

Abstract
Mutations in the copper-transporter ATP7A lead to severe neurodegeneration in the mottled brindled hemizygous male (MoBr/y) mouse and human patients with Menkes disease. Our earlier studies demonstrated cell-type and stage-specific changes in ATP7A protein expression during postnatal neurodevelopment. Here we examined copper and cuproenzyme levels in MoBr/y mice to search for compensatory responses. While all MoBr/y neocortical subcellular fractions had decreased copper levels, the greatest decrease (8-fold) was observed in cytosol. Immunostaining for ATP7A revealed decreased levels in MoBr/y hippocampal pyramidal and cerebellar Purkinje neurons. In contrast, an upregulation of ATP7A protein occurred in MoBr/y endothelial cells, perhaps to compensate for a lack of copper in the neuropil. MoBr/y astrocytes and microglia increased their physical association with the blood-brain barrier. No alterations in ATP7A levels were observed in ependymal cells, arguing for specificity in the alteration observed at the blood-brain barrier. The decreased expression of ATP7A protein in MoBr/y Purkinje cells was associated with impaired synaptogenesis and dramatic cytoskeletal dysfunction. Immunoblotting failed to reveal any compensatory increase in levels of ATP7B. While total levels of several cuproenzymes (peptide amidating monooxygenase, SOD1, SOD3) were unaltered in the MoBr/y brain, levels of amidated cholecystokinin (CCK8) and amidated pituitary adenylate cyclase-activating polypeptide (PACAP38) were reduced in a tissue-specific fashion. The compensatory changes observed in the neurovascular unit provide insight into the success of copper injections within a defined neurodevelopmental period.
INTRODUCTION
Dietary and genetic copper deficiency indicates that copper is required for normal development and function of the nervous system (Prohaska, 2000; Shim and Harris, 2003). Copper is a potent reductant essential for the catalytic activity of cuproenzymes such as peptidylglycine α-amidating monooxygenase, dopamine β-monooxygenase, copper-zinc superoxide dismutase and ceruloplasmin. However, reduced copper (Cu+) reacts with hydrogen peroxide (H2O2) to generate hydroxyl radical (OH-), a deleterious reactive oxygen species that damages DNA, lipid and protein. Impaired copper homeostasis is implicated in the pathogenesis of numerous neurodegenerative diseases including Alzheimer disease (Bush, 2003), familial amyotrophic lateral sclerosis (Selverstone et al., 2005) and prion disease (Brown, 2004).
Copper transporters and chaperones have evolved to minimize free intracellular copper while ensuring delivery to specific subcellular destinations. Mice with mutations in these proteins show substantial developmental neuropathology. Mice lacking CTR1, a high-affinity plasma membrane copper transporter, do not survive beyond E8.5, exhibiting aberrant neuroepithelial layering and impaired neural tube closure (Lee et al., 2001; Kuo et al., 2001). Genetic elimination of ATOX-1, a cytosolic copper chaperone, results in postnatal lethality due to connective tissue and nervous system dysfunction (Hamza et al., 2001). ATOX-1 delivers copper to ATP7A, a P-type ATPase localized to the trans-Golgi network at steady state and responsible for copper delivery to secretory pathway curproenzymes (Francis et al., 1998; La Fontaine et al., 1998; Petris et al., 2000). In neurons, ATP7A is found in axons and dendrites and its intracellular trafficking is responsive to copper and N-methyl-D-aspartate receptor activation (Schlief et al., 2005).
Mottled mouse strains with different mutations in ATP7A display diverse phenotypes, replicating the varied clinical severity of Menkes disease. The mottled brindled (MoBr/y) mouse provides a model of classic Menkes disease, with postnatal lethality and neurological deficits. MoBr/y ATP7A has only limited ability to transport copper but is expressed at close to normal levels (La Fontaine et al., 1999; Steveson et al., 2003). Postnatal copper injections of MoBr/y mice and human patients attenuate the neurological deficiencies and can sometimes extend survival into adulthood. It is assumed that the residual activity of mutant ATP7A transports copper to critical brain targets.
We recently described cell-type specific, developmentally-regulated changes in the expression and localization of ATP7A in neurons and glia (Niciu et al., 2006). In the current study, we examined ATP7A expression in the CNS of MoBr/y mice to search for compensatory alterations that may occur in response to copper deficiency. Several studies have reported impaired cuproenzyme activity in MoBr/y brain (Kunz et al., 1999; Steveson et al., 2003), but deficiencies in copper content and cuproenzyme activities in different subcellular compartments have not been compared. An analysis of compensatory alterations will increase our understanding of the efficacy of parenteral copper injections and assist in the design of new therapies for Menkes disease.
MATERIALS AND METHODS
Animals
A colony of mottled-brindled (MoBr) mice was maintained by mating adult female heterozygous mottled brindled (C57BL/6J MoBr/+) mice with wild-type male C57BL/6J mice (Jackson Laboratory; Bar Harbor, ME). The mice were bred and maintained on a 12 h light, 12 h dark cycle in the animal care facility at the University of Connecticut Health Center (UCHC). All animal protocols were approved by the UCHC Animal Care and Use Committee prior to experimentation. In this study, both postnatal male (+/y) and female (+/+) mice were used as controls depending on animal availability. The sex of the pups was determined by the greater anogenital distance in males relative to females. Early postnatal animals were killed by decapitation after anesthetizing on ice. A dissecting microscope was utilized to facilitate separation of brain areas.
Immunoblotting
Brain areas (neocortex, hippocampus, olfactory bulb, cerebellum and hypothalamus) and liver from postnatal wild-type and MoBr/y mice were homogenized in denaturing buffer with protease inhibitors, protein measured, and equal aliquots of protein subjected to Western blot analysis as described (Niciu et al., 2006). Antisera directed against the indicated proteins were incubated with membranes: 1:1000 (in Tween-20 and Tris-buffered saline containing 0.5% nonfat dry milk), rabbit polyclonal antiserum CT77, an anti-peptide antibody raised against the intracellular C-terminus of ATP7A (Steveson et al., 2003); 1:5000, neuronal class III β-tubulin mouse monoclonal antibody TuJ1 (Covance Research Products, Berkeley, CA, USA); 1:500, ATP7B rabbit polyclonal antiserum NB 100-360 (Novus Biologicals, Littleton, CO) overnight at 4°C; 1:5000, type I Cu-Zn superoxide dismutase (SOD1) rabbit polyclonal antiserum (Stressgen, Victoria, British Columbia, Canada) for 2 h at RT; 1:500, type III Cu-Zn superoxide dismutase (SOD3) rabbit polyclonal antiserum (Stressgen) overnight at 4°C.
ATP7B signal specificity was verified by preincubating a 1:100 dilution of ATP7B antiserum with 0.1 mg/ml immunizing peptide or an irrelevant peptide (MNK2, part of the intracellular N-terminus of ATP7A) for 30 min at RT before diluting 5-fold. These mixtures were then incubated overnight at 4°C with PVDF membranes containing purified ATP7B from baculovirus-infected Sf9 cells (kindly provided by Dr. Svetlana Lutsenko, Oregon Health Science University, Portland, OR) as a positive control. Membranes were also exposed to a 1:500 dilution of preimmune ATP7B antiserum as an additional specificity control. After rinsing, the membranes were visualized with HRP-tagged secondary antibodies as described (Niciu et al., 2006).
Subcellular Fractionation by Differential Centrifugation
P4 wild-type (+/+ or +/y) and MoBr/y neocortices were homogenized in 10 volumes (wt/vol) 0.32 M sucrose, 10 mM HEPES buffer-NaOH, pH 7.0 containing protease inhibitors using five strokes with a motorized Potter-Elvehjem homogenizer. Homogenates were fractionated by differential centrifugation. Briefly, homogenates were centrifuged at 1,000 × gmax for 5 min to remove cell debris and nuclei; the pellet (P1) was resuspended in 500 μl homogenization buffer containing protease inhibitors and saved for analysis. The supernatant (S1) was centrifuged at 4,000 × gmax for 15 min, and the resulting pellet (P2) was resuspended in 250 μl as above. This supernatant (S2) was then centrifuged at 37,000 × gmax for 15 min, and the resulting pellet (P3) was resuspended in 250 μl. Finally, this supernatant (S3) was centrifuged at ~413,000 × gmax for 15 min to separate a microsomal membrane pellet (P4) from cytosolic proteins (S4); P4 was resuspended in 250 μl.
Characterization of Subcellular Fractions
The protein concentrations of the initial homogenate, resuspended pellets (P1-P4) and cytosolic fraction were determined. For copper measurements, fractions were incubated with 0.5% Triton X-100 for 2 h at RT and centrifuged at 1,000 × gmax for 5 min to eliminate debris. The copper content in each subcellular fraction was determined in triplicate via atomic absorption spectroscopy using an AA600 furnace spectrometer (Perkin-Elmer A Analyst 600, Wellesley, MA) and normalized to total protein and to neocortical wet weight. Protein (20 μg) from each fraction was subjected to Western blot analysis as described above. The membranes were probed with the following antisera at the indicated concentrations in TTBS and 0.5% milk: 1:1000, CT77; 1:5000, type 1 Cu-Zn superoxide dismutase (SOD1) rabbit polyclonal antiserum (Stressgen); 1:1000, Rho-GDI mouse monoclonal antibody (BD Transduction Laboratories). PHM assays were performed as described (Mair et al., 2004).
Radioimmunoassays (RIAs) of Amidated Neuropeptides
P8 and P12 wild-type and MoBr/y neocortices, hippocampi, olfactory bulbs, cerebella and hypothalami were extracted in ice-cold 5 N acetic acid with 30 μg/ml PMSF, clarified by centrifugation at 4°C for 20 min at 16,000 × gmax, lyophilized overnight and reconstituted in 20 mM Na N-Tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid (TES), 10 mM mannitol, 1% Triton X-100, pH 7.4 and 30 μg/ml PMSF. Samples were again clarified by centrifugation at 4°C for 20 min at 16,000 × gmax before analysis of immunoreactive CCK-8-NH2 and PACAP-38-NH2. Polyclonal C-terminal amidated CCK-specific antibody [Ab 8007; kindly provided by Dr. Jens Rehfeld (Department of Clinical Biochemistry, Rigshospitalet, University of Copenhagen, Denmark)] was used at a working dilution of 1:400,000 (midpoint typically 9 pg); synthetic CCK-8 (CCK 26-33-NH2) was used to generate a standard curve with 13,000 dpm [125I]-CCK-8 (Peninsula Laboratories, Belmont, CA). Rabbit polyclonal C-terminal amidated PACAP38-specific antibody (T-4473; Peninsula) was used at 1:80,000 (midpoint typically 12 pg); synthetic PACAP(31-38)-NH2 was used to generate a standard curve with 4,000 dpm [125I]-PACAP(31-38)-NH2 (Peninsula). BCA assays were utilized to normalize amidated peptide content to total protein.
