole human blood, hemolytic activity was minimal even at millimolar concentrations (data not shown). Daily s.c. administration of EVK-203 to mice at a dose of 40 mg/kg per day for five days (note that full protection against LPS-induced lethality was conferred with a single dose of 2 mg/kg) resulted in no clinically observable signs of apparent toxicity. Importantly, at the end of the experiment (Day 6) there were no demonstrable signs of nephro- or hepatotoxicity as assessed by clinical chemistry (data not shown).
Discussion
Sepsis, or “blood poisoning” in lay terminology, is a common and serious clinical problem. While fewer than 100 cases were reported prior to 1920,40 it is now the thirteenth leading cause of overall mortality,41 and the number one cause of deaths in the intensive care unit42 accounting for some 200,000 fatalities in the US annually.43 While the incidence continues to rise in the US7 and worldwide44 due to increased invasive procedures, immunosuppression and cytotoxic chemotherapy, mortality has essentially remained unchanged at about 45%45 despite tremendous strides in antimicrobial chemotherapy, due to the lack of specific therapy aimed at the pathophysiology of sepsis.
The primary trigger in the Gram-negative septic shock syndrome is endotoxin, a constituent of the outer membrane of all Gram-negative bacteria. Total synthesis of the structurally highly conserved lipid A has been shown to be the active moiety of LPS,46 and is thus a logical therapeutic target for neutralization. The anionic amphiphilic nature of lipid A enables it to interact with a variety of cationic hydrophobic ligands.47-49
Polymyxin B (PMB), a cationic amphiphilic cyclic decapeptide antibiotic isolated from Bacillus polymyxa50 has long been recognized to bind lipid A,51 and neutralize its toxicity in animal models of endotoxemia.52-54 Although PMB is a commonly-used topical antibiotic, it is potently nephro- and oto-toxic, which, while precluding its utility as an LPS-neutralizer in patients with sepsis, has stimulated the search for nontoxic PMB analogs,55;56 PMB derivatives,57-59 as well as other structurally diverse cationic amphiphilic peptides56;60-63 as candidate LPS-binding agents.
Our long-term goal has been to utilize the structural information in the LPS-PMB complex to rationally design non-peptide, small-molecule LPS-sequestrants. We have elected to focus on targeting the lipid A moiety of LPS rather than on downstream inflammatory processes which have all met with failure.64;65 We initially evaluated peptides, both naturally occurring,12;66;67 and de novo synthesized,12;68 testing specific hypotheses pertaining to structural correlates of lipid A binding, and later extended those design principles to small molecules.14;15 This approach firstly led to the definition of a crucial pharmacophore that determined LPS-recognition and –neutralization properties in small molecules,14;15 and, subsequently, to the discovery of a novel lipopolyamine lead,69;70 which was shown to be an effective LPS-neutralizer.18 In an ongoing effort to understand in detail the structure-activity relationships in such molecules, we have systematically evaluated congeneric series of compounds, examining first the contribution of the lipid tail, and then the backbone itself. Several iterative design-and-test cycles of SAR studies ultimately helped converge and focus our efforts on evaluating compounds with C16-N-alkyl polyamine motifs.
The experimental studies described in this paper establish that EVK-203 binds LPS and attenuates its toxicity with a potency comparable to that of PMB. The inhibition of early cellular activation events, namely, p38MAPK phosphorylation and NFκB translocation, in conjunction with the inhibition of the more distal response events of cytokine and NO production, as well as the lack of activity against non-LPS stimuli clearly shown that the mechanism of action is via sequestration of LPS. EVK-203 was found to exhibit significant anti-endotoxin activities not only in whole human blood ex vivo, but also in a murine model of endotoxic shock.
Particularly instructive are the results of the time course experiment in mice (Fig. 7B) which show that if LPS-sequestering compounds such as EVK-203 are ever to find utility in the clinic, they will have to be used as prophylactic agents, rather than to treat established sepsis for once the inflammatory cascades are set in motion, LPS sequestrants would be of no value. This may indeed be feasible and, indeed desirable, since not only have many of the therapeutic strategies that target downstream processes such as blockade of TNF-α or IL-1β have failed64;65, but also because the predisposing factors for septic shock are very well recognized71;72. It may be possible to institute LPS-sequestrant therapy as an adjunct to conventional antimicrobial chemotherapy. It is relevant to note in this context that analogues similar to EVK-203 have been shown to not only possess intrinsic antimicrobial activity, but also potentiates the activity of otherwise impermeable hydrophobic antibiotics such as rifampin.73
Obligatory requirements in the development of compounds such as EVK-203 for the prophylaxis of Gram-negative sepsis would be a high therapeutic index, and an acceptable pharmacokinetic profile. It is encouraging to have observed the absence of any apparent toxicity in murine models administered multiples of therapeutic doses over several days. Further toxicity testing with escalating dose regimens are currently ongoing. Although we are yet to establish circulating half-lives of EVK-203 in a range of animal models, the pharmacodynamic profile that we have ascertained in the murine model would suggest a half-life of about 4 hours. We now have an LC-MS method in place for assaying plasma levels of EVK-203, and pharmacokinetic experiments are planned in rodent and non-rodent species. In summary, we have described in this paper a detailed characterization of EVK-203, a novel alkylpolyamine, exhibiting true LPS-sequestering and -neutralizing properties in a panel of in vitro assays as well is in a murine model of endotoxic shock. The potency of EVK-203 rivals that of PMB and, in some key assays, including a murine model of endotoxic shock, is superior to PMB. The favorable pharmacodynamic and toxicity profile of EVK-203 renders this compound an attractive candidate for further preclinical development.
Experimental
All of the solvents and reagents used were obtained commercially and used as such unless noted otherwise. Moisture or air sensitive reactions were conducted under argon atmosphere in oven dried (120°C) glass apparatus. Anhydrous THF was obtained by distillation over sodium benzophenone ketyl, prior to use. Solvents were removed under reduced pressure using standard rotary evaporators. Flash column chromatography was carried out using Silica gel 60 (230−400 mesh), while thin layer chromatography (tlc) was carried out on Silica Gel HLF, precoated glass plates. All yields reported refer to isolated material judged to be homogeneous by tlc and NMR spectroscopy. Unless noted otherwise, NMR spectra were recorded with the chemical shifts (d) reported in ppm relative to Me4Si (for 1H) and CDCl3 (for 13C) as internal standards respectively. Following a literature procedure,74 hexadecanal (1) was prepared by oxidation of commercially available 1-hexadecanol, while the tetra-Boc-polyamines 3 and 14 were synthesized following our earlier reported procedure.21
Compound 2.
