

Abstract
Compound S4 [S-3-(4-acetylamino-phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamide] is a novel nonsteroidal selective androgen receptor modulator that demonstrates tissue-selective androgenic and anabolic effects. The purpose of this in vitro study was to identify the phase I metabolites, potential species differences in metabolism, and the cytochromes P450 (P450s) involved in the phase I metabolism of S4 using 14C-S4, recombinant P450s, and other liver enzyme preparations from human, rat, and dog. The major phase I metabolism pathways of S4 in humans were identified as deacetylation of the B-ring acetamide group, hydrolysis of the amide bond, reduction of the A-ring nitro group, and oxidation of the aromatic rings, with deacetylation being the predominant pathway observed with most of the enzyme preparations tested. Among the major human P450 enzymes tested, CYP3A4 appeared to be one of the major phase I enzymes that could be responsible for the phase I metabolism of S4 [Km = 16.1 μM, Vmax = 1.6 pmol/(pmol · min)] in humans and mainly catalyzed the deacetylation, hydrolysis, and oxidation of S4. In humans, the cytosolic enzymes mainly catalyzed the hydrolysis reaction, whereas the microsomal enzymes primarily catalyzed the deacetylation reactions. Similar phase I metabolic profiles were observed in rats and dogs as well, except that the amide bond hydrolysis seemed to occur more rapidly in rats. In summary, these results showed that the major phase I reaction of S4 in human, rat, and dog is acetamide group deacetylation.
Selective androgen receptor modulators (SARMs) have been proposed as a new class of androgen receptor (AR) ligands with unique tissue selectivity that could be used as safer and more beneficial treatment in male hormone replacement therapy (Negro-Vilar, 1999). Structural modifications of the nonsteroidal AR ligand, bicalutamide, turned this “pure antagonist” into a new series of AR agonists (Dalton et al., 1998). As shown in our previous studies (He et al., 2002; Yin et al., 2003b), the thio linkage and B-ring substitution are critical structural features required for the agonist activity of the ligand. Although it is not clear how the structural modifications affect the ligand-receptor interaction, the modifications did significantly change the metabolic profile of the ligands.
The initial change of the B-ring substitution from fluoro group to acetamide group, and the sulfonyl linkage to thio group, greatly improved the binding affinity and in vitro agonist activity of acetothiolutamide (Yin et al., 2003c). However, it also made the molecule more susceptible to metabolism, resulting in a short half-life (less than 1 h in rat) and loss of in vivo pharmacological activity. Further modification changed the thio linkage to an ether linkage [compound S4, S-3-(4-acetylamino-phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamide) (Yin et al., 2003a), which maintained the agonist activity of the ligand and increased the in vivo half-life to 3 to 4 h in rat (Kearbey et al., 2004). Even so, the ideal half-life required for once a day administration of SARM, as proposed by Negro-Vilar (1999), is yet to be achieved. Better understanding of the hepatic metabolism profile of S4 will be helpful to guide additional structural modification.
Our previous studies (Gao et al., 2004) have shown that S4 could not be metabolized in AR target tissues by either 5α-reductase or aromatase. However, the formation of active metabolites in vivo represents another possible mechanism for the observed tissue selectivity of SARMs (Yin et al., 2003a). As a matter of fact, the pharmacological activity of another nonsteroidal AR antagonist, flutamide, is mediated by its active metabolite, hydroxyflutamide (Katchen and Buxbaum, 1975) (Fig. 1). Although S4 is not a substrate for either 5α-reductase or aromatase, it is unclear whether any active hepatic metabolite(s) could contribute to the pharmacologic activity of S4. The studies described herein were designed to address these issues.
Fig. 1.
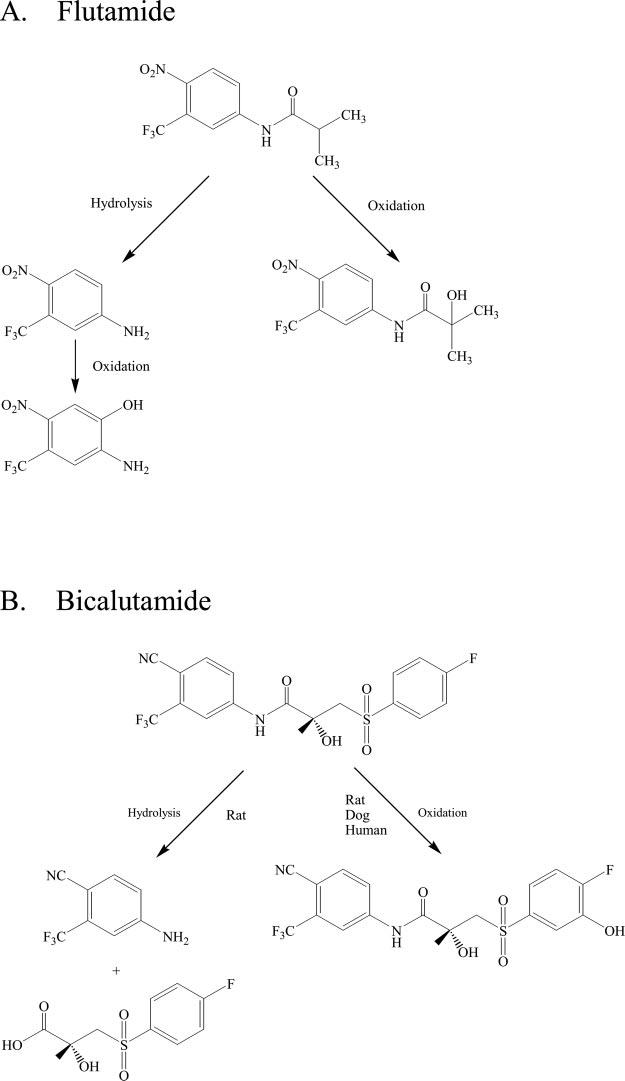
Phase I metabolism of flutamide (A) (Katchen and Buxbaum, 1975) and bicalutamide (B) (Boyle et al., 1993; McKillop et al., 1993).