Immunohistochemistry
Wild-type and MoBr/y mice were anesthetized and perfusion-fixed. Brain sections (15 μm) were cut sagittally on a cryostat and stored at -80°C until use. Double label immunofluorescence analysis with primary antisera raised in different species was performed. Sections were blocked in 1% bovine serum albumin (Sigma, minimum 98% as determined electrophoretically), 10% normal donkey serum (Jackson Immunoresearch, West Grove, PA), 10% normal goat serum, and 0.025% Triton X-100 in 1X PBS, pH 7.4 for 1 h at RT. Sections were then incubated in the following rabbit polyclonal antisera overnight at 4°C: 1:1000, CT77; 1:300, anti-β-IV spectrin (courteous gift of Dr. Matthew Rasband, UCHC). Slides were co-stained with the indicated dilution of the following monoclonal antisera: 1:500, anti-glial fibrillary acidic protein (GFAP) mouse monoclonal antibody MAB360 (Chemicon, Temecula, CA); 1:500, anti-CD11b rat monoclonal antibody MCA711G (Serotec, Raleigh, NC); 1:500, anti-MAP2 mouse monoclonal antibody AP-20 (Sigma); 1:200, anti-bassoon mouse monoclonal antibody SAP7F407 (Stressgen); 1:250, anti-PSD-95 mouse monoclonal antibody K43/28 (Upstate, Lake Placid, NY); 1:200, anti-GM130 mouse monoclonal antibody (BD Transduction Laboratories, San Jose, CA); 1:100, anti-neurofilament 200 (NF200) mouse monoclonal antibody N52 (Sigma). After rinsing three times in 1X PBS, slides were incubated in a 1:1000 dilution of Cy3-conjugated donkey anti-rabbit IgG and a 1:500 dilution of FITC-conjugated goat anti-rat or donkey anti-mouse IgG (Jackson ImmunoResearch). Slides were rinsed and counterstained by incubation in 1 μM TO-PRO-3 (Invitrogen-Molecular Probes, Eugene, OR) in 1X PBS for 30 min at RT. After a final rinse, slides were then mounted in ProLong® Gold antifade reagent (Molecular Probes) and visualized with a LSM510 confocal microscope (Center for Cell Analysis and Monitoring at UCHC; Carl Zeiss, Thornwood, NY).
For analysis of brain endothelium, blocked sections were incubated in a 1:200 dilution of biotinylated Ricinis communis agglutinin-1 (RCA-1) (Vector Laboratories, Burlingame, CA). After rinsing three times, the sections were incubated in 1:200, FITC-conjugated avidin D (Vector Laboratories) in 1X PBS and processed as above. Brain endothelium was alternately identified with the following primary antisera: 1:100, rat monoclonal antibody MEC 13.3 to mouse CD31/PECAM-1 (BD Biosciences Pharmingen, San Jose, CA), which labels all endothelial cells regardless of vessel diameter; 1:100, FITC-conjugated mouse monoclonal antibody 1A4 to alpha-smooth muscle actin (Sigma-Aldrich, St. Louis, MO), which labels smooth muscle surrounding large-diameter blood vessels and pericytes. Antibody specificity was determined as described previously (Niciu et al., 2006).
RESULTS
ATP7A and ATP7B protein levels in MoBr/y brain
Deficits in ATP7A function in MoBr/y mice might be compensated by increased expression of the mutant protein or a closely related P-type ATPase, ATP7B. The levels of mutant ATP7A protein in MoBr/y brain were first investigated by immunoblotting. Mutant ATP7A protein was present in each brain region examined at levels equal to or slightly higher than those in wild-type mice (Fig. 1A). No significant differences in expression of the neuronal marker βIII-tubulin were observed across brain areas (Fig. 1A).
Figure 1.
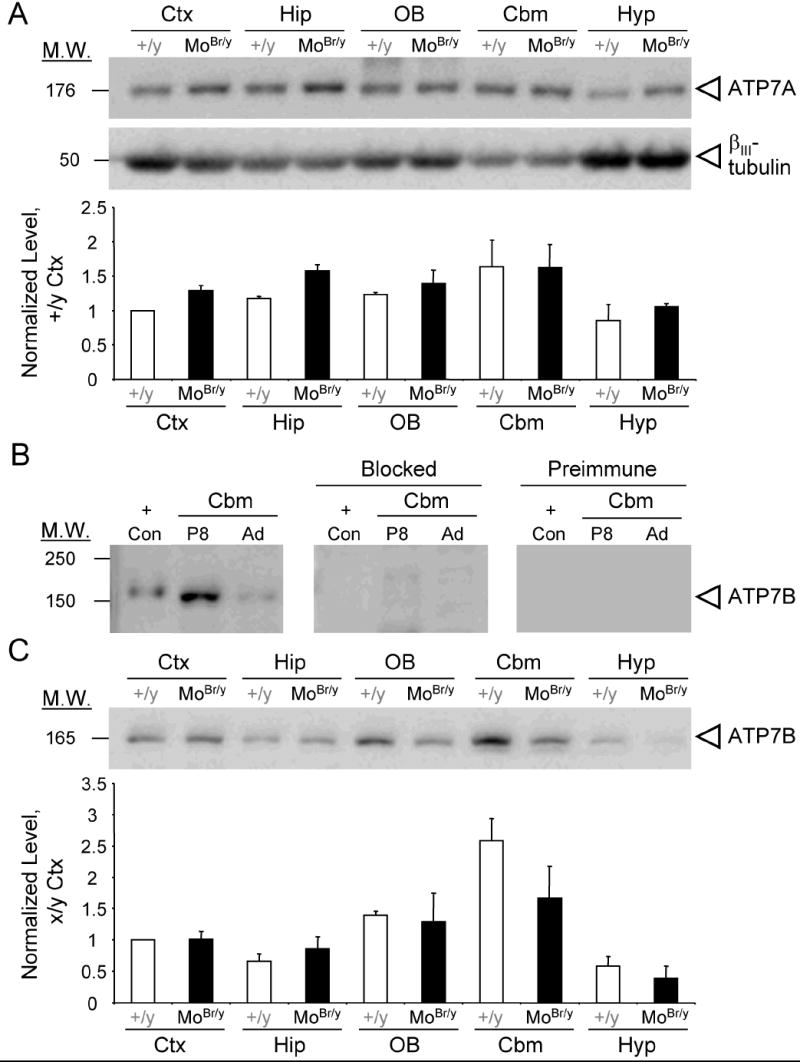
ATP7A and ATP7B levels in MoBr/y brain. A. SDS-solubilized protein (20 μg) prepared from the indicated tissues from P8 wild-type male and MoBr/y mice was fractionated by SDS-PAGE: neocortex (Ctx), hippocampus (Hip), olfactory bulb (OB), cerebellum (Cbm) and hypothalamus (Hyp). The membrane was probed with antisera directed against ATP7A and βIII-tubulin. Quantification of ATP7A signal intensity is shown. The error bars indicate the range from duplicate experiments, each was normalized to P8 wild-type neocortex. B. Validation of ATP7B antiserum via immunoblotting. Extracts of baculovirus-infected Sf9 cells expressing purified ATP7B (+Con) and homogenates (40 μg total protein) from P8 and adult cerebellum were fractionated and the membrane was incubated with a 1:500 dilution of ATP7B antiserum, blocked antiserum or preimmune serum. C. ATP7B immunoblot of SDS-solubilized homogenates from P8 wild-type and MoBr/y brain. Quantification of ATP7B signal intensity is shown. The error bars indicate the range from duplicate experiments normalized to wild-type neocortex.
ATP7B, the homologous copper-transporter mutated in Wilson disease, has enzymatic properties similar to those of ATP7A and might substitute for the mutant enzyme in MoBr/y brain. To assess this possibility, purified ATP7B was analyzed along with SDS lysates from P8 and adult cerebellum, which are known sites of ATP7B expression (Barnes et al., 2005). A 165 kDa band, slightly smaller than the purified ATP7B control (170 kDa), was more prevalent in P8 than adult cerebellum (Fig. 1B). Immunoreactivity was eliminated by preadsorption of the antiserum with the immunizing peptide or using preimmune serum (Fig. 1B). The difference in molecular mass between ATP7B expressed in insect cells and endogenous ATP7B may reflect different posttranslational modifications.
Extracts prepared from P8 wild-type and MoBr/y tissues were probed for ATP7B levels, with highest expression in the cerebellum and lowest in the hypothalamus (Fig. 1C). No compensatory increase in ATP7B levels was observed in MoBr/y tissues via immunoblotting (Fig. 1C). Elevated levels of ATP7B mRNA in the postnatal cerebella of wild-type and MoBr/y mice were verified using two independent ATP7B-specific primer pairs for RT-PCR (data not shown). The consistency between ATP7B mRNA and protein levels confirms that ATP7B is expressed at high levels in the postnatal cerebellum (Barnes et al., 2005).
MoBr/y neocortical cytosol is more copper-depleted than particulate fractions
To further characterize the effects of mutant ATP7A expression in the MoBr/y brain, crude subcellular fractions were prepared. The P4 neocortex was analyzed since expression of wild-type ATP7A peaks at this time (Niciu et al., 2006). A decrease in the overall copper content of MoBr/y neocortex was expected based on previous studies (Camakaris et al., 1979). Each MoBr/y subcellular fraction displayed decreased copper content whether normalized to total protein (Fig. 2A) or tissue wet weight (data not shown). The copper content (μg/g protein) of MoBr/y cytosol was reduced 8-fold compared to wild-type cytosol. Although a deficit in ATP7A activity might be expected to have a greater effect on luminal copper than on cytosolic copper, the copper content of the particulate fractions was reduced only about 3-fold (Fig. 2A). Copper transport in the MoBr/y brain likely occurs from the residual activity of mutant ATP7A and/or the activity of ATP7B.
Figure 2.