A mixture of hexadecanal (1)3 (1.2 g, 5 mmol) and anhydrous AlCl3 (2.6 g, 20 mmol) in pyridine (80 mL) was refluxed for 30 min. After cooling to room temperature, the reaction mixture was diluted with diethyl ether (250 mL), the precipitated solid was removed by filtration and the filtrate concentrated under reduced pressure. The residue was purified by flash column chromatography (Hexane/EtOAc = 99/1) to yield the product 2 as a low melting solid (1.05g, 45%). 1H NMR (400 MHz, CDCl3) δ 0.90 (t, J = 7.04 Hz, 6H), 1.28 (br s, 48H), 1.47−1.55 (m, 2H), 2.22−2.31 (m, 2H), 2.34−2.39 (m, 2H), 6.45 (t J = 7.44 Hz, 1H), 9.35 (s, 1H); MS (ESI) calcd for C32H62O m/z 462.4, found 463.5 (MH)+.
Compound 4.
To a solution of the amine 32 (0.2 g, 0.3 mmol) in anhydrous methanol (8 mL) at room temperature were added anhydrous MgSO4 (ca. 1g) and a solution of the aldehyde 2 (0.2 g, 0.43 mmol) in anhydrous THF (4 mL). The resulting mixture was stirred at room temperature overnight, followed by addition of methanol (4 mL) and NaBH4 (60 mg, 1.6 mmol). After stirring for another 4 hours, the reaction mixture was concentrated under reduced pressure, the residue was dissolved in CHCl3 (50 mL) and washed with water (2×5 mL). The organic layer was dried over Na2SO4, solvent removed under vacuum and the residue purified by flash column chromatography (CH2Cl2:MeOH:NH4OH = 95:5:1) affording the product 4 (0.14 g, 42%) as an oily liquid. 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.6 Hz, 6H), 1.27 (br s, 50H), 1.46 (s, 38H), 1.70−1.74 (br m, 6H), 2.02−2.14 (br m, 6H), 2.71(br s, 2H), 3.05−3.35 (br m, 17H), 5.29 (br t, J = 6.68 Hz, 1H); 13C NMR (100.6 MHz, CDCl3); 14.0, 22.6, 25.9, 27.5, 28.3, 28.4, 28.5, 28.8, 29.2, 29.4, 29.5, 29.6, 29.9, 31.8, 31.9, 37.2, 43.8, 44.1, 44.6, 46.4, 46.7, 55.3, 78.8, 79.2, 126.3, 137.5, 155.3, 155.4, 156.0; MS(ESI) calcd for C65H127N5O8 m/z 1106.7 found 1107.0 (MH)+.
Compound 5.
Removal of the Boc-protecting groups were carried out by treatment of compound 4 (50 mg, 0.045 mmol) with an excess of trifluoroacetic acid (6 mL) and stirring the solution at room temperature overnight. Concentration of the reaction mixture under reduced pressure, followed by trituration of the residual liquid with methylene chloride provided the product 5 as a white powder (35 mg, 61%). 1H NMR (400 MHz, DMSO-d6) δ 0.85 (t, J = 6.6Hz, 6H), 1.25 (br s, 50H), 1.55−1.65 (br s, 4H), 1.85−2.12 (br m, 10H), 2.88−3.05 (br m, 18H), 5.55 (br s, 1H), 7.85 (br s, 2H), 8.75−9.05 (br m, 4H); 13C NMR (100.6 MHz, DMSO-d6) 13.1, 22.2, 22.4, 22.6, 28.6, 28.7, 29.0, 31.3, 36.2, 43.5, 43.8, 44.1, 54.4, 54.8, 130.7, 133.1; MS (FAB) calcd for C45H95N5 (free amine) m/z 705.7 found 706.9 (MH)+.
Compound 6.
To a room temperature solution of the N-alkenyl polyamine 4 (139 mg, 0.126 mmol) in methanol (10 mL), was added the Pearlman catalyst (200 mg), and the mixture stirred under H2 atmosphere (balloon) overnight. The solution was filtered through celite and the residue washed thoroughly with methanol. The combined filtrate was concentrated and the residue purified by flash column chromatography (CH2Cl2:MeOH:NH4OH = 95:5:1) affording the product 6 (0.1 g, 72%) as an oily liquid. 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 6.2 Hz, 6H), 1.26 (br s, 52H), 1.45 (s, 43H), 1.59−1.82 (br m, 6H), 1.93−2.19 (br m, 2H), 2.62−2.83 (m, 4H), 3.04−3.43 (br m, 18H), 4.91 (br s, 1H); 13C NMR (100.6 MHz, CDCl3); 14.1, 22.7, 25.5, 26.1, 28.4, 28.5, 28.6, 29.4, 29.6, 29.7, 29.9, 31.4, 31.9, 37.3, 43.3, 44.6, 45.9, 46.8, 52.9, 78.9, 79.5, 80.8, 155.5, 155.4, 156.1; MS(ESI) calcd for C65H129N5O8 m/z 1108.7 found 1109.0 (MH)+.
Compound 7.
A solution of the tetra-Boc-amine 6 (32 mg, 0.028 mmol) in trifluoroacetic acid (6 mL) was stirred at room temperature overnight. Removal of the solvent under reduced pressure, followed by trituration of the residual oily liquid with methylene chloride provided the product 7 as a white powder (27 mg, 77%). 1H NMR (400 MHz, DMSO-d6) δ 0.85 (br s, 6H), 1.24 (br s, 56H), 1.63 (br s, 5H), 1.89−2.02 (m, 6H), 2.72−3.0 (br m, 18H), 7.94 (br s, 2H), 8.59 (br s, 1H), 8.69 (br s, 1H), 8.90 (br s, 1H), 9.13 (br s, 1H); 13C NMR (100.6 MHz, DMSO-d6) 13.9, 22.1, 22.2, 22.4, 22.6, 23.8, 25.2, 28.7, 28.8, 28.9, 29.0, 30.2, 31.3, 34.6, 36.1, 43.9, 44.0, 44.1, 44.7, 46.1, 50.9; MS (FAB) calcd for C45H97N5 (free amine) m/z 707.7 found 708.8 (MH)+.
Compound 8.