Both flutamide and bicalutamide are structural analogs of S4. The metabolic profiles of flutamide and bicalutamide have been well characterized (Katchen and Buxbaum, 1975; Boyle et al., 1993; McKillop et al., 1993). Hydrolysis of the amide bond and oxidation of the aromatic rings are the major phase I metabolism pathways for both flutamide and bicalutamide (Fig. 1). Species differences were observed in bicalutamide metabolism (Boyle et al., 1993; McKillop et al., 1993) (Fig. 1B). Hydrolysis of the amide bond of bicalutamide was only observed in rats, but not in humans and dogs, which explains the prolonged half-life of bicalutamide in humans (McKillop et al., 1993). In comparison, although the previous compound (Yin et al., 2003c), acetothiolutamide, also undergoes extensive oxidation of the thio group and aromatic rings in rat, amide bond hydrolysis was not observed. Modification of the thio linkage in acetothiolutamide to the ether linkage in S4 was intended to eliminate the possibility of linker oxidation, while maintaining the stability of the amide bond.
In the current study, the metabolic profile and potential species differences in the metabolism of S4 were characterized using recombinant human cytochrome P450 (P450) enzymes and pooled liver enzyme preparations from rats, dogs, and humans.
Materials and Methods
Materials
Compounds S4 and M1, and 14C-S4 were synthesized in our laboratories. Recombinant human P450 enzyme and insect cell control (Supersomes), pooled human, rat, and dog liver microsome, cytosol, and S9 preparations were purchased from BD Gentest (Woburn, MA). 4′-Hydroxy-diclofenac, 4′-hydroxy-mephenytoin, mephenytoin, 1′-hydroxy-bufuralol, and bufuralol were also purchased from BD Gentest. EcoLite(+) scintillation cocktail was purchased from MP Biomedicals (Irvine, CA). All other chemicals and reagents were purchased from Sigma-Aldrich (St Louis, MO). All analytical columns were purchased from Waters (Milford, MA). NHS-Biotin (N-hydroxysuccinimidobiotin) was purchased from Pierce Chemical (Rockford, IL).
In Vitro Metabolism Reactions
In vitro enzyme reactions were conducted according to the instructions provided by BD Gentest. All phase I reactions were conducted at 37°C in the presence of 1 mM NADPH in 100 mM phosphate buffer (pH 7.4) for various times. For liver microsomes, cytosol, or liver S9 preparations, a total protein concentration of 2 mg/ml was used in the reaction, and the recombinant P450 enzymes were used with the final concentration of 200 pmol/ml. In general, the incubation time was 2 h for metabolite identification, whereas incubation time for the determination of enzyme kinetics parameters was 10 min. Incubation conditions were optimized so that the rate of metabolism was linear with respect to incubation time and microsomal protein concentration. The kinetic parameters, Km and Vmax, were determined by nonlinear regression analysis using WinNonlin (version 4.0; Pharsight, Mountain View, CA) and the Michaelis-Menten kinetics model. For experiments using recombinant P450 enzymes, insect cell control Supersomes were included as a control for native activities from the expression system, and comparable protein concentrations were used. Reactions were stopped by adding ice-cold acetonitrile (v/v, 1:1) containing internal standard (CM-II-87, a structural analog of S4) for HPLC analysis. Protein present in the reaction mixture was precipitated by centrifugation (>16,000g, 30 min at 4°C), and the supernatant was either diluted with appropriate mobile phase or directly used for HPLC analysis. S4 was separated on a reversed-phase column (Symmetry C8, 3.9 × 150 mm) with a mobile phase of 45% acetonitrile and 50 mM phosphate buffer (pH 4.8) in deionized water, at a flow rate of 1.0 ml/min, and was detected by its UV absorbance at 230 nm.
Identification of the Phase I Metabolite of S4
14C-S4 (2 μM) was incubated with human, rat, or dog liver preparations at 37°C for 2 h. After precipitation of the protein with acetonitrile (v/v, 1:1), the organic phase left in the supernatant was evaporated under nitrogen, and the resulting concentrated samples were used for HPLC analysis. 14C-S4 and its metabolites were separated using a reversed-phase column (NovaPak C18, 3.0 × 300 mm) with a mobile phase of acetonitrile in deionized water at a flow rate of 1 ml/min. The initial mobile phase contained 0% acetonitrile and was maintained at this composition for 3 min. The percentage of acetonitrile was increased to 5% linearly over the next 7 min and was further increased to 30% in the following 5 min. The percentage of acetonitrile was again slowly increased to 35% over the next 15 min, then rapidly changed to 95% in another 5 min, and was maintained at this composition for 5 min. Finally, the percentage of acetonitrile was returned to 0% in the last 5 min. The eluted fractions (30 s per fraction) from the HPLC were collected, and the total radioactivity (dpm) in each fraction was counted in EcoLite(+) scintillation cocktail.
A similar experiment was conducted using nonradiolabeled S4. In this case, the eluted fractions containing possible metabolites were analyzed using negative-ion electrospray ionization (ESI) mass spectrometry (Thermo Electron LCQ DECA ion trap mass spectrometer; Thermo Electron Corporation, Waltham, MA) as previously described (Wu et al., 2004). For the MS system, the heated capillary temperature was set at 200°C, spray voltage was 3.5 kV, and the sheath gas and auxiliary gas flow rates were 60 and 20 ml/min, respectively. All other parameters were set to the optimized conditions for ionization and detection of S4. Data acquisition was controlled by Xcalibur software (Revision B; Thermo Electron Corporation). For MS2 analysis, S4 or metabolite ions were isolated with a width of 1.5 m/z and fragmented using a fragmentation energy ranging from 15 to 50%.
Biotinylation of the Hydrolysis Metabolites
The HPLC eluate from the first 3 min of the gradient run, as described above, was collected and concentrated under nitrogen. NHS-Biotin was dissolved in dimethyl sulfoxide and was added to the concentrated HPLC eluate (1:10 v/v) to a final concentration of 20 mM. The biotinylation reaction was conducted at room temperature for 1 h. The reaction mixture was then centrifuged, and the supernatant was subjected to HPLC analysis using the same condition as described above.