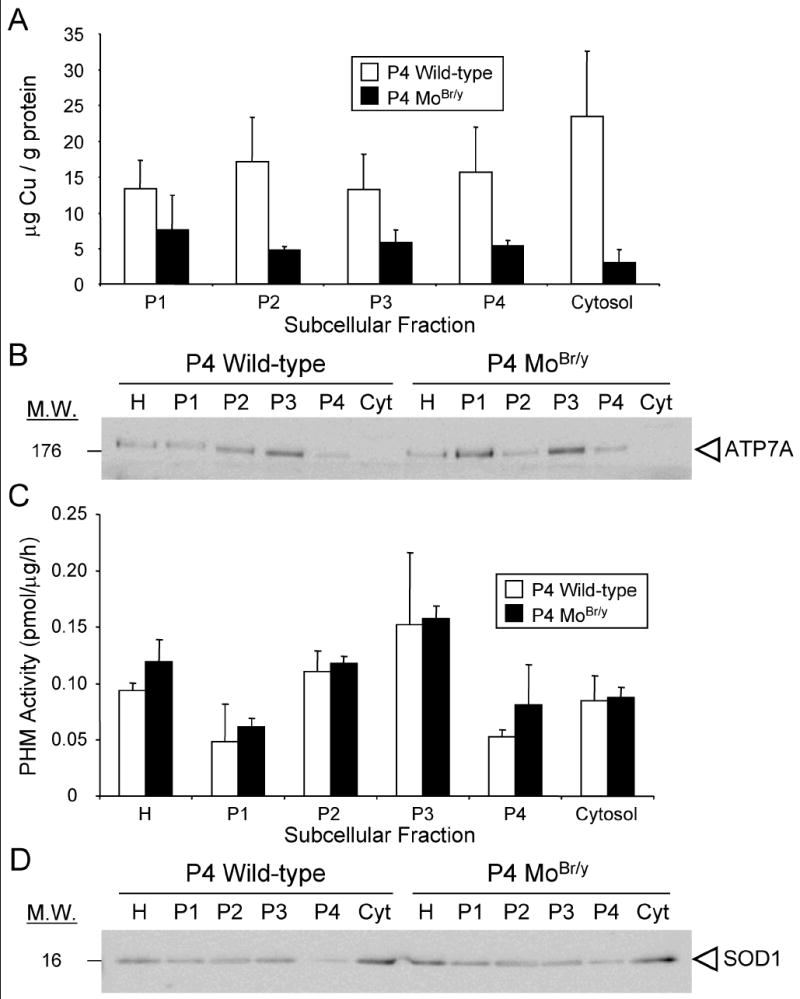
Copper content of MoBr/y cytosol is reduced more than particulate fractions. Pools of P4 wild-type and MoBr/y neocortex were fractionated by differential centrifugation as described in Materials and Methods. Copper concentrations were measured by atomic absorption spectroscopy. Two independent sets of samples were analyzed. A. Copper content of subcellular fractions normalized to total protein. P1, the initial low speed pellet, contains cell debris and a complex mixture of organelles. B. ATP7A immunoblot (20 μg protein) of homogenate and subcellular fractions. C. PHM activity of subcellular fractions normalized to total protein. D. SOD1 immunoblot (20 μg protein) of homogenate and subcellular fractions. Blots shown are representative of at least two experiments.
The subcellular distribution of ATP7A and two cuproenzymes, peptidylglycine α-amidating monooxygenase (PAM) and type 1 copper-zinc superoxide dismutase (SOD1) was next investigated. Analyzing equal amounts of protein from each subcellular fraction, wild-type and MoBr/y ATP7A were most concentrated in P3, a fraction enriched in trans-Golgi network (TGN) and secretory granules (Fig. 2B). The subcellular distribution of PAM, a representative secretory pathway cuproenzyme, was determined using an enzyme assay for its copper-dependent monooxygenase domain. PAM post-translationally modifies ~50% of peptide hormones with the addition of a C-terminal amide group that is essential for full peptide bioactivity. PAM null mice die midway through gestation (Czyzyk et al., 2005). When assayed in vitro in the presence of optimal amounts of exogenous copper, the monooxygenase activity in wild-type and MoBr/y neocortical fractions was indistinguishable, and the specific activity of PHM was highest in P3 as anticipated (Fig. 2C).
Finally, SOD1, the only identified primarily cytosolic cuproenzyme, was examined (Okado-Matsumoto and Fridovich, 2001). As expected, in both wild-type and MoBr/y samples, SOD1 was most enriched in the cytosol (Fig. 2D) as was another primarily cytosolic protein, Rho-guanine nucleotide dissociation inhibitor (Rho-GDI) (data not shown). The total levels of SOD1 and SOD3, a differentially glycosylated secreted cuproenzyme encoded by a separate gene, in brain and other tissues were also examined for compensatory changes given the reduction in cytosolic copper concentration relative to particulate fractions. No difference in the expression of either isoform was observed when comparing wild-type and MoBr/y extracts (Suppl. Fig. 1).
Amidation is differentially impaired in a peptide and area-specific fashion in MoBr/y brain
Neither in vitro PHM activity (Fig. 2C) nor in vivo PAM protein levels (Steveson et al., 2003) display compensatory alterations in MoBr/y mice. However, copper does not bind tightly to the catalytic core of PHM and enzymatic assays performed in the absence of exogenous copper reveal almost no activity (data not shown). CCK8 and PACAP38 were selected for study due to the essential role that amidation plays in determining their full biological activity, their abundance throughout the brain, and their roles in the proliferation, protection, differentiation and migration of neuronal subpopulations known to degenerate in Menkes disease patients and mouse models of the disease (Vaudry et al., 1999; Giacobini et al., 2004).
As reported previously for neocortex, CCK8 amidation is also impaired in the MoBr/y hippocampus, olfactory bulb and hypothalamus (Fig. 3A) (Steveson et al., 2003). The decrement in CCK8 amidation is region-specific; at P8, CCK8 amidation declined 10-fold in the olfactory bulb but only 2-fold in the hypothalamus (Fig. 3A). Impairment of PACAP38 amidation is also region-specific; MoBr/y neocortex and hypothalamus display significant decreases in PACAP38 amidation at P8 and P12, while no difference in PACAP38 amidation was observed in the MoBr/y cerebellum (Fig. 3B). Interestingly, peptide amidation in MoBr/y brain is impaired to a greater extent than anticipated based upon the observed two-fold decrease in copper content of the particulate fractions (Fig. 2A).
Figure 3.
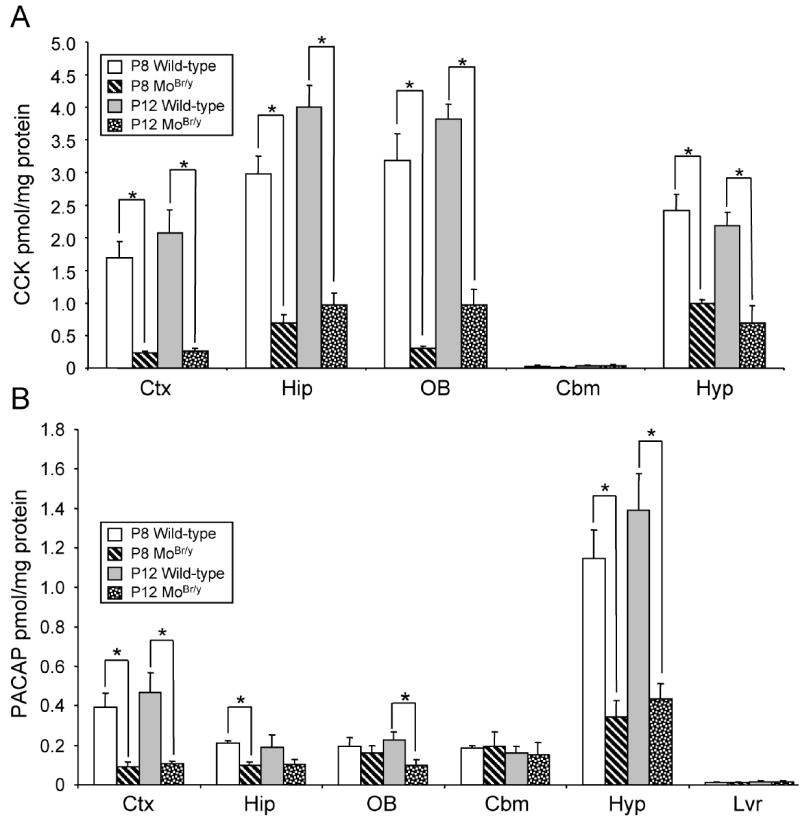
Peptide amidation is differentially impaired in MoBr/y brain. A. CCK8-NH2 radioimmunoassay from P8 and P12 wild-type and MoBr/y neocortex (Ctx), hippocampus (Hip), olfactory bulb (OB), cerebellum (Cbm) and hypothalamus (Hyp). B. PACAP38-NH2 radioimmunoassay from P8 and P12 wild-type and MoBr/y brain and liver (Lvr, negative control). Peptide levels were normalized to total protein. Each radioimmunoassay was performed in triplicate with pooled samples from three individual animals. *, p < 0.05 as determined by two-tailed student’s t test.
Mutant ATP7A displays subtle compensatory alterations in MoBr/y brain
Immunohistochemical analysis was used to search for compensatory changes that might explain the differential sensitivity of peptide amidation to reduced ATP7A function. ATP7A expression was examined in postnatal wild-type and MoBr/y neocortex, hippocampus and cerebellum; these areas are known to degenerate in Menkes disease patients and mouse models of this disorder. In P8 wild-type hippocampus, ATP7A protein is most abundantly expressed in large-diameter CA1/2 pyramidal neurons and scattered interneurons in the stratum oriens, stratum radiatum and polymorph cells of the dentate gyrus (Fig. 4A, wild-type) (Schlief et al., 2005; Niciu et al., 2006). In MoBr/y hippocampus, a striking decrease in ATP7A immunoreactivity in pyramidal neurons is observed (Fig. 4A, MoBr/y).
Figure 4.