To solution of the amine 3 (138 mg, 0.2 mmol) and 16-hentriacontanone (113 mg, 0.25 mmol) in anhydrous benzene (10 mL), two drops of glacial acetic acid was added and the solution refluxed overnight. After cooling the reaction mixture to room temperature, methanol (5 mL) was added to it, followed by addition of sodium borohydride (37 mg, 1 mmol). After stirring the resulting mixture at room temperature for 4h, solvent was removed under reduced pressure. The residue was dissolved in chloroform (50 mL), washed with water, dried over anhydrous Na2SO4 and concentrated. Purification of the residue by flash column chromatography (CH2Cl2:MeOH:NH4OH = 95:5:1) afforded the product 8 (129 mg, 59%) as an oily liquid. 1H NMR (500 MHz, CDCl3) δ 0.88 (t, J = 6.6 Hz, 6H), 1.25 (br s, 52H), 1.44 (s, 42H), 1.61−1.83 (br m, 8H), 1.95−2.56 (2m, 3H), 3.09−3.33 (br m, 15H), 5.34 (br s, 1H); 13C NMR (125.7 MHz, CDCl3); 14.1, 22.7, 25.8, 28.4, 28.5, 29.4, 29.5, 29.6, 29.7, 31.9, 32.8, 37.3, 44.7, 46.4, 46.8, 54.4, 60.4, 63.0, 79.4, 80.1, 155.4, 156.1; MS(FAB+) calcd for C64H127N5O8 m/z 1094.7 found 1095.1 (MH)+.
Compound 9.
A solution of the tetra-Boc-amine 8 (120 mg, 0.11 mmol) in trifluoroacetic acid (10 mL) was stirred at room temperature overnight. Removal of the solvent under reduced pressure, followed by trituration of the residual oily liquid with methylene chloride provided the product 9 as a white powder (113 mg, 81%). 1H NMR (400 MHz, DMSO-d6) δ 0.86 (t, J = 6.6 Hz, 6H), 1.24 (br s, 54H), 1.45−1.77 (m, 7H), 1.84−2.03 (m, 5H), 2.74−3.11 (m, 17H), 7.98 (br s, 2H), 8.70 (br s, 1H), 8.81−9.31 (br m, 3H); MS (FAB+) calcd for C44H95N5 (free amine) m/z 693.7 found 694.8 (MH)+.
Compound 10.
To a solution of the amine 3 (0.40 g, 0.61 mmol) and acetic acid (5 drops) in dry methanol (20 mL) was added hexadecylaldehyde (0.073 g, 0.30 mmol), followed by sodium cyanoborohydride (0.02 g, 0.30 mmol) in one portion. The colorless solution was stirred at room temperature for 24 hours. Concentrated hydrochloric acid (2 drops, pH = 2) was added, and the solution was basified to pH = 12 (solid sodium hydroxide). Methanol was removed under reduced pressure. The basic residue was extracted with ether (3 × 50 mL), and the organic layers were combined and washed with saturated aqueous sodium chloride and dried over sodium sulfate. After purification by flash column chromatography (CH2Cl2:MeOH:NH4OH = 95:5:1 as eluent), pure 10 was obtained as a colorless oil (0.22 g, 79%). 1H NMR (CDCl3, 400 MHz) δ 0.89 (t, J = 8.0 Hz, 3H), 1.32−1.27 (m, 26H), 1.46 (br s, 42H), 1.74−1.66 (m, 6H), 2.61 (br s, 4H), 3.25−3.16 (m, 15H); 13C NMR (CDCl3, 125.7 MHz) δ 14.1, 22.7, 27.3, 28.5, 29.4, 29.5, 29.6, 29.7, 31.9, 37.4, 44.2, 44.8, 46.8, 50.0, 79.3, 155.5, 156.1; HRMS (ESI) calcd for C49H98N5O8 (MH+) m/z 884.7515, found 884.7525.
Compound 11.
The Boc-protected lipopolyamine 10 (150 mg, 0.17 mmol) was dissolved in trifluoroacetic acid (15 ml) and stirred at room temperature for 20 hours. Excess solvent was removed under reduced pressure and the residue was thoroughly triturated with dichloromethane and diethyl ether to obtain the product 11 as a white flaky solid (157 mg, 88%). 1H NMR (DMSO-d6, 400 MHz) δ 0.86 (t, J = 8.0 Hz, 3H), 1.26 (br s, 26H), 1.64−1.57 (m, 6H), 1.96−1.90 (m, 6H), 2.99−2.89 (m, 18H), 7.93 (br s, 2H), 8.97−8.67 (m, 4H); 13C NMR (DMSO-d6, 125 MHz) δ 14.2, 22.8, 23.0, 24.2, 25.8, 26.3, 28.9, 29.2, 29.3, 29.4, 31.7, 36.6, 39.5, 39.7, 40.1, 40.4, 40.6, 44.3, 44.5, 46.5, 47.2; HRMS (ESI) calculated for C29H66N5 (MH+) m/z 484.5318, found 484.5326.
Compound 12.
To a solution of the monoalkylated amine 10 (0.23 g, 0.26 mmol) in dry benzene (8 mL) at room temperature was added hexadecyl iodide (60 mg, 0.17 mmol) and the mixture refluxed for 36 h. After removal of the solvent under reduced pressure, the residue was purified by flash column chromatography (hexane:EtOAc = 3:1) to afford the N,N-dialkylated product 12 (132 mg 46%) as a colorless oil . 1H NMR (400 MHz, CDCl3) δ 0.86 (t, J = 6.6 Hz, 6H), 1.26 (br s, 54H), 1.45 (br s, 42H), 1.65−1.74 (br m, 6H), 2.39 (br s, 4H), 3.1−3.3 (m, 16H); 13C NMR (100.6 MHz, CDCl3) 14.1, 22.7, 25.5, 25.8, 26.8, 27.6, 28.4, 28.5, 29.2, 29.6, 31.9, 43.7, 44.1, 44.7, 46.4, 46.8, 51.5, 54.0, 79.2, 79.5, 155.4, 155.6; MS (ESI) calcd for C65H129N5O8 m/z 1108.7 found 1109.0 (MH)+.
Compound 13.
The tetra-Boc-dialkylated amine 12 (44 mg, 0.04 mmol) was dissolved in trifluoroacetic acid (5 mL) and stirred at room temperature overnight. Excess solvent was removed under reduced pressure and the residue was triturated with dichloromethane to afford the N,N-dialkylated lipoplyamine 13 as a white flaky solid (38 mg, 75%). 1H NMR (400 MHz, DMSO-d6) δ 0.85 (br s, 6H), 1.25 (br s, 48H) 1.5−1.65 (m, 10H) 1.85−2.05 (br m, 8H), 2.80−3.10 (br m, 20H), 7.90−8.01 (br s, 1H), 8.81−9.12 (br m, 3H) ; 13C NMR (100.6 DMSO-d6); 13.5, 23.7, 24.6, 25.1, 25.3, 25.8, 28.7, 28.8, 29.0, 31.2, 36.1, 40.6, 43.7, 46.1, 51.9, 54.8; MS (FAB) calcd for C45H97N5 (free amine) m/z 708.2 found 709.0 (MH)+.
Compound 15.