Androgen Receptor Binding Assay
The binding affinity of the deacetylated metabolite, M1, was determined using cytosolic AR prepared from ventral prostates of castrated male Sprague-Dawley rats (about 250 g), as described previously (Mukherjee et al., 1996).
Transcriptional Activation Assay
The ability of the compounds to influence AR-mediated transcriptional activation was examined using a cotransfection system, as described previously (Yin et al., 2003b). Transcriptional activation by M1 was measured at multiple concentrations, ranging from 0.1 to 1000 nM, and was reported as a percentage of the transcriptional activation observed with 0.1 nM DHT. Emax and EC50 were determined by nonlinear regression analysis using WinNonlin and the Sigmoidal Emax model.
Results
Identification of the Phase I Metabolites of S4
14C-S4 (2 μM) was incubated with liver S9 at 37°C for 2 h, and the 14C-metabolites were separated on HPLC. The concentration was chosen according to the hepatic concentration of S4 at pharmacologically relevant doses as determined by total body autoradiography in rats (data not shown). A similar experiment was repeated using nonradiolabeled S4, and the fractions corresponding to the 14C-metabolites were collected and subjected to MS analysis.
Four major metabolism pathways were identified (Fig. 2A): 1) deacetylation of the B-ring acetamide group, 2) hydrolysis of the amide bond, 3) reduction of the A-ring nitro group, and 4) oxidation of the B aromatic ring. Radiochromatographs corresponding to the identified metabolites are illustrated in Fig. 2B. MS2 spectra and the proposed fragmentation pattern of S4 and its metabolites are shown in Figs. 3, 4, and 5.
Fig. 2.
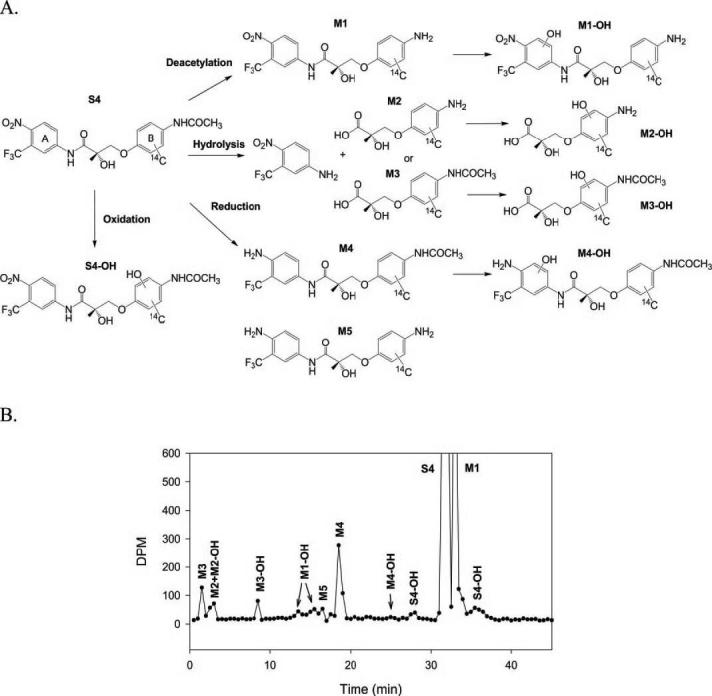
Summary of phase I metabolism pathway of S4. A, phase I metabolism pathways of 14C-S4 (uniformly labeled B-ring) as determined in human, rat, and dog liver preparations. B, radiochromatogram displaying the metabolism of 14C-S4 by pooled human liver S9 fraction.
Fig. 3.
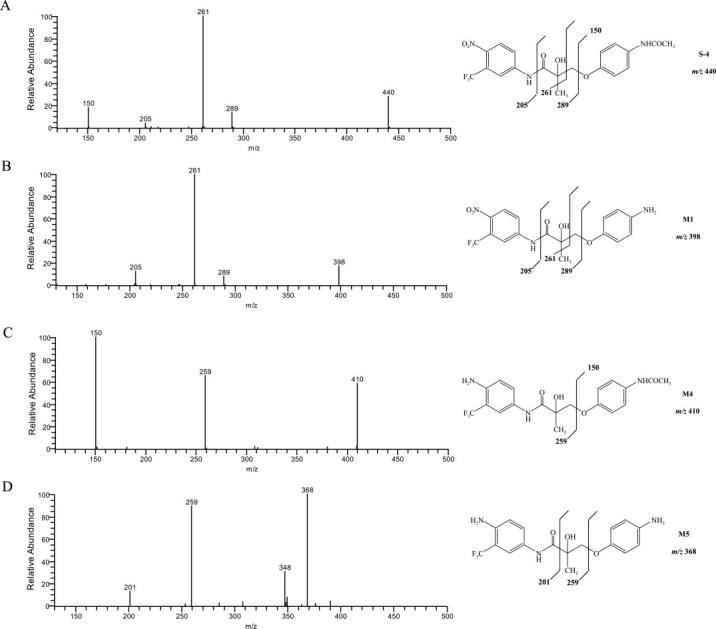
MS2 spectra (ESI negative ion mode) of S4 and the reduction and deacetylation products M1, M4, and M5.
Fig. 4.