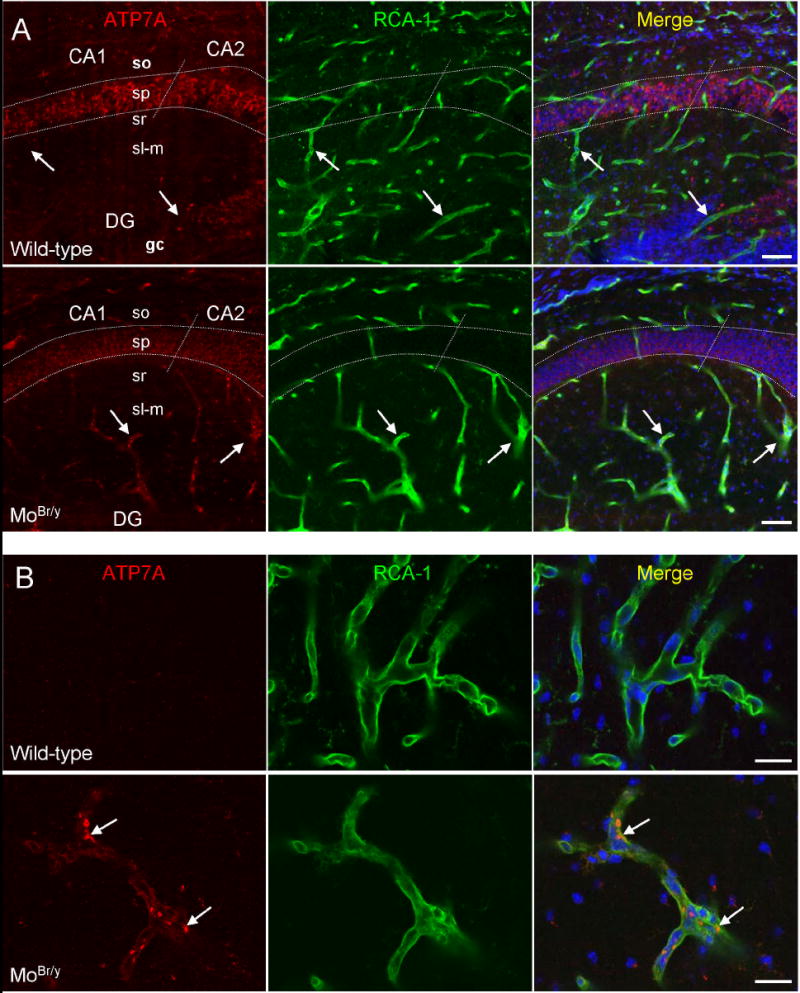
ATP7A expression in MoBr/y hippocampus is decreased in pyramidal neurons and increased in adjacent microvessels. Sagittal sections from the CA1/2 region of P8 wild-type and MoBr/y hippocampus were stained simultaneously for ATP7A (red), an endothelial cell marker [Ricinus communis agglutinin-1 (RCA-1); green] and DNA (TO-PRO-3; blue) and examined via laser scanning confocal microscopy. A. Low power images. In wild-type hippocampus, staining for ATP7A is apparent in CA1 and CA2 neuronal cell bodies with no detectable immunoreactivity in the RCA-1 positive microvasculature (arrows). In MoBr/y hippocampus, ATP7A immunoreactivity is decreased in the pyramidal cell layer but increased in the surrounding capillary endothelium (vessels ≤ 10 μm in diameter) (arrows). The borders of stratum pyramidale (sp) are outlined with white dotted lines, and the demarcation between CA1 and CA2 is indicated with diagonal intersections. B. High power images. Little ATP7A immunoreactivity is apparent in brain microvessels of capillary diameter from wild-type hippocampus. ATP7A is detected in a perinuclear location (arrows) in MoBr/y microvessels; DG, dentate gyrus; gc, granule cell layer of the dentate gyrus; sl-m, stratum lucidum-moleculare; so, stratum oriens; sr, stratum radiatum; Scale bars: A, 50 μm; B, 20 μm.
Consistent with the fact that Western blot analysis did not reveal a decrease in total ATP7A levels in the MoBr/y hippocampus, increased expression was observed outside of the pyramidal cell layer in structures exhibiting a vascular profile (Fig. 4A,B, MoBr/y). Biotinylated Ricinis communis agglutinin-1 (RCA-1) was used to identify cerebral microvessels. In the MoBr/y brain, ATP7A expression increases dramatically in endothelial cells lining small diameter microvessels having the appearance of capillaries (≤10 μm diameter) (Fig. 4A,B, MoBr/y). This result was confirmed by colocalization of ATP7A and PECAM-1/CD31, a pan-endothelial cell marker (data not shown). The mutant ATP7A protein is largely localized to the perinuclear, TGN region of the endothelial cells (Fig. 4B). In the MoBr/y hippocampus, the decrease in ATP7A immunoreactivity in large-diameter neurons, together with a corresponding increase in capillary endothelial cells, likely accounts for similiar levels observed via immunoblotting (Fig. 1A).
Unlike small-diameter vessel endothelium, wild-type and mutant ATP7A are expressed at equivalent levels in endothelial cells of relatively larger diameter cerebral vessels, as indicated by co-localization analyses with the smooth muscle/pericyte marker protein, α-smooth muscle actin (Fig. 5A). Additionally, mutant ATP7A is abundantly expressed, but displays no compensatory alterations, in ependymal cells/choroid plexus (Fig. 5B). Thus, mutant ATP7A levels are specifically upregulated in endothelial cells lining microvessels, presumably capillaries, which are the recognized site of the blood-brain barrier. However, no compensatory alterations in ATP7A expression are observed at other contact interfaces like the blood-cerebrospinal fluid (CSF) barrier.
Figure 5.
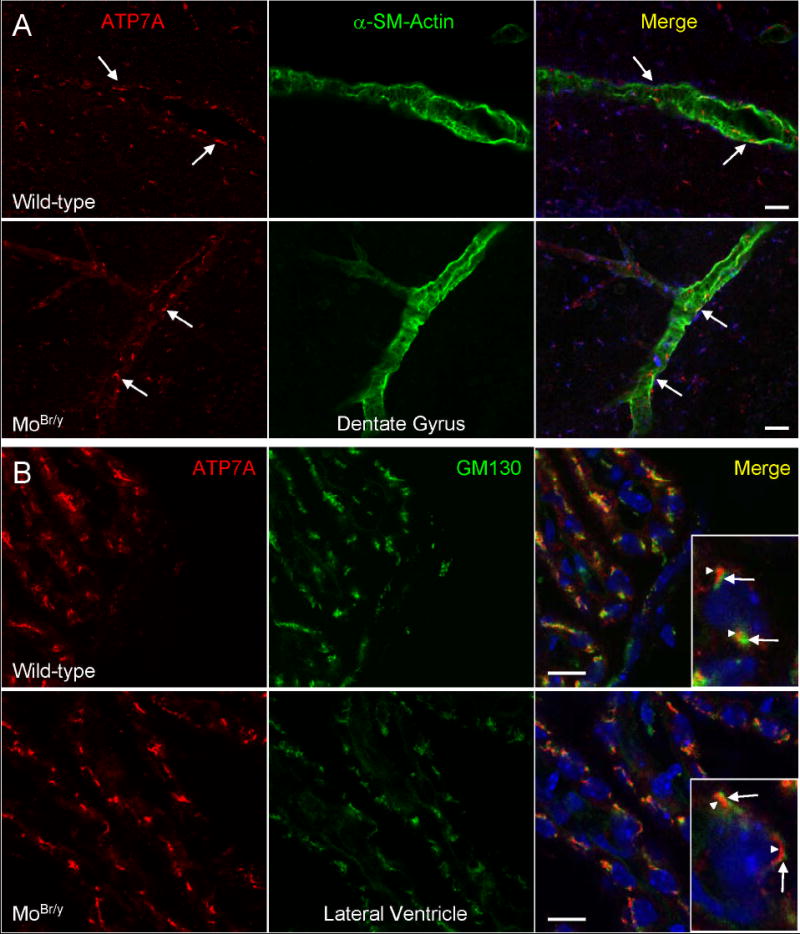
ATP7A expression is unaltered in larger diameter blood vessels and ependymal cells/choroid plexus. A. Sagittal sections from the dentate gyrus of P8 wild-type and MoBr/y hippocampus were stained simultaneously for ATP7A (red), the vascular smooth muscle/pericyte marker α-smooth muscle actin (green) and DNA (blue). ATP7A is present at equal levels in wild-type and MoBr/y endothelial cells lining larger diameter vessels. B. Sagittal sections of P8 wild-type and MoBr/y lateral ventricle were stained for ATP7A (red), the cis-to-medial Golgi marker GM130 (green) and DNA (blue). ATP7A is expressed at highest levels in wild-type and MoBr/y ependyma and segregates to a perinuclear compartment (arrowheads in insets) distinct from GM130 (arrows in insets); Scale bars: A, 20 μm; B, 10 μm.
Increased neurovascular unit activity in the MoBr/y cerebrovasculature
Co-localization analyses were performed with glial cell markers to investigate further alterations observed at the blood-brain barrier. In wild-type postnatal brain, ATP7A is heterogeneously expressed in astrocytes (Niciu et al., 2006). As expected, astrocytosis was observed in the MoBr/y neocortex, hippocampus and cerebellum (Shafit-Zagardo et al., 1988). MoBr/y neocortical, hippocampal and cerebellar astrocytes upregulate mutant ATP7A levels (Fig. 6A). Closer analysis of these regions revealed increased GFAP-positive astrocytic end-feet that contact the MoBr/y cerebrovasculature (Fig. 6A). Recruitment of astrocytic end-feet to the MoBr/y cerebrovasculature was also observed at P11, but was not examined at ages earlier than P8 (data not shown).
Figure 6.