To a room temperature solution of the tetra-Boc poctected diamine 142 (100 mg, 0.14 mmol) in dry THF (15 mL) were added MgSO4 (ca. 0.6 g) and the α,β unsaturated aldehyde 2 (258 mg, 0.5 mmol). After stirring the mixture for 8 h, methanol (10 mL) and NaBH4 (206 mg, 5.4 mmol) were added and stirring continued for another 5 h. After removal of the solvent, the residue was dissolved in CHCl3, washed with water and the organic layer was dried over Na2SO4. Removal of solvent under reduced pressure and purification of the residue by flash column chromatography (CH2Cl2: MeOH: NH4OH) (95:5:1) afforded the bis-alkenyl amine 15 as an oily liquid (92 mg, 41%). 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 7 Hz, 12H), 1.27 (br s, 96H), 1.46 (br m, 44H), 1.60−1.75 (m, 12H), 2.00−2.04 (br m, 6H), 2.30−2.80 (br m, 4H), 3.36 (br m, 20H), 5.29−5.46 (m, 2H); 13C NMR (100.6 MHz, CDCl3) δ 14.0, 22.6, 25.9, 27.5, 28.3, 28.3, 28.5, 28.8, 29.2, 29.3, 29.5, 29.52, 29.6, 29.8, 31.8, 37.2, 44.6, 46.3, 46.7, 55.3, 78.7, 79.2, 126.3, 137.4, 155.3, 155.4, 156.0; MS (FAB) calculated for C100H196N6O8; m/z 1609.5 found 1610.7 (MH)+.
Compound 16.
The Boc-protected dialkenyl amine 15 (50 mg, 0.03 mmol) was dissolved in trifluoroacetic acid (6 mL) and stirred for 18 h at ambient temperature. Excess solvent was removed under reduced pressure and the resulting sticky residue was washed thoroughly with dry methylene chloride to obtain the desired amine 16 as a white flaky solid (38 mg, 68%). 1H NMR (400 MHz, CDCl3) δ 0.85 (br s, 12H), 1.23 (br s, 94H), 1.72 (br m, 10H), 1.91−2.12 (br m, 16H), 2.85−3.15 (br m, 24H), 5.52 (br t, 2H) 8.7−9.0 (br m, 6H); 13C NMR (125.7 MHz, DMSO-d6) δ 13.9, 22.0, 22.1, 22.2, 22.3, 23.0, 24.0, 27.5, 27.8, 28.6, 28.7, 28.9, 29.0, 31.2, 36.0, 43.6, 43.9, 46.1, 51.4, 130.7, 133.1. MS (ESI-TOF) calcd for C80H164N6 (free amine) m/z 1209.302; found 1210.321 (MH)+.
Compound 17.
To a solution of the tetra-Boc poctected diamine 14 (156 mg, 0.21 mmol) in dry benzene (8 mL) at room temperature were added 16-hentriacontanone (245 mg, 0.542 mmol) and 2 drops of glacial acetic acid. The resulting solution was refluxed overnight. After cooling to room temperature, methanol (10 mL) and NaBH4 (38o mg, 10 mmol) were added to the reaction mixture and stirring continued at room temperature for another 3 h. After removal of solvent, the residue was dissolved in CHCl3, washed with water, and the organic layer was dried over Na2SO4. After removal of solvent, the residue was purified by flash column chromatography (CH2Cl2: MeOH: NH4OH = 95:2:1) to afford the branched chain dialkylated product 17 as an oily liquid (226 mg, 68%). 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 7 Hz, 12H), 1.26 (br s, 108H), 1.45 (br s, 44H), 1.61−1.74 (br m, 8H), 2.56−2.58 (br m, 6H), 3.1−3.3 (br m, 16H), 3.62 (br s, 2H); 13C NMR (100.6 MHz, CDCl3) δ 14.1, 22.7, 25.6, 25.7, 28.5, 29.3, 29.6, 29.6, 29.7, 30.0, 31.9, 34.0, 37.4, 44.6, 57.7, 72.0, 79.3, 155.5; MS (FAB+) calculated for C98H196N6O8 m/z 1586.6 found 1587.7 (MH)+.
Compound 18.
The Boc-protected bis alkyl polyamine derivative 17 (115 mg, 0.072 mmol) was dissolved in 6 ml of trifluoroacetic acid and stirred for 12 h at ambient temperature. Excess solvent was removed under reduced pressure and the resulting sticky residue was washed thoroughly with dry methylene chloride to obtain the desired alkyl branched lipopolyamine 18 as a white flaky solid (91 mg, 67%). 1H NMR (400 MHz, CDCl3) δ 0.83 (br s, 12H), 1.17 (br s, 102H), 1.55−1.71 (br m, 12H), 1.82−1.95 (br m, 10H), 2.98−3.12 (br m, 22H), 9.09−9.24 (br m, 6H); 13C NMR (125.7 MHz, DMSO-d6) δ 14.3, 22.4, 23.0, 24.3, 29.0, 29.1, 29.3, 29.4, 31.6, 37.1, 42.1, 44.2, 44.3, 46.5, 57.6; MS (FAB+) calculated for C78H164N6 1186.18 found 1186.8.
Binding affinity measurements:
The BC displacement assay for quantifying binding affinity has been described previously 22;29;30. Briefly, to the first column (16 wells) of a Corning Nonbinding Surface 384-well flat-bottom black fluorescence microplate were added 80 μl aliquots of 1 mM stock solutions of DS-96 or polymyxin B (reference compound) in quadruplicates, and were serially diluted two-fold across the remaining 23 columns in 50 mM Tris buffer, pH 7.4, using a Precision 2000 automated microplate pipetting system, achieving a final dilution of 0.112 nM in a volume of 40 μl. 40 μl aliquots of a mixture of 50 μg/ml of LPS and 5 μM BC in buffer were added to each well of the plate using the Precision 2000 instrument. Fluorescence measurements were made at 25 °C on a SpectraMax M2 multifunction plate reader (Molecular Devices, Sunnyvale, CA) with excitation and emission wavelengths at 580 and 620 nm, respectively. Relative binding affinities were measured as the effective displacement of 50% of bound probe (ED50) using standard four-parameter logistic curve fitting subroutines in Origin version 7.0 (OriginLab, Northampton, MA). E. coli O111:B4 smooth LPS was used as the ligand in the BC displacement experiment, and as stimuli in all in vitro activity assays.
In vitro XTT cytotoxicity assay:
The determination of cell viability was accomplished by the addition of an XTT75 solution to murine macrophage J774A.1 cultures treated with graded concentrations of the test compounds. Cell culture and plating procedures were performed as described below for nitric oxide measurement. Cytotoxicity was measured the following day by the addition of 80 μl/well XTT/ Phenazine methosulfate (PMS) solution (XTT solution, 2 mM in PBS, pH 7.4, pH adjusted to 6.0−6.5; PMS solution, 0.92 mg/ml in PBS, pH 7.4; solutions mixed at a ratio of 8 ml XTT solution to 200 μl PMS solution) followed by an incubation time of 1.5h at 37 °C. Absorbance was read at 490 nm with scatter correction at 690 nm.