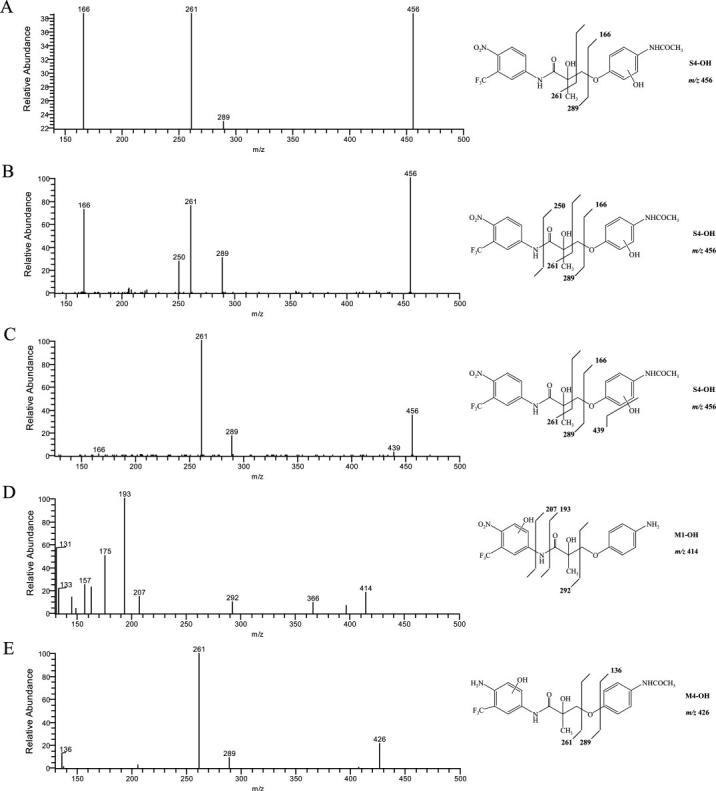
MS2 spectra (ESI negative ion mode) of the oxidation products S4-OH, M1-OH, and M4-OH. Three different S4 oxidation metabolites with different fragmentation patterns (A, B, and C) were identified.
Fig. 5.
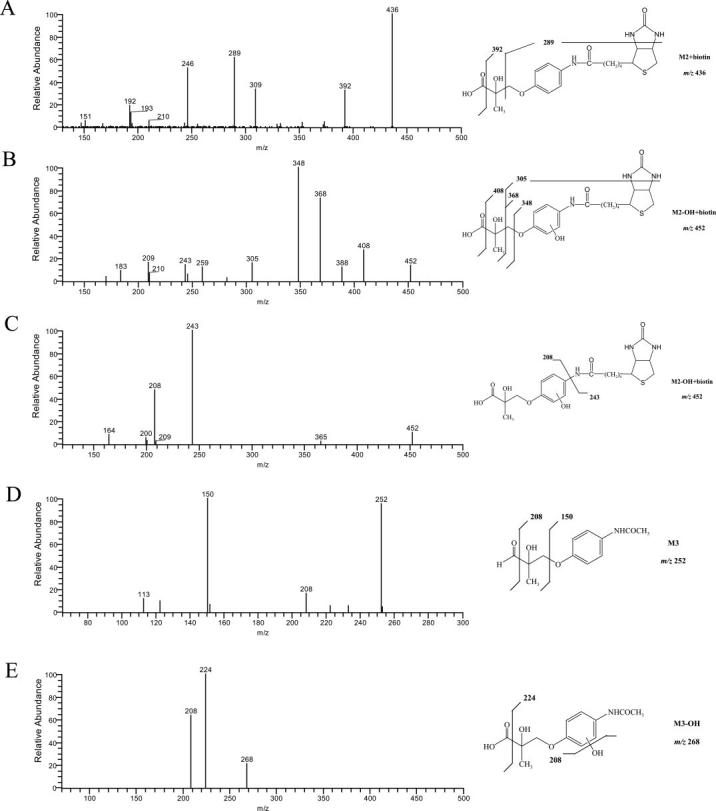
MS2 spectra (ESI negative ion mode) of the amide bond hydrolysis products M3, M3-OH, and the biotinylated M2 and M2-OH (B, C).
Deacetylation product M1, hydrolysis products M2, M2-OH, and M3, and reduction product M4 were the major in vitro metabolites of S4 in liver S9 fractions. S4 and M1 shared very similar fragmentation patterns (Fig. 3), except that the fragment ion at 150 m/z was not present in the M1 MS2 spectrum due to the deacetylation of the B-ring acetamide group. A similar pattern was observed for M4 and M5 as well.
Besides these major metabolites, the corresponding oxidation products, S4-OH, M1-OH, and M4-OH were also observed. As shown in Fig. 2A and Fig. 4, A through C, multiple oxidation products of S4 were identified, which could be due to differences in hydroxylation position on the B-ring. No A-ring hydroxylation product of S4 was observed. However, unlike S4, oxidation of M1 and M4 was only observed in the A-ring, not the B-ring. The fragmentation pattern of M1-OH and M4-OH changed dramatically as well, as shown in Fig. 4, D and E. Also, two different M1 oxidation products were observed as separated by HPLC (Fig. 2B). However, two products shared the same fragmentation pattern as shown in Fig. 4D.
Among all the metabolites identified, the hydrolysis products were the most hydrophilic due to the presence of the carboxyl group, and most of them coeluted with the solvent front (Fig. 2B). M2, particularly, contains an amine group on the B-ring, which mimics the structure of an amino acid and makes the analysis of the metabolite more difficult. To facilitate the separation and detection of the molecule, we collected and concentrated the corresponding HPLC eluate, and modified the aromatic amine group. NHS-Biotin is a commonly used protein labeling reagent that specifically modifies primary amine groups (Fig. 6A). Biotinylation of the B-ring amine group greatly facilitated the HPLC separation of the hydrolysis metabolites that contained the aromatic amine group, as shown in the chromatogram in Fig. 6B. On other hand, M3, the hydrolysis product with an intact acetamide group on the B-ring, was not modified and still coeluted with the solvent front. The MS2 fragmentation patterns for M3, M3-OH, and the biotinylated metabolite of M2 are shown in Fig. 5.
Fig. 6.