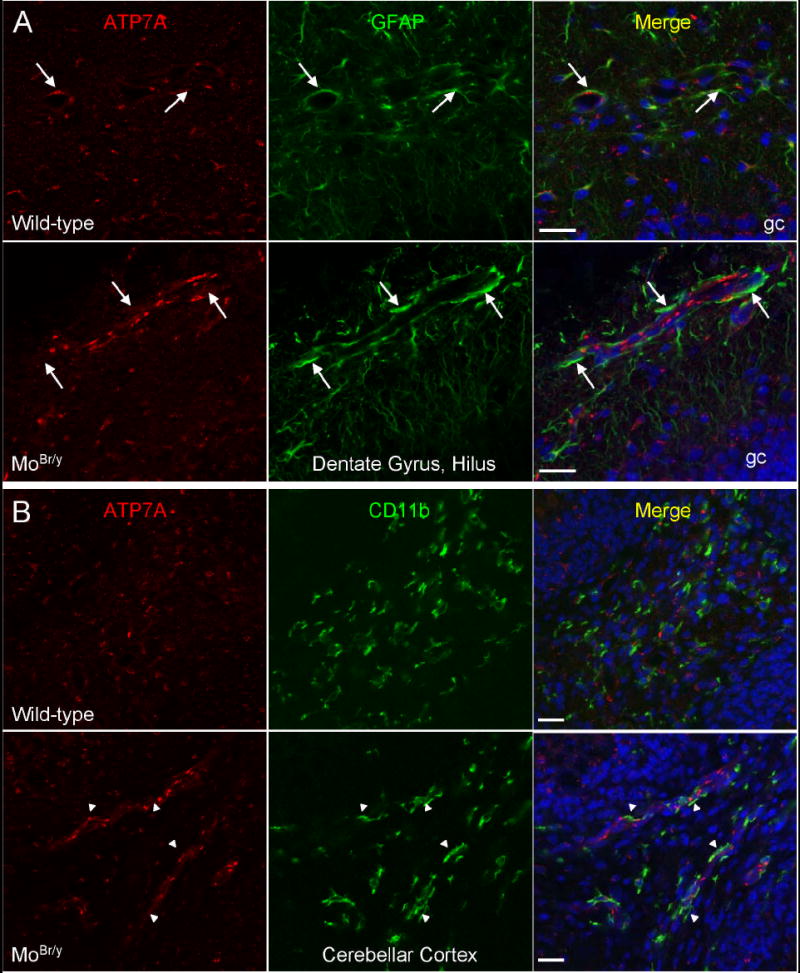
Increased neurovascular unit activity in MoBr/y brain. A. P8 wild-type and MoBr/y hippocampi were stained simultaneously for ATP7A (red), the astrocyte marker GFAP (green) and DNA (blue). In wild-type hippocampus, astrocytic end-feet contact the basal surface of endothelial cells (arrows). In MoBr/y hippocampus, astrocytic end-feet are recruited to the cerebrovasculature as displayed by enhanced levels of GFAP immunoreactivity at these basal contacts (arrows). B. P8 wild-type and MoBr/y cerebella were stained simultaneously for ATP7A (red), the microglial marker CD11b (green) and DNA (blue). In wild-type mice, there is minimal contact of quiescent microglia with the cerebrovasculature. In contrast, in MoBr/y mice, contact of perivascular microglia with the cerebrovasculature is enhanced (arrowheads). These observations are typical of the three P8 wild-type and three MoBr/y animals examined; similar observations were noted in P11 wild-type and MoBr/y brain (data not shown); gc, granule cell layer of the dentate gyrus; Scale bars: 20 μm.
CD11b, a microglial marker, and ATP7A colocalization studies were next performed. Perivascular microglia contact the wild-type cerebrovasculature only minimally (Fig. 6B, wild-type). Like the astrocytic end-feet in MoBr/y mice, the interdigitating processes of CD11b-positive perivascular microglia display increased association with the cerebrovasculature (Fig. 6B, MoBr/y). CD11b-positive microglia are also recruited to MoBr/y cerebrovasculature in the hippocampus, but to a lesser degree than in the cerebellum (data not shown). Similar observations were made at P11 (data not shown).
Postsynaptic dysfunction in MoBr/y Purkinje cells
Although cerebellar Purkinje neurons are particularly sensitive to ATP7A mutation (Yamano and Suzuki, 1985; Robain et al., 1988), cerebellar levels of amidated PACAP are not diminished in MoBr/y mice (Fig. 3B). The number of Purkinje cells is not drastically reduced, but severe disorganization is often observed, in particular in the midline floccus (Fig. 7, MoBr/y). To better understand the causes of Purkinje neuron dysfunction, expression of ATP7A was compared in wild-type and MoBr/y cerebellum. In P11 wild-type cerebellum, co-localization with a somatodendritic marker, microtubule-associated protein 2 (MAP-2), demonstrates that Purkinje neurons express ATP7A in a perinuclear location, with extension of ATP7A-positive tubuloreticular structures into proximal dendrites (arrow) (Fig. 7A, wild-type). In contrast, ATP7A expression is barely detectable in the somatodendritic compartment of MoBr/y Purkinje neurons (Fig. 7A, MoBr/y). Even though cerebellar granule cell number is reduced, ATP7A levels are unaltered in the remaining MoBr/y granule cells. Interestingly, the major neuropathological finding in MoBr/y granular cell layers is failed migration from the external granular layer (EGL) to their more internal destination (internal granular layer, IGL) in the mature cerebellar cortex (Figs. 7A, 8A – cf. wild-type vs. MoBr/y EGL thickness). Expression of mutant ATP7A also declines dramatically in hippocampal pyramidal neurons (Fig. 4A) and neocortical layer 5 pyramidal neurons (data not shown).
Figure 7.
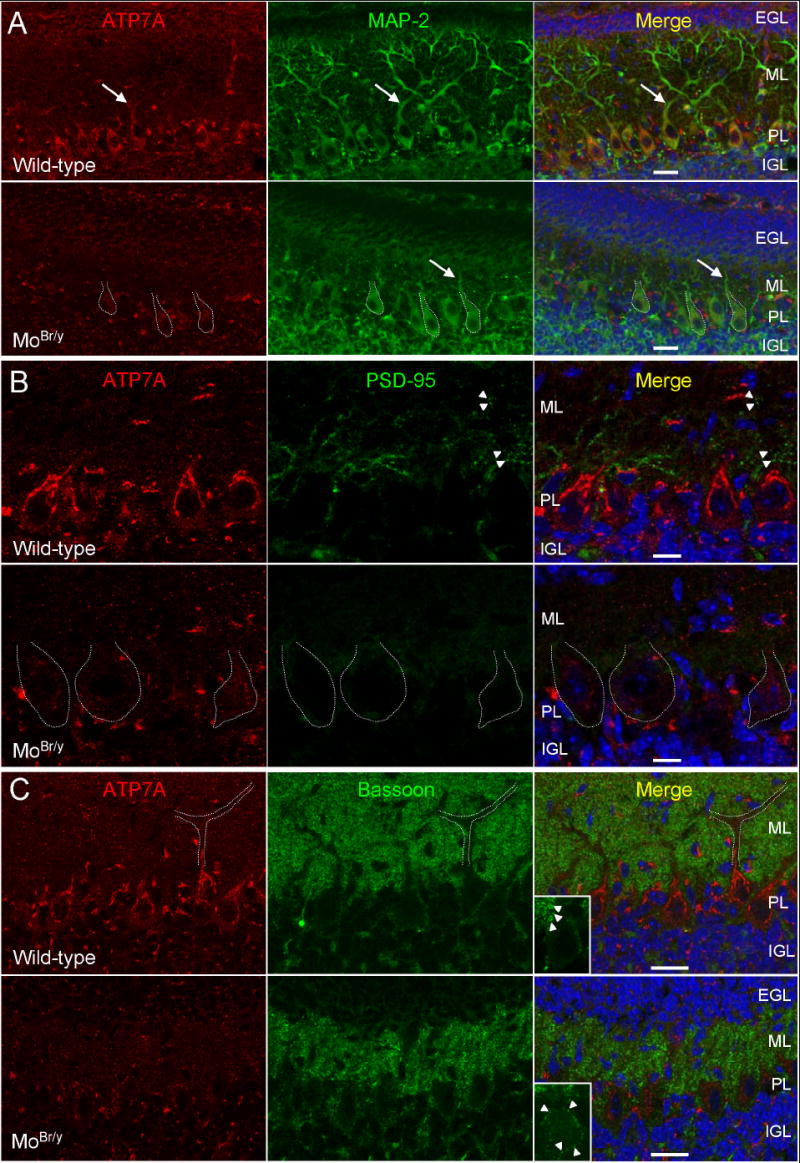
Decreased expression of mutant ATP7A and postsynaptic dysfunction in MoBr/y cerebellar Purkinje neurons. A. Wild-type and MoBr/y cerebella were stained simultaneously for ATP7A (red), MAP2 (green) and DNA (blue). In wild-type tissue, ATP7A immunoreactivity is intense in Purkinje neurons in a perinuclear location and extends into proximal dendrites (arrow). Mutant ATP7A expression is reduced in MoBr/y Purkinje cells; MoBr/y Purkinje cell somata are outlined in white dotted lines. B. Wild-type and MoBr/y cerebella were stained simultaneously for ATP7A (red), PSD-95 (green) and DNA (blue). The punctate PSD-95 distribution apparent in wild-type tissue (arrowheads) is absent from MoBr/y Purkinje cell dendrites. C. Wild-type and MoBr/y cerebella were stained simultaneously for ATP7A (red), bassoon (green) and DNA (blue). Bassoon staining is most evident in the molecular layer, outlining the dendritic arbor of wild-type Purkinje cells (white outline; arrowheads in inset). In MoBr/y cerebellum, bassoon immunoreactivity surrounding Purkinje cell bodies is evident (arrowheads in inset). Three wild-type and three MoBr/y animals were examined at P11; similar observations were noted in three wild-type and MoBr/y animals at P8 (data not shown); EGL, external granular layer; IGL, internal granular layer; ML, molecular layer; PL, Purkinje layer; Scale bars: A,C, 50 μm; B, 10 μm.
Figure 8.