Hemolytic Activity:
Hemolysis was quantified using extremely diluted, aged human whole blood such that the effects of the compounds binding to plasma proteins would be negligible, and the hemolytic activity would be magnified because of increased osmotic fragility of the erythrocytes as a consequence of depleted Na+ K+ ATPase activity.76 Dilute erythrocyte suspensions were prepared by diluting one-week-old whole blood obtained by venipuncture from healthy human volunteers 1:1000 in isotonic (0.9 g/100 ml) saline solution to which was added graded doses of compound. Absorptimetric determination of hemoglobin released from such a dilute erythrocyte suspension was not reliable. The samples were therefore examined with a Beckman-Coulter Vi-Cell™ Cell Viability Analyzer (Beckman-Coulter, Hialeah, FL). This instrument implements an automated intravital trypan blue exclusion method using real-time automated video microscopy. Measurement parameters for erythrocytes were gated appropriately on control erythrocytes to specify thresholds of cell recognition and viability. Data on total number of cells/ml and viable cells/ml were collected through 50 captured images per sample with a counting accuracy of +/− 3%.
Measurement of nitric oxide release in murine macrophages:
Nitric oxide (NO) was measured as total nitrite in murine macrophage J774.A1 cells using the Griess reagent system 77;78 as described previously 21;22. J774.A1 cells were grown in RPMI-1640 cell-culture medium containing L-glutamine and sodium bicarbonate and supplemented with 10% fetal bovine serum, 1% L-glutamine-penicillin-streptomycin solution, and 200 μg/ml L-arginine at 37°C in a 5% CO2 atmosphere, and plated at ∼105 cells/ml in a volume of 80 μl/well, in 384-well, flat-bottomed, cell culture-treated microtiter plates until confluency and subsequently stimulated with 10 ng/ml lipopolysaccharide (LPS). Concurrent to LPS stimulation, serially diluted concentrations of test compounds were added to the cell medium and left to incubate overnight for 16h. Polymyxin B was used as reference compound in each plate. Positive-(LPS stimulation only) and negative-controls (J774.A1 medium only) were included in each experiment. Nitrite concentrations were measured adding 50 μl of supernatant to equal volumes of Griess reagents (50 μl/well; 0.1% NED solution in ddH2O and 1% sulfanilamide, 5% phosphoric acid solution in ddH2O) and incubating for 15 minutes at room temperature in the dark. Absorbance at 535 nm was measured using a Molecular Devices Spectramax M2 multifunction plate reader. Nitrite concentrations were interpolated from standard curves obtained from serially diluted sodium nitrite standards.
Multiplexed cytokine assay ex vivo in human blood and in in vivo murine blood:
100 μl aliquots of fresh whole blood, anticoagulated with EDTA, obtained by venipuncture from healthy human volunteers with informed consent and as per guidelines approved by the Human Subjects Experimentation Committee, was exposed to an equal volume of 20 ng/ml of E. coli 0111:B4 LPS, with graded concentrations of test compounds diluted in saline for 4h in a 96-well microtiter plate as described previously 21;22. The effect of the compounds on modulating cytokine production was examined using a FACSArray multiplexed flow-cytometric bead array (CBA) system (Becton-Dickinson-Pharmingen, San Jose, CA). The system uses a sandwich ELISA-on-a-bead principle 79;80, and is comprised of 6 populations of microbeads that are spectrally unique in terms of their intrinsic fluorescence emission intensities (detected in the FL3 channel of a standard flow cytometer). Each bead population is coated with a distinct capture antibody to detect six different cytokines concurrently from biological samples (the human inflammation CBA kit includes the following analytes: TNF-α, IL-1β, IL-6, IL-8, IL-10, and IL-12p70). The beads are incubated with 30 μl of sample, and the cytokines of interest are first captured on the bead. After washing the beads, a mixture of optimally paired second antibodies conjugated to phycoerythrin is added which then forms a fluorescent ternary complex with the immobilized cytokine, the intensity (measured in the FL2 channel) of which is proportional to the cytokine concentration on the bead. The assay was performed according to protocols provided by the vendor. Standard curves were generated using recombinant cytokines provided in the kit. The data were analyzed in the CBA software suite that is integral to the FACSArray system. The CBA multiplexed assay was also used to quantify cytokine production in mouse blood samples (see below) using the mouse inflammation CBA kit which includes the following analytes: TNF-α, IL-6, IL-10, macrophage chemotactic protein-1 (MCP-1), IFN-γ, and IL-12p70.
Inhibition of LPS-induced NF-κB induction:
The inhibition of induction of NF-κB (a key transcripitional activator of the innate immune system) was quantified using human embryonic kidney 293 cells cotransfected with TLR4 (LPS receptor), CD14 and MD2 (co-receptors), available from InvivoGen, Inc. (HEK-Blue™, San Diego, CA), as described elsewhere 81. Stable expression of secreted alkaline phosphatase (seAP) under control of NF-κB/AP-1 promoters is inducible by LPS, and extracellular seAP in the supernatant is proportional to NF-κB induction. HEK-4 cells were incubated at a density of ∼105 cells/ml in a volume of 80 μl/well, in 384-well, flat-bottomed, cell culture-treated microtiter plates until confluency was achieved, and subsequently stimulated with 10 ng/ml lipopolysaccharide (LPS). Concurrent to LPS stimulation, serially diluted concentrations of test compounds were added to the cell medium using a rapid-throughput, automated protocol employing a Bio-Tek P2000 liquid handler as described above, and left to incubate overnight.. Polymyxin B was used as reference compound in each plate. Positive- (LPS stimulation only) and negative-controls (HEK-detection medium only) were included in each experiment. seAP was assayed spectrophotometrically using an alkaline phosphatase-specific chromogen (present in HEK-detection medium as supplied by the vendor) at 620 nm.
Phosflow™ flow cytometric assay for p38MAPK:
1 ml aliquots of fresh whole blood, anticoagulated with heparin (obtained by venipuncture from healthy human volunteers with informed consent and as per guidelines approved by the Human Subjects Experimentation Committee) were incubated with 25 μl of a mix of 8 μg/ml of E. coli 0111:B4 LPS and graded concentrations of test compounds diluted in saline (typically serially diluted from 800 μM) for 15 minutes at 37°C. This resulted in final concentrations of 100 ng/ml of LPS and 200 μM of compound (at the lowest dilution). Positive (LPS alone) and negative (saline) controls were included in each experiment. Erythrocytes were lysed and leukocytes were fixed in one step by mixing 200 μl of the samples in 4 ml pre-warmed Whole Blood Lyse/Fix Buffer (Becton-Dickinson Biosciences, San Jose, CA). After washing the cells at 500 g for 8 minutes in CBA buffer, the cells were permeabilized in ice-cold methanol for 30 min, washed twice in CBA buffer and transferred to a Millipore MultiScreen BV 1.2μ filter plate and stained with either phycoerythrin (PE)-conjugated mouse anti-p38MAPK (pT180/pY182; BD Biosciences) mAb, or a matched PE-labeled mouse IgG1 κ isotype control mAb for 60 minutes. The cells were washed twice in the plate by aspiration as per protocols supplied by the vendor. Cytometry was performed using a BD FACSArray instrument in the single-color mode for PE acquisition on 20,000 gated events. Post-acquisition analyses were performed using FlowJo v 7.0 software (Treestar, Ashland, OR).