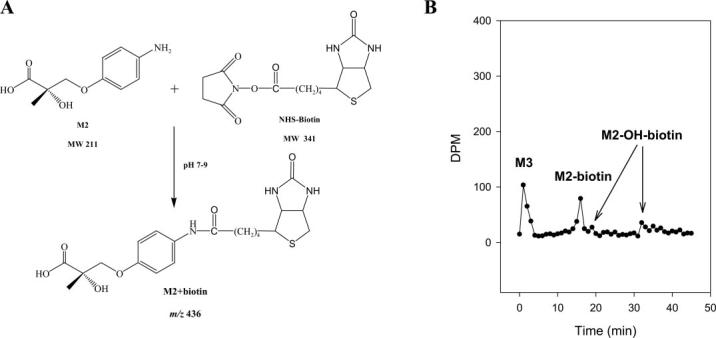
Derivatization and separation of M2. A, biotinylation reaction of S4 by NHS-Biotin. B, radiochromatogram displaying the separation of biotinylated 14C-M2 and 14C-M2-OH from 14C-M3.
Although M3 cannot be modified, the acidic condition could decrease the ionization of the carboxyl group and increase its retention time on column, whereas the hydrolysis products with the amine group would be highly ionized and eluted with the solvent front. The HPLC elution (first 3 min) containing M3, as separated under neutral pH (as described above), was collected and concentrated under nitrogen. M3 was further separated from the solvent front using a similar HPLC condition with pH 4.0 (0.2% acetic acid), and was eluted at 15 min (data not shown).
Comparison of the Phase I Metabolites of S4 in Different Species
The metabolic profile of S4 in different species was estimated and compared using human, rat, and dog liver S9 fraction. A similar total protein concentration (2 mg/ml) was used for all reactions. The relative abundance of each fraction in different reaction mixtures is presented in Fig. 7A as a percentage of 14C-S4 metabolized. Similarly, all four metabolic pathways were observed in the species tested. After a 2-h incubation, the predominant metabolite, M1, accounted for 61%, 44%, and 58% of the S4 metabolized in human, rat, and dog S9 fraction, respectively. However, the relative contribution of other pathways was quite different in these three species.
Fig. 7.
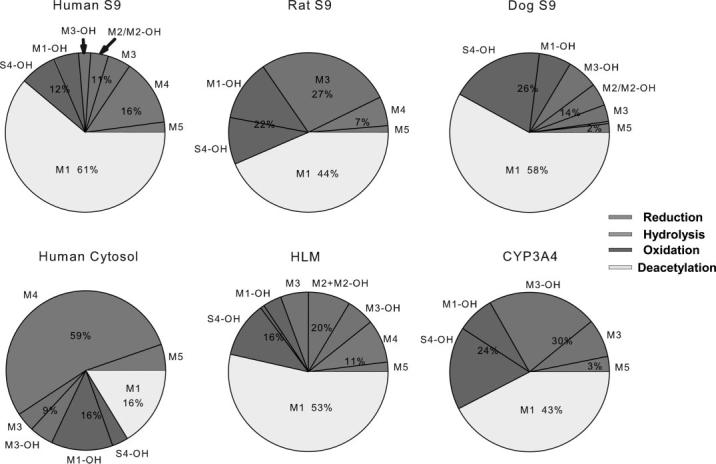
The relative abundance of the major in vitro metabolites of 14C-S4 after incubation with different liver enzyme preparations. A, metabolic profile of 14C-S4 in the presence of human, rat, and dog liver S9 fractions. B, metabolic profile of 14C-S4 in the presence of different subcellular fractions of human liver. The reaction was carried out in the presence of different liver enzyme preparations for 2 h at 37°C.
In human S9 fraction, besides deacetylation, the other three metabolic pathways contributed very similarly to the metabolism of S4, with hydrolysis, reduction, and oxidation accounting for 11%, 16%, and 12% of the S4 metabolized, respectively. However, reduction of S4 in human liver S9 fraction contributed to a relatively larger extent in S4 metabolism (16%), compared with those observed in rat (7%) and dog (2%) S9 fractions.
In contrast, hydrolysis seemed to play a larger role in S4 metabolism by rat S9 fraction, accounting for 27% of the S4 metabolized, as compared with 11% and 14% in human and dog S9 fractions. More interestingly, M3 was the only hydrolysis product observed in rat S9 fraction, whereas the hydrolysis products by human and dog S9 fractions contained M2, M3, and their oxidized products, which indicated that hydrolysis might be a faster process than deacetylation or oxidation in rat S9 fraction.
Also, oxidation of S4 and M1 accounted for more than 20% of the S4 metabolized by rat and dog S9 fractions, but only 12% of the S4 metabolized by human S9 fraction. Considering the fact that more oxidized hydrolysis metabolites were observed in dog S9 fraction incubations compared with rat S9 fraction, oxidation might contribute more significantly to S4 metabolism in dog S9 fraction.
In general, the major metabolic pathways observed in the three species tested were similar: deacetylation product M1 was the primary phase I metabolite of S4, with hydrolysis, reduction, and oxidation products observed in all three species as well.
Comparison of the Phase I Metabolism of S4 in Different Subcellular Fractions
The phase I metabolism of S4 was further estimated in incubations with different subcellular fractions of human liver preparations, including pooled human liver microsomes (HLMs) and human liver cytosol. After 2 h of incubation, the metabolic profiles of S4 after incubation with HLMs and human liver cytosol fractions were dramatically different (Fig. 7B). Because the majority of the phase I enzymes are located in HLMs, the metabolic profile observed in the reactions with HLMs was very similar to that observed with human liver S9 fraction (Fig. 7A), as described above. However, in the presence of human liver cytosol, deacetylation product M1 was no longer the primary metabolite observed, accounting for only 16% of the S4 metabolized, compared with 53% as observed in HLM incubations. On the contrary, reduction product M4 counted for more than 50% of the S4 metabolized by human liver cytosol, compared with 11% as observed in HLM incubations.
Although the amide bond hydrolysis reaction was commonly believed to be catalyzed by esterase present in the cytosolic fraction, hydrolysis of S4 seemed to contribute to S4 metabolism by HLMs to a much larger extent, 20%, as compared with 9% by the cytosolic fraction. Also, since the deacetylation reaction was preferred in the microsomal fraction, M3 turned out to be the major hydrolysis product in human liver cytosol incubates, whereas both acetylated (M3) and deacetylated hydrolysis products (M2 and M2-OH) of similar amount were observed in HLM incubates.