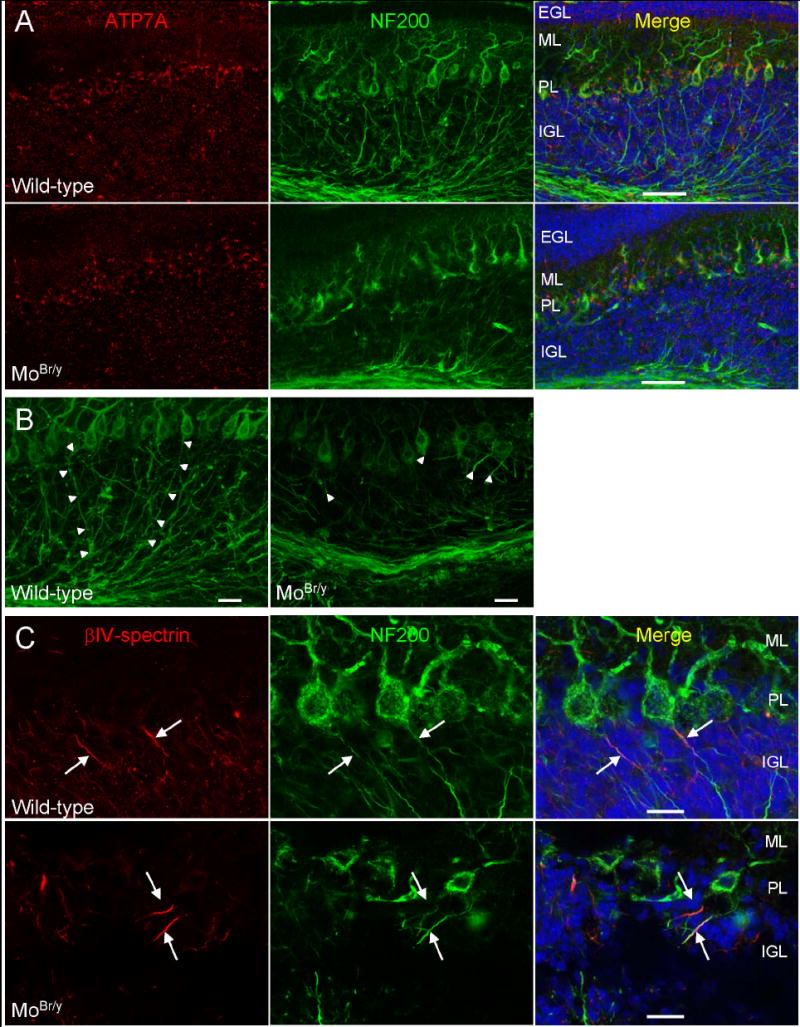
Decreased expression of mutant ATP7A and axonal pathology in MoBr/y cerebellar Purkinje neurons. A. Sections of P11 wild-type and MoBr/y cerebellum were stained simultaneously with antisera to ATP7A (red), NF200 (green; to identify Purkinje cell axons) and DNA (blue). B. Maximum projection images of a 0.3 μm Z-step series representing the total section thickness of 15 μm. Purkinje cell axons are indicated by arrowheads. C. P11 wild-type and MoBr/y cerebellar sections were stained simultaneously for βIV-spectrin (red), NF200 (green) and nuclei (blue). Axon initial segments are identified by arrows; Scale bars: A, 50 μm; B,C, 20 μm.
As expected, MAP-2 immunostaining of MoBr/y cerebellum reveals simplification of the extensive dendritic arbor characteristic of wild-type Purkinje cells (arrow) (Fig. 7A, MoBr/y). In order to characterize the molecular effects of this dendritic pathology, colocalization analyses were undertaken with synaptic markers. In P11 wild-type cerebellum, clustered PSD-95 immunoreactivity is observed in spine-like structures along Purkinje cell dendrites (arrowheads) (Fig. 7B, wild-type), indicating that synaptogenesis has commenced in the molecular layer. Despite the fact that PSD-95 levels were equivalent in wild-type and MoBr/y cerebellar homogenates based on immunoblotting (data not shown), clustered PSD-95 puncta are not detected in the truncated dendrites of the MoBr/y molecular layer (Fig. 7B, MoBr/y). Aberrant sprouting of spine-like structures from Purkinje cell somata has been reported in Menkes disease patients and MoBr/y cerebellum (Purpura et al., 1976; Yamano and Suzuki, 1985). However, no clustered PSD-95 expression was observed in these spine-like structures in MoBr/y Purkinje cell bodies (Fig. 7B, MoBr/y).
To explore the fate of glutamatergic presynaptic endings normally targeted to Purkinje cell dendrites, P11 wild-type and MoBr/y cerebella were stained for bassoon, a cytoplasmic matrix protein localized to the presynaptic active zone. Bassoon immunoreactivity from putative parallel fibers and intra-molecular layer basket and stellate cell contacts outlines Purkinje cell dendrites in the wild-type molecular layer (Fig. 7C, wild-type). Although the width of the molecular layer is decreased in MoBr/y cerebellum, bassoon staining is still clustered into presynaptic puncta (Fig. 7C, MoBr/y). Interestingly, some presynaptic endings are mistargeted to Purkinje cell bodies (Fig. 7C, MoBr/y, arrowheads in inset). These aberrant bassoon-positive structures are not likely to form functional synapses on spine-like structures sprouting from MoBr/y Purkinje cell bodies as they lack clustered PSD-95 (Fig. 7B, MoBr/y).
MoBr/y Purkinje cell axons show morphological deficits
After observing these defects in dendritic morphogenesis, axonal morphology in MoBr/y Purkinje neurons was examined using an antibody to neurofilament protein 200, N52. This phosphorylation state-independent antibody stains both the axonal and somatodendritic compartments of Purkinje neurons, facilitating accurate cell body-to-axon designation (Fig. 8). In P11 wild-type cerebellum, Purkinje cell axons, the sole output from the cerebellar cortex, project the entire distance of the granular layer (arrowheads) (Fig. 8A,B, wild-type). In contrast, most MoBr/y Purkinje cell axons fail to project through the entire granular layer, often becoming tortuous and/or exhibiting bulbous deformities before abruptly terminating (arrowheads) (Fig. 8A,B, MoBr/y).
We sought a separate measure of axonal pathology. The axon initial segment is the site of action potential initiation in Purkinje neurons (Khaliq and Raman, 2006); βIV-spectrin serves as a marker for the axon initial segment (Berghs et al., 2000). P11 cerebellar sections from wild-type and MoBr/y tissue were stained simultaneously with antisera for NF200 and βIV-spectrin (Fig. 8C). βIV-spectrin staining is clustered at the proximal segment of wild-type Purkinje cell axons, defining the axon initial segment (arrows) (Fig. 8C, wild-type). Despite the clear pathology apparent in MoBr/y Purkinje cell axons, βIV-spectrin staining remains clustered in axon initial segments (arrowheads) (Fig. 8C, MoBr/y). Normal axon initial segment formation suggests that deficits in Purkinje cell function are selective and do not reflect a simple response to decreased energy stores.
DISCUSSION
Subcellular copper-depletion and differential impairment of cuproenzyme activity in MoBr/y brain
The decrease in brain copper content in Menkes disease patients and mouse models of this disorder is well-documented. In MoBr/y brain, copper concentration is decreased 2-4 fold and the difference is exacerbated with age (Camakaris et al., 1979; Phillips et al., 1986). However, copper levels in different subcellular compartments have not been reported. Unexpectedly, the MoBr/y neocortical cytosol, with contributions from all cell-types, is more severely copper-depleted than particulate fractions (Fig. 2A). Although ATP7B can transport copper into the secretory pathway (Barnes et al., 2005), no compensatory increase in ATP7B levels is observed (Fig. 1C). Total levels of ATP7A, PAM, SOD1 and SOD3 were also unaltered (Steveson et al., 2003) (Figs. 1A, 2B-D, 3; Suppl. Fig. 1). Similarly, SOD1 levels were unaltered in copper-deficient 4 week old murine brain (Prohaska et al., 2003). In contrast, SOD3 levels increased in the aortas of the mottled blotchy Atp7a mutant mouse while SOD1 levels were unaltered (Qin et al., 2006). The mechanisms through which the particulate fractions sequester copper in MoBr/y brain are not clear. Although metallothioneins are cytosolic, most cuproenzymes and brain copper binding proteins such as prion protein and amyloid precursor protein reside in the secretory pathway (Inestrosa et al., 2005). The luminal copper-binding domains of these proteins could retain the small amounts of copper transported via mutant ATP7A and/or ATP7B.
In a previous study, we found reduced levels of amidated CCK in MoBr/y neocortex (Steveson et al., 2003). CCK expression is restricted to peptidergic neurons throughout the brain (Cain et al., 2003). The decrease in expression of ATP7A observed in hippocampal pyramidal neurons in MoBr/y mice would contribute to the observed decrease in amidated CCK (Fig. 4). In addition to neurons, PAM is expressed in astrocytes and oligodendrocytes (Rhodes et al., 1990; Klein and Fricker, 1992). ATP7A expression in a subpopulation of astrocytes is increased in the MoBr/y mouse; consistent with this, amidation of PACAP, which is expressed in both neurons and astrocytes, is diminished to a lesser extent than amidation of CCK (Fig. 4). The relative resistance of hypothalamic peptide amidation to diminished ATP7A function may reflect the high levels of PAM expression in hypothalamic neurons (Schafer et al., 1992). The differential impaired amidation of these and other neuropeptides may contribute to the selective neurodegeneration seen in MoBr/y brain.
Sites of ATP7A expression are dramatically altered in MoBr/y brain
Expression of mutant ATP7A protein in the brains of mouse models of Menkes disease or human patients has not been investigated. Previous in situ hybridization studies revealed no change in levels or localization of transcripts encoding ATP7A in the macular mouse (Iwase et al., 1996; Murata et al., 1997). In contrast, in the MoBr/y brain, transcript levels were diminished in Purkinje cells and hippocampal pyramidal neurons, the cells most susceptible to neurodegeneration (Iwase et al., 1996). Although only slight differences were observed in total ATP7A protein when homogenates of wild-type and MoBr/y brain were examined via immunoblotting (Fig. 1), dramatic differences in the cell-types expressing ATP7A were identified using immunofluorescence.
Interestingly, using a modified silver sulfide/trichloroacetic acid stain to detect copper histochemically in macular and MoBr/y brain, the neuronal populations with diminished ATP7A levels were shown to have decreased copper content (Yoshimura, 1994; Murata et al., 1998). In contrast, mutant ATP7A levels increase dramatically in the endothelial cells of MoBr/y brain capillaries (Fig. 4). Copper levels are elevated in cerebral endothelial cells in macular and MoBr/y brain (Yoshimura, 1994; Murata et al., 1998). The positive correlation between ATP7A levels and intracellular copper content in diverse cell-types suggests that copper may regulate Atp7a gene expression in a manner similar to iron, another transition metal. A copper requirement for ATP7A gene expression early in development would contribute to the observed critical period for copper replenishment (Fujii et al., 1990).
Compensatory responses in MoBr/y cerebrovasculature
Copper transport across the blood-brain barrier is believed to require ATP7A (Qian et al., 1998). Wild-type and mutant ATP7A are equivalently expressed in larger-diameter arterioles and pial blood vessels (Fig. 5), which are not believed to function in a barrier capacity. ATP7A with the brindled mutation retains some ability to transport copper (La Fontaine et al., 1999). The increased expression of mutant ATP7A in MoBr/y small-diameter blood vessels may serve as a compensatory mechanism to facilitate copper transport across the blood-brain barrier, contributing to the time window during which copper-injections have therapeutic efficacy.