Murine in vivo experiments:
Dose-response and time-course experiments in a D-galactosamine sensitized mouse model of endotoxic shock was performed as described elsewhere21;22 using a supralethal (twice the LD100 dose, i.e., 200 ng/mouse) of LPS. All animal experiments were carried out with prior institutional IACUC approval. Female, outbred, 9- to 11-week-old CF-1 mice (Charles River, Wilmington, MA) weighing 22−28 g were used in all studies. Experiments designed to analyze the temporal profile of in vivo cytokine levels in animals treated with compound were performed as follows. Cohorts of 5 animals per group were administered graded doses of EVK-203 (or vehicle) subcutaneously at time = −1 h. At t = 0 h, either 200 ng LPS in saline (twice the LD100 dose), or vehicle alone was administered intraperitoneally. Following lethal LPS challenge, animals were anesthetized with halothane and blood samples were obtained by terminal cardiac puncture at t = 1, 2, and 3 h. Plasma samples were processed for cytokine analyses using the CBA assay (Mouse Inflammation CBA Kit, BD Biosciences) as described above. For toxicity studies, mice received EVK-203 (40 mg/kg) dissolved in 0.2 ml isotonic saline containing human serum albumin at 66 mg/ml subcutaneously in the lower flank in a volume of 0.2 ml, each day for 5 days (200 mg/kg EVK-203 total dose) and monitored for abnormal external characteristics. Blood was harvested on day 6 via cardiac puncture under anesthesia into heparin tubes, centrifuged at 4000 rpm for 10 minutes. Plasma samples were submitted for testing to University of Missouri Research Animal Diagnostic Laboratory (Columbia, MO).
Acknowledgment:
This work was supported from NIH grant 1R01 AI50107.
Glossary
Abbreviations:
- LPS
lipopolysaccharide
- IL-1β
interleukin-1β
- IL-6
interleukin 6
- PMB
polymyxin B
- CBA
Cytometric bead array
- BC
BODIPY-TR-cadaverine (5-(((4-(4,4-difluoro-5-(2-thienyl)-4-bora-3a,4a-diaza-s-indacene-3-yl) phenoxy)acetyl)amino)pentylamine hydrochloride )
- ED50
effective displacement to 50% of bound probe
- p38MAPK
p38 mitogen activated protein kinase
- PMN
polymorphonuclear cells
- s.c.
subcutaneous
- i.p.
intraperitoneal
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zähringer U, Seydel U, Di Padova F, et al. FASEB J. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 2.Hurley JC. Drug Saf. 1995;12:183–195. doi: 10.2165/00002018-199512030-00004. [DOI] [PubMed] [Google Scholar]
- 3.Prins JM, van Agtmael MA, Kuijper EJ, van Deventer SJ, Speelman P. J.Infect.Dis. 1995;172:886–891. doi: 10.1093/infdis/172.3.886. [DOI] [PubMed] [Google Scholar]
- 4.Ulevitch RJ. Immunol.Res. 2000;21:49–54. doi: 10.1385/IR:21:2-3:49. [DOI] [PubMed] [Google Scholar]
- 5.Dinarello CA. Curr.Top.Microbiol.Immunol. 1996;216:133–165. doi: 10.1007/978-3-642-80186-0_7. [DOI] [PubMed] [Google Scholar]
- 6.Bone RC. Clin.Chest Med. 1996;17:175–181. doi: 10.1016/s0272-5231(05)70307-5. [DOI] [PubMed] [Google Scholar]
- 7.Martin GS, Mannino DM, Eaton S, Moss M. N.Engl.J.Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 8.Holst O, Ulmer AJ, Brade H, Rietschel ET. On the chemistry and biology of bacterial endotoxic lipopolysaccharides. In: Masihi N, editor. Immunotherapy of infections. Vol. 94. Marcel Dekker; New York, Basel, Hong Kong: pp. 281–308. [Google Scholar]
- 9.Vesentini S, Soncini M, Zaupa A, Silvestri V, Fiore GB, Redaelli A. Int.J.Artif.Organs. 2006;29:239–250. doi: 10.1177/039139880602900210. [DOI] [PubMed] [Google Scholar]
- 10.Vincent JL, Laterre PF, Cohen J, Burchardi H, Bruining H, Lerma FA, Wittebole X, De Backer D, Brett S, Marzo D, Nakamura H, John S. Shock. 2005;23:400–405. doi: 10.1097/01.shk.0000159930.87737.8a. [DOI] [PubMed] [Google Scholar]
- 11.Vincent JL, Laterre PF, Cohen J, Burchardi H, Bruining H, Lerma FA, Wittebole X, De Backer D, Brett S, Marzo D, Nakamura H, John S. Shock. 2005;23:400–405. doi: 10.1097/01.shk.0000159930.87737.8a. [DOI] [PubMed] [Google Scholar]
- 12.Bhattacharjya S, David SA, Mathan VI, Balaram P. Biopolymers. 1997;41:251–265. [Google Scholar]
- 13.David SA. J.Molec.Recognition. 2001;14:370–387. doi: 10.1002/jmr.549. [DOI] [PubMed] [Google Scholar]
- 14.David SA, Bechtel B, Annaiah C, Mathan VI, Balaram P. Biochim.Biophys.Acta. 1994;1212:167–175. doi: 10.1016/0005-2760(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 15.David SA, Mathan VI, Balaram P. J.Endotoxin.Res. 1995;2:325–336. [Google Scholar]
- 16.Blagbrough IS, Geall AJ, David SA. Bioorg.Med.Chem.Lett. 2000;10:1959–1962. doi: 10.1016/s0960-894x(00)00380-2. [DOI] [PubMed] [Google Scholar]
- 17.David SA, Perez L, Infante MR. Bioorg.Med.Chem.Lett. 2002;12:357–360. doi: 10.1016/s0960-894x(01)00749-1. [DOI] [PubMed] [Google Scholar]