To further identify the major P450 enzymes responsible for phase I metabolism of S4, five major human P450 enzymes, CYP1A2, 2C9, 2C19, 2D6, and 3A4, were tested by measuring the disappearance of S4 after 2h of incubation with S4 (2 μM) at 37°C. Among the five enzymes tested, CYP3A4 was identified as one of the major P450 enzymes that might be responsible for S4 metabolism at 2 μM (data not shown). When incubated with 14C-S4 (2 μM), the metabolic profile of S4 by CYP3A4 was similar to that observed after incubation with HLMs (Fig. 7B). Recombinant human CYP3A4 mainly catalyzed deacetylation, hydrolysis, and oxidation reactions, which accounted for 43%, 30%, and 24%, respectively, of S4 metabolized. No significant metabolism of S4 was observed after incubation with insect cell control microsome, which does not contain the recombinant P450 enzymes.
Kinetics of S4 Metabolism by Recombinant Human CYP3A4
The disappearance of S4 was determined as an initial measure to estimate the enzyme kinetics parameters of CYP3A4 (Fig. 9). S4 showed similar affinity to CYP3A4 (16.1 μM) as testosterone (13 μM), but a lower Vmax [1.6 pmol/(pmol · min)] compared with testosterone [7.6 pmol/(pmol · min); data not shown].
Fig. 9.
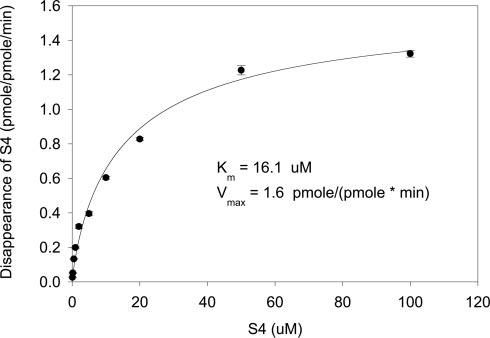
Enzyme kinetics of S4 metabolism by CYP3A4 as determined by measuring the disappearance of S4. The reaction was carried out in the presence of 200 pmol/ml CYP3A4 for 10 min at 37°C.
Characterization of M1 as an Active Metabolite of S4
S4 deacetylation product M1 maintained the core structure of the pharmacophore, which suggests that it could also interact with AR and act as an active metabolite. We previously showed that other B-ring amine derivatives showed moderate binding affinity toward AR (Yin et al., 2003b). In vitro receptor binding assay showed that M1 bound the AR with relatively lower affinity (Ki, 24.6 nM) compared with S4 (Ki, 4.0 nM). Furthermore, it behaved as a partial agonist in an in vitro transcriptional activation assay (Fig. 8), with relative agonist activity of 42% at 1 μM, compared with 0.1 nM DHT. These results suggested that M1 could also activate AR and might contribute to the pharmacological activity of S4 in vivo. More detailed studies of M1 are reported separately (Gao et al., 2006).
Fig. 8.
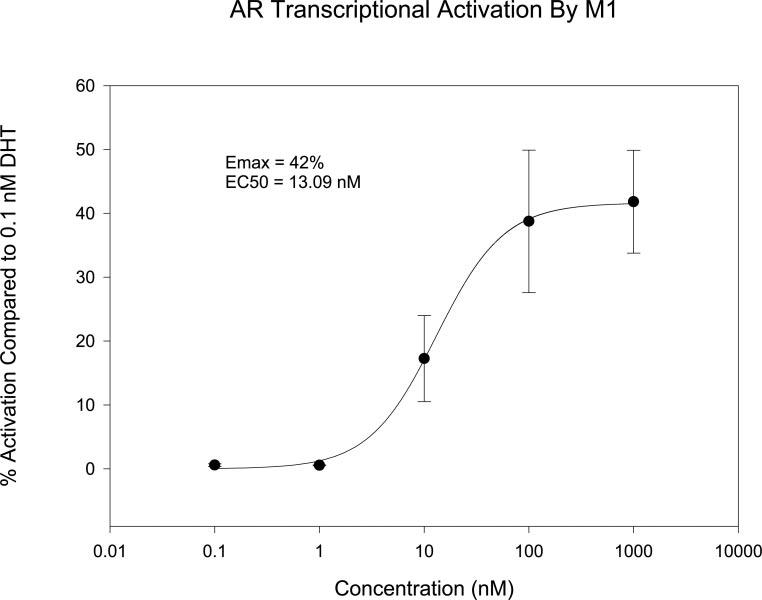
In vitro AR transcriptional activation by M1 using a cotransfection assay. The activation by M1 is presented as a percentage of the activation obtained in the presence of 0.1 nM DHT.
Discussion
The major phase I metabolism pathways of S4 in rats, dogs, and humans were identified using different liver enzyme preparations: deacetylation of the B-ring acetamide group, hydrolysis of the amide bond, reduction of the A-ring nitro group, and oxidation of the aromatic rings. Compared with our previous compound, acetothiolutamide, the introduction of the ether linkage in S4 eliminated the oxidation of the previous thio-ether linkage and reduced oxidation of the B-ring. However, it also decreased the overall stability of the amide bond, which was susceptible to extensive hydrolysis, particularly in rat. The cyano group on the A-ring was replaced by the nitro group in S4, considering the fact that the nitro group might improve the binding affinity of the ligand to AR, as shown in our previous study (Yin et al., 2003b). However, the nitro group was susceptible to reduction, which made the A-ring a metabolically labile site as well. Similar to acetothiolutamide, deacetylation of the B-ring acetamide group continues to play a prominent role in S4 metabolism.
Based on the structure-activity relationship established in our previous studies (He et al., 2002; Yin et al., 2003b), the introduction of the ether linkage and/or the acetamide substitution on the para-position of the B-ring was crucial to the agonist activity of this class of ligands. The ether linkage, particularly, is required for the high binding affinity of the ligand to AR. Therefore, modification of other metabolically labile sites would be necessary to improve the metabolic stability of the compound while maintaining its agonist activity: 1) the nitro substitution on the A-ring can be replaced by the cyano group; and 2) the acetamide group on the B-ring can be substituted by halogens, like compound S1, as presented in our previous pharmacologic study (Yin et al., 2003a), which will not only avoid the deacetylation reaction, but also reduce B-ring oxidation by reducing its electron density, since halogens are stronger electron-withdrawn groups.