In addition to up-regulation of mutant ATP7A expression in MoBr/y endothelium, astrocytic end-feet are recruited to small-diameter vessels (Fig. 6). The mechanism(s) responsible for astrocytic recruitment is/are unknown. Brain endothelial cells and the surrounding perivascular cells are separated from astrocytic end-feet by a basal lamina. Astrocytes and endothelial cells communicate by secreting diffusible factors that modulate neurovascular unit activity. Astrocytes secrete proteins such as glial-derived neurotrophic factor (Utsumi et al., 2000) and transforming growth factor-β (Tran et al., 1999) to alter blood-brain barrier properties. Endothelial cells can induce astrocyte differentiation by releasing factors such as leukemia inhibitory factor (Mi et al., 2001). Previous studies in MoBr/y and postmortem Wilson disease brains demonstrate that astrocytes are also responsive to copper levels (Shafit-Zagardo et al., 1988; Bertrand et al., 2001).
Microglia adopt a reactive morphology in MoBr/y brain (Ohno et al., 1992). Additionally, these cells express high levels of metallothioneins I and II, proteins implicated in metal homeostasis (Penkowa et al., 1999). The increase in CD11b-positive cells contacting the MoBr/y cerebrovasculature (Fig. 7) could represent bone-marrow derived perivascular microglia/pericytes or juxtavascular CNS-resident microglia. The microglia-endothelium interaction is bidirectional. Perivascular cell contacts are necessary for vessel wall integrity during development, especially at capillary beds devoid of encircling smooth muscle. These endothelium-associated microglia regulate the growth and development of endothelial cells by direct contact and/or secretion of growth factors including basic fibroblast growth factor (Watanabe et al., 1997).
Decreased expression of ATP7A is associated with neuronal pathology
In contrast to the compensatory increase of mutant ATP7A expression in MoBr/y endothelium, mutant protein levels were dramatically reduced in cerebellar Purkinje neurons, hippocampal pyramidal neurons and surrounding interneurons (Figs. 4, 7, 8). A key question to be addressed in future studies is why expression of ATP7A declines in these subsets of neurons while remaining normal in others. The Purkinje cell degeneration in Menkes disease patients and mouse models of the disorder involves severe cytoskeletal abnormalities which cannot be directly attributed to the impaired catalytic activity of known cuproenzymes: perisomatic sprouting of spine-like structures; focal swelling of stem dendrites with microtubule disruption; the presence of Hirano bodies (eosinophilic rod-like structures immunopositive for filamentous actin) in proximal dendrites (Peterson et al., 1986; Onaga et al., 1987; Robain et al., 1988). Copper administration attenuates, but does not completely reverse, Purkinje cell cytoskeletal pathology (Yamano et al., 1985; Kawasaki et al., 1988; Yamano et al., 1988), suggesting neuropathology that involves mutant ATP7A in a role independent of copper transport. Recent experimental evidence in Drosophila and zebrafish supports a cell-autonomous role for ATP7A in nervous system development (Mendelsohn et al., 2006; Norgate et al., 2006). Additionally, MoBr/y hippocampal neurons in vitro and in vivo are more sensitive to N-methyl-D-aspartate receptor mediated excitotoxic insults potentially mediated by a cuproenzyme-independent mechanism (Schlief et al., 2006).
Axonal pathology in Menkes disease patients and mouse models of this disorder has not been described in detail. Examination of MoBr/y Purkinje axons revealed significant cytoskeleton disruption, with tortuous, bulbous axons terminating abruptly in the cerebellar cortex (Fig. 8). Despite this, βIV-spectrin was properly clustered in the axon initial segment, the anatomical site of action potential initiation (Fig. 8). Although MoBr/y Purkinje neurons may be capable of initiating action potentials, their inhibitory signals would fail to propagate to postsynaptic targets outside of the cerebellar cortex. Recent evidence from our laboratory demonstrates that ATP7A also plays a critical role in synaptogenesis and axonal outgrowth in olfactory receptor neurons (El Meskini et al., 2007), yet the copper-dependence of these phenomenona remains to be investigated. Determining whether exogenous copper supplementation to pregnant dams or their pups in the early postnatal period attenuates the neuropathological insults observed in this work will require further study.
Supplementary Material
SOD1 and SOD3 levels are unchanged in MoBr/y brain. A. SOD1 immunoblot of SDS-solubilized homogenates from the indicated P8 wild-type and MoBr/y tissues. B. Quantification of SOD1 signal intensity; error bars, range of duplicates. C. SOD3 immunoblot of the indicated homogenates; lung and kidney (Kdy) were included as positive controls (Folz et al., 1997; Ookawara et al., 1998). Mouse SOD3 (NP_035565) has a predicted molecular mass of 27.3 kDa; 35 and 33 kDa bands are detected in tissue, indicative of glycosylation (Ookawara et al., 1997; Ookawara et al., 1998)
Acknowledgments
This study was supported by grants from the National Institutes of Health DK-32949 and GM-08607. Amide-specific polyclonal CCK-antiserum was kindly provided by Dr. Jens Rehfeld [University of Copenhagen (Denmark), Rigshospitalet, Department of Clinical Biochemistry]. Samples of ATP7B were kindly provided by Dr. Svetlana Lutsenko (Oregon Health Science University, Portland, OR). βIV-spectrin polyclonal antiserum was graciously provided by Dr. Matthew Rasband (UCHC). We would also like to thank Ms. Darlene D’Amato for assistance with mouse husbandry and the remainder of the Neuropeptide Laboratory for their suggestions and engaging discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Barnes N, Tsivkovskii R, Tsivkovskaia N, Lutsenko S. The copper-transporting ATPases, menkes and wilson disease proteins, have distinct roles in adult and developing cerebellum. J Biol Chem. 2005;280:9640–9645. doi: 10.1074/jbc.M413840200. [DOI] [PubMed] [Google Scholar]
- Berghs S, Aggujaro D, Dirkx R, Jr, Maksimova E, Stabach P, Hermel JM, Zhang JP, Philbrick W, Slepnev V, Ort T, Solimena M. betaIV spectrin, a new spectrin localized at axon initial segments and nodes of ranvier in the central and peripheral nervous system. J Cell Biol. 2000;151:985–1002. doi: 10.1083/jcb.151.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand E, Lewandowska E, Szpak GM, Hoogenraad T, Blaauwgers HG, Czlonkowska A, Dymecki J. Neuropathological analysis of pathological forms of astroglia in Wilson’s disease. Folia Neuropathol. 2001;39:73–79. [PubMed] [Google Scholar]
- Brown DR. Metallic prions. Biochem Soc Symp. 2004:193–202. doi: 10.1042/bss0710193. [DOI] [PubMed] [Google Scholar]
- Bush AI. The metallobiology of Alzheimer’s disease. Trends Neurosci. 2003;26:207–214. doi: 10.1016/S0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- Cain BM, Connolly K, Blum A, Vishnuvardhan D, Marchand JE, Beinfeld MC. Distribution and colocalization of cholecystokinin with the prohormone convertase enzymes PC1, PC2, and PC5 in rat brain. J Comp Neurol. 2003;467:307–325. doi: 10.1002/cne.10924. [DOI] [PubMed] [Google Scholar]
- Camakaris J, Mann JR, Danks DM. Copper metabolism in mottled mouse mutants: copper concentrations in tissues during development. Biochem J. 1979;180:597–604. doi: 10.1042/bj1800597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czyzyk TA, Ning Y, Hsu MS, Peng B, Mains RE, Eipper BA, Pintar JE. Deletion of peptide amidation enzymatic activity leads to edema and embryonic lethality in the mouse. Dev Biol. 2005;287:301–313. doi: 10.1016/j.ydbio.2005.09.001. [DOI] [PubMed] [Google Scholar]
- El Meskini R, Crabtree KL, Cline LB, Mains RE, Eipper BA, Ronnett GV. ATP7A (Menkes protein) functions in axonal targeting and synaptogenesis. Mol Cell Neurosci. 2007;34:409–421. doi: 10.1016/j.mcn.2006.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folz RJ, Guan J, Seldin MF, Oury TD, Enghild JJ, Crapo JD. Mouse extracellular superoxide dismutase: primary structure, tissue-specific gene expression, chromosomal localization, and lung in situ hybridization. Am J Respir Cell Mol Biol. 1997;17:393–403. doi: 10.1165/ajrcmb.17.4.2826. [DOI] [PubMed] [Google Scholar]
- Francis MJ, Jones EE, Levy ER, Ponnambalam S, Chelly J, Monaco AP. A Golgi localization signal identified in the Menkes recombinant protein. Hum Mol Genet. 1998;7:1245–1252. doi: 10.1093/hmg/7.8.1245. [DOI] [PubMed] [Google Scholar]
- Fujii T, Ito M, Tsuda H, Mikawa H. Biochemical study on the critical period for treatment of the mottled brindled mouse. J Neurochem. 1990;55:885–889. doi: 10.1111/j.1471-4159.1990.tb04574.x. [DOI] [PubMed] [Google Scholar]
- Giacobini P, Kopin AS, Beart PM, Mercer LD, Fasolo A, Wray S. Cholecystokinin modulates migration of gonadotropin-releasing hormone-1 neurons. J Neurosci. 2004;24:4737–4748. doi: 10.1523/JNEUROSCI.0649-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza I, Faisst A, Prohaska J, Chen J, Gruss P, Gitlin JD. The metallochaperone Atox1 plays a critical role in perinatal copper homeostasis. Proc Natl Acad Sci U S A. 2001;98:6848–6852. doi: 10.1073/pnas.111058498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inestrosa NC, Cerpa W, Varela-Nallar L. Copper brain homeostasis: role of amyloid precursor protein and prion protein. IUBMB Life. 2005;57:645–650. doi: 10.1080/15216540500264620. [DOI] [PubMed] [Google Scholar]