- 18.David SA, Silverstein R, Amura CR, Kielian T, Morrison DC. Antimicrob.Agents Chemother. 1999;43:912–919. doi: 10.1128/aac.43.4.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nabel GJ, Yang ZY, Nabel EG, Bishop K, Marquet M, Felgner PL, Gordon D, Chang AE. Ann.N.Y.Acad.Sci. 1995;772:227–231. doi: 10.1111/j.1749-6632.1995.tb44748.x. [DOI] [PubMed] [Google Scholar]
- 20.Rosenberg SA, Blaese RM, Brenner MK, Deisseroth AB, Ledley FD, Lotze MT, Wilson JM, Nabel GJ, Cornetta K, Economou JS, Freeman S, Riddell SR, Oldfield E, Gansbacher B, Dunbar C, Walker RE, Schuening FG, Roth JA, Crystal RG, Welsh MJ, Culver K, Heslop HE, Simons J, Wilmott RW, Tiberghien P. Hum.Gene Ther. 1996;7:2287–2313. doi: 10.1089/hum.1996.7.18-2287. [DOI] [PubMed] [Google Scholar]
- 21.Miller KA, Suresh Kumar EVK, Wood SJ, Cromer JR, Datta A, David SA. J.Med.Chem. 2005;48:2589–2599. doi: 10.1021/jm049449j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burns MR, Wood SJ, Miller KA, Nguyen T, Cromer JR, David SA. Bioorg.Med.Chem. 2005;13:2523–2536. doi: 10.1016/j.bmc.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 23.Burns MR, Jenkins SA, Wood SJ, Miller K, David SA. J.Comb.Chem. 2006;8:32–43. doi: 10.1021/cc0500755. [DOI] [PubMed] [Google Scholar]
- 24.Guo JX, Wood SJ, David SA, Lushington GH. Bioorg.Med.Chem.Lett. 2006;16:714–717. doi: 10.1016/j.bmcl.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 25.Raetz CR, Garrett TA, Reynolds CM, Shaw WA, Moore JD, Smith DC, Jr., Ribeiro AA, Murphy RC, Ulevitch RJ, Fearns C, Reichart D, Glass CK, Benner C, Subramaniam S, Harkewicz R, Bowers-Gentry RC, Buczynski MW, Cooper JA, Deems RA, Dennis EA. J.Lipid Res. 2006;47:1097–1111. doi: 10.1194/jlr.M600027-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Burns MR, Jenkins SA, Kimbrell MR, Balakrishna R, Nguyen TB, Abbo BG, David SA. J.Med.Chem. 2007 Feb 22;50:877–888. doi: 10.1021/jm061198m. [DOI] [PubMed] [Google Scholar]
- 27.Khownium K, Wood SJ, Miller KA, Balakrishna R, Nguyen TB, Kimbrell MR, Georg GI, David SA. Bioorg.Med.Chem.Lett. 2006 Mar 1;16:1305–1308. doi: 10.1016/j.bmcl.2005.11.059. [DOI] [PubMed] [Google Scholar]
- 28.Robert F, Hériter J, Quiquirez J, Simian H, Blank I. J.Agric.Food Chem. 2004;52:3525–3529. doi: 10.1021/jf0498968. [DOI] [PubMed] [Google Scholar]
- 29.Wood SJ, Miller KA, David SA. Comb.Chem.High.Throughput.Screen. 2004;7:239–249. doi: 10.2174/1386207043328832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wood SJ, Miller KA, David SA. Comb.Chem.High.Throughput.Screen. 2004;7:733–743. doi: 10.2174/1386207043328229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weisz A, Oguchi S, Cicatiello L, Esumi H. J.Biol.Chem. 1994;269:8324–8333. [PubMed] [Google Scholar]
- 32.David SA, Balaram P, Mathan VI. J.Endotoxin.Res. 1995;2:99–106. [Google Scholar]
- 33.Fenton MJ, Golenbock DT. J.Leukoc.Biol. 1998;64:25–32. doi: 10.1002/jlb.64.1.25. [DOI] [PubMed] [Google Scholar]
- 34.Beamer LJ, Carroll SF, Eisenberg D. Biochem.Pharmacol. 1999 Feb 1;57:225–229. doi: 10.1016/s0006-2952(98)00279-2. [DOI] [PubMed] [Google Scholar]
- 35.Rossignol DP, Wasan KM, Choo E, Yau E, Wong N, Rose J, Moran J, Lynn M. Antimicrob.Agents Chemother. 2004;48:3233–3240. doi: 10.1128/AAC.48.9.3233-3240.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pages G, Pouyssegur J. Methods Mol.Biol. 2004;250:155–166. doi: 10.1385/1-59259-671-1:155. [DOI] [PubMed] [Google Scholar]
- 37.Brunet A, Pouyssegur J. Essays Biochem. 1997;32:1–16. [PubMed] [Google Scholar]
- 38.Khownium K, Wood SJ, Miller KA, Balakrishna R, Nguyen TB, Kimbrell MR, Georg GI, David SA. Bioorg.Med.Chem.Lett. 2006 Mar 1;16:1305–1308. doi: 10.1016/j.bmcl.2005.11.059. [DOI] [PubMed] [Google Scholar]
- 39.Wasan KM, Sivak O, Cote RA, MacInnes AI, Boulanger KD, Lynn M, Christ WJ, Hawkins LD, Rossignol DP. Antimicrob.Agents Chemother. 2003;47:2796–2803. doi: 10.1128/AAC.47.9.2796-2803.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Felty AR, Keefer CS. JAMA. 1924;82:1430–1433. [Google Scholar]
- 41.Gelfand JA, Shapiro L. New Horizons. 1993;1:13–22. [PubMed] [Google Scholar]
- 42.Gasche Y, Pittet D, Sutter P. Outcome and prognostic factors in bacteremic sepsis. In: Sibbald WJ, Vincent JL, editors. Clinical trials for treatment of sepsis. Vol. 95. Springer-Verlag; Berlin: pp. 35–51. [Google Scholar]
- 43.MMWR. 1990;39:31–34. [Google Scholar]
- 44.Moss M, Martin GS. Intensive Care Med. 2004;30:527–529. doi: 10.1007/s00134-004-2182-z. [DOI] [PubMed] [Google Scholar]
- 45.Cross A, Opal SM. J.Endotoxin Res. 1994;1:57–59. [Google Scholar]
- 46.Rietschel ET, Brade H, Brade L, Brandenburg K, Schade UF, Seydel U, Z,,hringer U, Galanos C, Lüderitz O, Westphal O, Labischinski H, Kusumoto S, Shiba T. Prog.Clin.Biol.Res. 1987;231:25–53. [PubMed] [Google Scholar]