Although the precise mechanisms by which the ether linkage improves the agonist activity of the ligand but decreases the metabolic stability of the amide bond is unclear, the recently solved bicalut-amide-bound AR LBD mutant (W741L) crystal structure (Bohl et al., 2005) suggested that an intramolecular hydrogen bond between the oxygen atom from the ether linkage and the hydrogen atom from the amide bond might be related to the susceptibility of the amide bond to hydrolysis. Furthermore, the electron density of the oxygen atom is greatly affected by the para-substituents on the B-ring, suggesting that compounds incorporating different para-substituents might show a differing degree of hydrolysis. In the case of bicalutamide, the oxygen atom from the sulfonyl linkage could also form a similar intramolecular hydrogen bond, which could be related to the extensive hydro-lysis of its amide bond in rats. However, this hypothesis needs to be further confirmed by more detailed studies using structural analogs containing different B-ring substitutions. Nevertheless, S4 appeared to undergo more extensive amide bond hydrolysis in rat liver S9 fraction than in human liver S9 fraction (Fig. 7A), suggesting that amide bond hydrolysis might not be the major metabolic pathway in humans as compared with rats, similar to bicalutamide.
S4 metabolism profiles in three different species were characterized and compared. Since the in vitro incubation was carried out for 2 h due to the low specific activity of the 14C-S4, the reported data only represented the “estimates” of relative rates of metabolism via different pathways and fractions. Among all three species tested, deacetylation accounted for about half of the S4 metabolized in vitro and is likely the major metabolic pathway in vivo. Furthermore, the deacetylated metabolite M1 bound to AR and initiated transcription activation in vitro, suggesting that it might contribute to the in vivo pharmacologic activity of S4 as well. Additional pharmacokinetic studies to examine the disposition of M1 are necessary to determine the in vivo activity of M1 as active metabolite.
Amide bond hydrolysis and acetamide deacetylation occurred in both cytosolic and microsomal fractions of the human liver preparation (Fig. 7B). The cytosolic enzymes mainly catalyzed the hydrolysis reaction, whereas the microsomal enzymes primarily catalyzed the deacetylation reactions, which suggested that the microsomal P450 enzymes might also be responsible for the hydrolysis and deacetylation of S4. CYP3A4 is responsible for the metabolism of more than 70% of marketed drugs (Casarett et al., 1996), with oxidation being the most common metabolic reaction. Similarly, CYP3A4 is responsible for the oxidation of S4 in vitro, and even bicalutamide oxidation in human, as recently described by Cockshott (2004). However, in our study, human CYP3A4 seemed to be able to catalyze hydrolysis reactions as well. CYP3A4-catalyzed hydrolysis reactions were rarely documented (Ji et al., 2004); however, they do exist. As described above (Fig. 7B), in the presence of NADPH, CYP3A4 not only catalyzed the oxidation of S4, but also catalyzed the hydrolysis of the amide bond and deacetylation of the acetamide group on the B-ring. The deacetylation reaction can also be considered as a hydrolysis reaction. Also, kinetics studies showed that S4 had similar affinity (Km) to CYP3A4 as testosterone (Fig. 9). At pharmacologically relevant concentrations, it is likely that CYP3A4 would be responsible for the deacetylation of S4 in humans.
Besides the identification of the major in vitro metabolites of S4 in three different species, the HPLC separation and MS analysis of the hydrolysis products, M2 and M3, were also a challenging part of our study. The presence of both carboxyl and amine groups in M2 made this molecule extremely hydrophilic. Similar to amino acids, M2 could not be separated from the solvent front under either acidic or basic conditions, as we tested, which is much different from M3. The intact acetamide group in M3 made it easier to separate under acidic conditions (pH 4.0). To facilitate the separation without using stringent conditions in sample preparation and/or separation, we used a derivatization method that is often used in amino acid analysis. NHS-Biotin is one of the most commonly used protein-labeling re-agents that specifically modifies primary amine groups. The aromatic amine group in M2 could serve as a perfect substrate for similar modification. The addition of the large biotin moiety not only increased the column retention time of the molecule, but also increased the ionization efficiency during MS analysis. The mild reaction condition (room temperature, neutral pH) excluded possible hydrolysis due to artificial effects (i.e., strong acidic condition). As shown in Fig. 6B, both M2 and M2-OH were modified and separated successfully from the void peak. This design might provide a unique strategy for analyzing highly hydrophilic metabolites that contain primary amine groups.
Besides the differences observed in the phase I metabolism of S4, potential species differences in the phase II metabolizing enzymes might also play a role in the in vivo metabolism of S4. As discussed above, the aromatic amine group in M1 and M2 is susceptible to modification; it might also be acetylated again by a phase II enzyme, like N-acetyltransferase, and be converted to S4. The interaction between M1 and N-acetyltransferase, and potential species differences in S4 metabolism, was studied in more detail in separate studies (Gao et al., 2006).
For the other hydrolysis product, M3, although it was easier to separate under acidic conditions (pH 4.0), the acidic condition is generally not recommended for MS analysis with negative ion mode. However, our previous study (Wu et al., 2004) has shown that lower concentrations of acetic acid can actually improve the MS response of S4 under negative ion mode. Similarly, the presence of 0.2% acetic acid during MS analysis did not suppress the MS response of M3 in this study, which further confirmed that mild acidic conditions could be used under negative ion mode as well.
In summary, in vitro metabolism studies showed that S4 was mainly metabolized by deacetylation of the B-ring acetamide group, hydrolysis of the amide bond, and reduction of the A-ring nitro group, with deacetylation being the major pathway in all three species tested, whereas amide bond hydrolysis was more significant in rat than in human and dog.