- 47.Peterson A, Hancock REW, McGroarty EJ. J.Bacteriol. 1985;164:1256–1261. doi: 10.1128/jb.164.3.1256-1261.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vaara M, Vaara T. Antimicrobial Agents and Chemotherapy. 1983;24:114–122. doi: 10.1128/aac.24.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rocque WJ, Fesik SW, Haug A, McGroarty EJ. Antimicrobial Agents and Chemotherapy. 1988;32:308–313. doi: 10.1128/aac.32.3.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Storm DR, Rosenthal K. Annual Reviews of Biochemistry. 1977;46:723–763. doi: 10.1146/annurev.bi.46.070177.003451. [DOI] [PubMed] [Google Scholar]
- 51.Morrison DC, Jacobs DM. Immunochemistry. 1976;13:813–818. doi: 10.1016/0019-2791(76)90181-6. [DOI] [PubMed] [Google Scholar]
- 52.Yao YM, Tian HM, Sheng ZY, Wang YP, Yu Y, Sun SR, Xu SH. J.Trauma. 1995;38:924–930. doi: 10.1097/00005373-199506000-00018. [DOI] [PubMed] [Google Scholar]
- 53.Durando MM, MacKay RJ, Linda S, Skelley LA. Am.J.Vet.Res. 1994;55:921–927. [PubMed] [Google Scholar]
- 54.Stokes DC, Shenep JL, Fishman ML, Hidner WK, Bysani GK, Rufus K. J.Infect.Dis. 1989;160:52–57. doi: 10.1093/infdis/160.1.52. [DOI] [PubMed] [Google Scholar]
- 55.Porro M, Rustici A, Velucchi M, Agnello D, Villa P, Ghezzi P. Prog.Clin.Biol.Res. 1998;397:315–325. [PubMed] [Google Scholar]
- 56.Rustici A, Velucchi M, Faggioni R, Sironi M, Ghezzi P, Quataert S, Green B, Porro M. Science. 1993;259:361–365. doi: 10.1126/science.8420003. [DOI] [PubMed] [Google Scholar]
- 57.Vaara M. FEMS Microbiology Letters. 1983;18:117–121. [Google Scholar]
- 58.Vaara M, Vaara T. Nature. 1983;303:526–528. doi: 10.1038/303526a0. [DOI] [PubMed] [Google Scholar]
- 59.Viljanen P, Matsunaga H, Kimura Y, Vaara M. J.Antibiot.Tokyo. 1991;44:517–523. doi: 10.7164/antibiotics.44.517. [DOI] [PubMed] [Google Scholar]
- 60.Iwagaki A, Porro M, Pollack M. Infect.Immun. 2000;68:1655–1663. doi: 10.1128/iai.68.3.1655-1663.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scott MG, Vreugdenhil AC, Buurman WA, Hancock RE, Gold MR. J.Immunol. 2000 Jan 15;164:549–553. doi: 10.4049/jimmunol.164.2.549. [DOI] [PubMed] [Google Scholar]
- 62.Jerala R, Porro M. Curr.Top.Med.Chem. 2004;4:1173–1184. doi: 10.2174/1568026043388079. [DOI] [PubMed] [Google Scholar]
- 63.Porro M. Trends.Microbiol. 1994;2:65–66. doi: 10.1016/0966-842x(94)90530-4. [DOI] [PubMed] [Google Scholar]
- 64.Zeni F, Freeman B, Natanson C. Crit.Care Med. 1997;25:1097–1100. doi: 10.1097/00003246-199707000-00001. [DOI] [PubMed] [Google Scholar]
- 65.Quezado ZMN, Banks SM, Natanson C. Trends.Biotech. 1995;13:56–63. doi: 10.1016/S0167-7799(00)88906-4. [DOI] [PubMed] [Google Scholar]
- 66.David SA, Balaram P, Mathan VI. Med.Microbiol.Lett. 1993;2:42–47. [Google Scholar]
- 67.David SA, Mathan VI, Balaram P. Biochim.Biophys.Acta. 1992;1123:269–274. doi: 10.1016/0005-2760(92)90006-h. [DOI] [PubMed] [Google Scholar]
- 68.David SA. J.Molec.Recognition. 2001;14:370–387. doi: 10.1002/jmr.549. [DOI] [PubMed] [Google Scholar]
- 69.Beamer LJ, Carroll SF, Eisenberg D. Protein Sci. 1998;7:906–914. doi: 10.1002/pro.5560070408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McCormack FX, Festa AL, Andrews RP, Linke M, Walzer PD. Biochemistry. 1997;36:8092–8099. doi: 10.1021/bi970313f. [DOI] [PubMed] [Google Scholar]
- 71.Bone RC. Clin.Microbiol.Rev. 1993;6:57–68. doi: 10.1128/cmr.6.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Opal SM, Yu RL. J. Drugs. 1998;55:497–508. doi: 10.2165/00003495-199855040-00002. [DOI] [PubMed] [Google Scholar]
- 73.Balakrishna R, Wood SJ, Nguyen TB, Miller KA, Suresh Kumar EV, Datta A, David SA. Antimicrob.Agents Chemother. 2006;50:852–861. doi: 10.1128/AAC.50.3.852-861.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haldar J, Kondaiah P, Bhattacharya S. J.Med.Chem. 2005;48:3823–3831. doi: 10.1021/jm049106l. [DOI] [PubMed] [Google Scholar]
- 75.Roehm NW, Rodgers GH, Hatfield SM, Glasebrook AL. J Immunol.Methods. 1991 Sep 13;142:257–265. doi: 10.1016/0022-1759(91)90114-u. [DOI] [PubMed] [Google Scholar]
- 76.Nagini S, Selvam S. Med.Sci.Res. 1997;25:119–121. [Google Scholar]
- 77.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Anal.Biochem. 1982;126:131. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 78.Greiss P. Chem.Ber. 1879;12:426–427. [Google Scholar]
- 79.Cook EB, Stahl JL, Lowe L, Chen R, Morgan E, Wilson J, Varro R, Chan A, Graziano FM, Barney NP. J.Immunol.Methods. 2001;254:109–116. doi: 10.1016/s0022-1759(01)00407-0. [DOI] [PubMed] [Google Scholar]
- 80.Funato Y, Baumhover H, Grantham-Wright D, Wilson J, Ernst D, Sepulveda H. Cytometry Res. 2002;12:93–103. [Google Scholar]
- 81.Khownium K, Wood SJ, Miller KA, Balakrishna R, Nguyen TB, Kimbrell MR, Georg GI, David SA. Bioorg.Med.Chem.Lett. 2006 Mar 1;16:1305–1308. doi: 10.1016/j.bmcl.2005.11.059. [DOI] [PubMed] [Google Scholar]
- 82.Ferguson AD, Hofmann E, Coulton J, Diedrichs K, Welte W. Science. 1998:2215–2220. doi: 10.1126/science.282.5397.2215. [DOI] [PubMed] [Google Scholar]