Acknowledgments
These studies were supported by grants from GTx Inc. (Memphis, TN) and the National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK59800) to J.T.D. and D.D.M. J.T.D. and D.D.M. are employees of GTx, Inc.
ABBREVIATIONS
- SARM
selective androgen receptor modulator
- AR
androgen receptor
- S4
S-3-(4-acetylamino-phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamide
- HPLC
high-performance liquid chromatography
- P450
cytochrome P450
- HLM
human liver microsome
- ESI
electrospray ionization
- MS
mass spectrometry
- MS2
tandem mass spectrometry
- DHT
dihydrotestosterone
References
- Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc Natl Acad Sci USA. 2005;102:6201–6206. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle GW, McKillop D, Phillips PJ, Harding JR, Pickford R, McCormick AD. Metabolism of Casodex in laboratory animals. Xenobiotica. 1993;23:781–798. doi: 10.3109/00498259309166784. [DOI] [PubMed] [Google Scholar]
- Casarett LJ, Klaassen CD, Amdur MO, Doull J. Casarett and Doull's Toxicology: The Basic Science of Poisons. McGraw-Hill Health Professions Division; New York: 1996. [Google Scholar]
- Cockshott ID. Bicalutamide: clinical pharmacokinetics and metabolism. Clin Pharmacokinet. 2004;43:855–878. doi: 10.2165/00003088-200443130-00003. [DOI] [PubMed] [Google Scholar]
- Dalton JT, Mukherjee A, Zhu Z, Kirkovsky L, Miller DD. Discovery of nonsteroidal androgens. Biochem Biophys Res Commun. 1998;244:1–4. doi: 10.1006/bbrc.1998.8209. [DOI] [PubMed] [Google Scholar]
- Gao W, Johnston JS, Miller DD, Dalton JT. Interspecies differences in pharmacokinetics and metabolism of S-3-(4-acetylamino-phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl-propionamide: the role of N-acetyltransferase. Drug Metab Dispos. 2006;34:254–260. doi: 10.1124/dmd.105.007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Kearbey JD, Nair VA, Chung K, Parlow AF, Miller DD, Dalton JT. Comparison of the pharmacological effects of a novel selective androgen receptor modulator (SARM), the 5{alpha}-reductase inhibitor finasteride and the antiandrogen hydroxyflutamide in intact rats: new approach for benign prostate hyperplasia (BPH) Endocrinology. 2004;145:5420–5428. doi: 10.1210/en.2004-0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Yin D, Perera M, Kirkovsky L, Stourman N, Li W, Dalton JT, Miller DD. Novel nonsteroidal ligands with high binding affinity and potent functional activity for the androgen receptor. Eur J Med Chem. 2002;37:619–634. doi: 10.1016/s0223-5234(02)01335-1. [DOI] [PubMed] [Google Scholar]
- Ji HY, Lee SS, Yoo SE, Kim H, Lee DH, Lim H, Lee HS. In vitro metabolism of a new neuroprotective agent, KR-31543 in the human liver microsomes: identification of human cytochrome P450. Arch Pharm Res. 2004;27:239–245. doi: 10.1007/BF02980112. [DOI] [PubMed] [Google Scholar]
- Katchen B, Buxbaum S. Disposition of a new, nonsteroid, antiandrogen, alpha,alpha,alpha-trifluoro-2-methyl-4′-nitro-m-propionotoluidide (Flutamide), in men following a single oral 200 mg dose. J Clin Endocrinol Metab. 1975;41:373–379. doi: 10.1210/jcem-41-2-373. [DOI] [PubMed] [Google Scholar]
- Kearbey JD, Wu D, Gao W, Miller DD, Dalton JT. Pharmacokinetics of S-3-(4-acetylamino-phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamide in rats, a nonsteroidal selective androgen receptor modulator. Xenobiotica. 2004;34:273–280. doi: 10.1080/0049825041008962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKillop D, Boyle GW, Cockshott ID, Jones DC, Phillips PJ, Yates RA. Metabolism and enantioselective pharmacokinetics of Casodex in man. Xenobiotica. 1993;23:1241–1253. doi: 10.3109/00498259309059435. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Kirkovsky L, Yao XT, Yates RC, Miller DD, Dalton JT. Enantioselective binding of Casodex to the androgen receptor. Xenobiotica. 1996;26:117–122. doi: 10.3109/00498259609046693. [DOI] [PubMed] [Google Scholar]
- Negro-Vilar A. Selective androgen receptor modulators (SARMs): a novel approach to androgen therapy for the new millennium. J Clin Endocrinol Metab. 1999;84:3459–3462. doi: 10.1210/jcem.84.10.6122. [DOI] [PubMed] [Google Scholar]
- Wu Z, Gao W, Phelps MA, Wu D, Miller DD, Dalton JT. Favorable effects of weak acids on negative-ion electrospray ionization mass spectrometry. Anal Chem. 2004;76:839–847. doi: 10.1021/ac0351670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin D, Gao W, Kearbey JD, Xu H, Chung K, He Y, Marhefka CA, Veverka KA, Miller DD, Dalton JT. Pharmacodynamics of selective androgen receptor modulators. J Pharmacol Exp Ther. 2003a;304:1334–1340. doi: 10.1124/jpet.102.040840. [DOI] [PubMed] [Google Scholar]
- Yin D, He Y, Perera MA, Hong SS, Marhefka C, Stourman N, Kirkovsky L, Miller DD, Dalton JT. Key structural features of nonsteroidal ligands for binding and activation of the androgen receptor. Mol Pharmacol. 2003b;63:211–223. doi: 10.1124/mol.63.1.211. [DOI] [PubMed] [Google Scholar]
- Yin D, Xu H, He Y, Kirkovsky LI, Miller DD, Dalton JT. Pharmacology, pharmacokinetics and metabolism of acetothiolutamide, a novel nonsteroidal agonist for the androgen receptor. J Pharmacol Exp Ther. 2003c;304:1323–1333. doi: 10.1124/jpet.102.040832. [DOI] [PubMed] [Google Scholar]