- Iwase T, Nishimura M, Sugimura H, Igarashi H, Ozawa F, Shinmura K, Suzuki M, Tanaka M, Kino I. Localization of Menkes gene expression in the mouse brain; its association with neurological manifestations in Menkes model mice. Acta Neuropathol (Berl) 1996;91:482–488. doi: 10.1007/s004010050455. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Yamano T, Iwane S, Shimada M. Golgi study on macular mutant mouse after copper therapy. Acta Neuropathol (Berl) 1988;76:606–612. doi: 10.1007/BF00689600. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Raman IM. Relative contributions of axonal and somatic Na channels to action potential initiation in cerebellar Purkinje neurons. J Neurosci. 2006;26:1935–1944. doi: 10.1523/JNEUROSCI.4664-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RS, Fricker LD. Cultured astrocytes express mRNA for peptidylglycine-alpha-amidating monooxygenase, a neuropeptide processing enzyme. Brain Res. 1992;596:202–208. doi: 10.1016/0006-8993(92)91548-s. [DOI] [PubMed] [Google Scholar]
- Kunz WS, Kuznetsov AV, Clark JF, Tracey I, Elger CE. Metabolic consequences of the cytochrome c oxidase deficiency in brain of copper-deficient Mo(vbr) mice. J Neurochem. 1999;72:1580–1585. doi: 10.1046/j.1471-4159.1999.721580.x. [DOI] [PubMed] [Google Scholar]
- Kuo YM, Zhou B, Cosco D, Gitschier J. The copper transporter CTR1 provides an essential function in mammalian embryonic development. Proc Natl Acad Sci U S A. 2001;98:6836–6841. doi: 10.1073/pnas.111057298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Fontaine S, Firth SD, Lockhart PJ, Brooks H, Camakaris J, Mercer JF. Intracellular localization and loss of copper responsiveness of Mnk, the murine homologue of the Menkes protein, in cells from blotchy (Mo blo) and brindled (Mo br) mouse mutants. Hum Mol Genet. 1999;8:1069–1075. doi: 10.1093/hmg/8.6.1069. [DOI] [PubMed] [Google Scholar]
- La Fontaine S, Firth SD, Lockhart PJ, Brooks H, Parton RG, Camakaris J, Mercer JF. Functional analysis and intracellular localization of the human menkes protein (MNK) stably expressed from a cDNA construct in Chinese hamster ovary cells (CHO-K1) Hum Mol Genet. 1998;7:1293–1300. doi: 10.1093/hmg/7.8.1293. [DOI] [PubMed] [Google Scholar]
- Lee J, Prohaska JR, Thiele DJ. Essential role for mammalian copper transporter Ctr1 in copper homeostasis and embryonic development. Proc Natl Acad Sci U S A. 2001;98:6842–6847. doi: 10.1073/pnas.111058698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair GR, Niciu MJ, Stewart MT, Brennan G, Omar H, Halton DW, Mains R, Eipper BA, Maule AG, Day TA. A functionally atypical amidating enzyme from the human parasite Schistosoma mansoni. FASEB J. 2004;18:114–121. doi: 10.1096/fj.03-0429com. [DOI] [PubMed] [Google Scholar]
- Mendelsohn BA, Yin C, Johnson SL, Wilm TP, Solnica-Krezel L, Gitlin JD. Atp7a determines a hierarchy of copper metabolism essential for notochord development. Cell Metab. 2006;4:155–162. doi: 10.1016/j.cmet.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Mi H, Haeberle H, Barres BA. Induction of astrocyte differentiation by endothelial cells. J Neurosci. 2001;21:1538–1547. doi: 10.1523/JNEUROSCI.21-05-01538.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y, Kodama H, Abe T, Ishida N, Nishimura M, Levinson B, Gitschier J, Packman S. Mutation analysis and expression of the mottled gene in the macular mouse model of Menkes disease. Pediatr Res. 1997;42:436–442. doi: 10.1203/00006450-199710000-00003. [DOI] [PubMed] [Google Scholar]
- Murata Y, Kodama H, Mori Y, Kobayashi M, Abe T. Mottled gene expression and copper distribution in the macular mouse, an animal model for Menkes disease. J Inherit Metab Dis. 1998;21:199–202. doi: 10.1023/a:1005383114315. [DOI] [PubMed] [Google Scholar]
- Niciu MJ, Ma XM, El Meskini R, Ronnett GV, Mains RE, Eipper BA. Developmental changes in the expression of ATP7A during a critical period in postnatal neurodevelopment. Neuroscience. 2006;139:947–964. doi: 10.1016/j.neuroscience.2006.01.044. [DOI] [PubMed] [Google Scholar]
- Norgate M, Lee E, Southon A, Farlow A, Batterham P, Camakaris J, Burke R. Essential roles in development and pigmentation for the Drosophila copper transporter DmATP7. Mol Biol Cell. 2006;17:475–484. doi: 10.1091/mbc.E05-06-0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Higashi Y, Suzuki K. Microglial cell response to neuronal degeneration in the brain of brindled mouse. Brain Res Dev Brain Res. 1992;67:37–45. doi: 10.1016/0165-3806(92)90023-P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J Biol Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- Onaga A, Kawasaki H, Yamano T, Shimada M, Nishimura M. Light and electron microscopic study on cerebellar cortex of macular mutant mouse as a model of Menkes kinky hair disease. Brain Dev. 1987;9:265–269. doi: 10.1016/s0387-7604(87)80043-8. [DOI] [PubMed] [Google Scholar]
- Ookawara T, Imazeki N, Matsubara O, Kizaki T, Oh-Ishi S, Nakao C, Sato Y, Ohno H. Tissue distribution of immunoreactive mouse extracellular superoxide dismutase. Am J Physiol. 1998;275:C840–C847. doi: 10.1152/ajpcell.1998.275.3.C840. [DOI] [PubMed] [Google Scholar]
- Ookawara T, Kizaki T, Ohishi S, Yamamoto M, Matsubara O, Ohno H. Purification and subunit structure of extracellular superoxide dismutase from mouse lung tissue. Arch Biochem Biophys. 1997;340:299–304. doi: 10.1006/abbi.1997.9912. [DOI] [PubMed] [Google Scholar]
- Penkowa M, Nielsen H, Hidalgo J, Bernth N, Moos T. Distribution of metallothionein I + II and vesicular zinc in the developing central nervous system: correlative study in the rat. J Comp Neurol. 1999;412:303–318. doi: 10.1002/(sici)1096-9861(19990920)412:2<303::aid-cne9>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Peterson C, Suzuki K, Kress Y, Goldman JE. Abnormalities of dendritic actin organization in the brindled mouse. Brain Res. 1986;382:205–212. doi: 10.1016/0006-8993(86)91331-4. [DOI] [PubMed] [Google Scholar]
- Petris MJ, Strausak D, Mercer JF. The Menkes copper transporter is required for the activation of tyrosinase. Hum Mol Genet. 2000;9:2845–2851. doi: 10.1093/hmg/9.19.2845. [DOI] [PubMed] [Google Scholar]
- Phillips M, Camakaris J, Danks DM. Comparisons of copper deficiency states in the murine mutants blotchy and brindled. Changes in copper-dependent enzyme activity in 13-day-old mice. Biochem J. 1986;238:177–183. doi: 10.1042/bj2380177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prohaska JR. Long-term functional consequences of malnutrition during brain development: copper. Nutrition. 2000;16:502–504. doi: 10.1016/s0899-9007(00)00308-7. [DOI] [PubMed] [Google Scholar]
- Prohaska JR, Geissler J, Brokate B, Broderius M. Copper, zinc-superoxide dismutase protein but not mRNA is lower in copper-deficient mice and mice lacking the copper chaperone for superoxide dismutase. Exp Biol Med (Maywood) 2003;228:959–966. doi: 10.1177/153537020322800812. [DOI] [PubMed] [Google Scholar]
- Purpura DP, Hirano A, French JH. Polydendritic Purkinje cells in X-chromosome linked copper malabsorption: a Golgi study. Brain Res. 1976;117:125–129. doi: 10.1016/0006-8993(76)90561-8. [DOI] [PubMed] [Google Scholar]
- Qian Y, Tiffany-Castiglioni E, Welsh J, Harris ED. Copper efflux from murine microvascular cells requires expression of the menkes disease Cu-ATPase. J Nutr. 1998;128:1276–1282. doi: 10.1093/jn/128.8.1276. [DOI] [PubMed] [Google Scholar]
- Qin Z, Itoh S, Jeney V, Ushio-Fukai M, Fukai T. Essential role for the Menkes ATPase in activation of extracellular superoxide dismutase: implication for vascular oxidative stress. FASEB J. 2006;20:334–336. doi: 10.1096/fj.05-4564fje. [DOI] [PubMed] [Google Scholar]
- Rhodes CH, Xu RY, Angeletti RH. Peptidylglycine alpha-amidating monooxygenase (PAM) in Schwann cells and glia as well as neurons. J Histochem Cytochem. 1990;38:1301–1311. doi: 10.1177/38.9.2387985. [DOI] [PubMed] [Google Scholar]
- Robain O, Aubourg P, Routon MC, Dulac O, Ponsot G. Menkes disease: a Golgi and electron microscopic study of the cerebellar cortex. Clin Neuropathol. 1988;7:47–52. [PubMed] [Google Scholar]
- Schafer MK, Stoffers DA, Eipper BA, Watson SJ. Expression of peptidylglycine alpha-amidating monooxygenase (EC 1.14.17.3) in the rat central nervous system. J Neurosci. 1992;12:222–234. doi: 10.1523/JNEUROSCI.12-01-00222.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlief ML, Craig AM, Gitlin JD. NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J Neurosci. 2005;25:239–246. doi: 10.1523/JNEUROSCI.3699-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlief ML, West T, Craig AM, Holtzman DM, Gitlin JD. Role of the Menkes copper-transporting ATPase in NMDA receptor-mediated neuronal toxicity. Proc Natl Acad Sci U S A. 2006;103:14919–14924. doi: 10.1073/pnas.0605390103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selverstone VJ, Doucette PA, Zittin PS. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem. 2005;74:563–593. doi: 10.1146/annurev.biochem.72.121801.161647. [DOI] [PubMed] [Google Scholar]
- Shafit-Zagardo B, Peterson C, Goldman JE. Rapid increases in glial fibrillary acidic protein mRNA and protein levels in the copper-deficient, brindled mouse. J Neurochem. 1988;51:1258–1266. doi: 10.1111/j.1471-4159.1988.tb03095.x. [DOI] [PubMed] [Google Scholar]
- Shim H, Harris ZL. Genetic defects in copper metabolism. J Nutr. 2003;133:1527S–1531S. doi: 10.1093/jn/133.5.1527S. [DOI] [PubMed] [Google Scholar]
- Steveson TC, Ciccotosto GD, Ma XM, Mueller GP, Mains RE, Eipper BA. Menkes protein contributes to the function of peptidylglycine alpha-amidating monooxygenase. Endocrinology. 2003;144:188–200. doi: 10.1210/en.2002-220716. [DOI] [PubMed] [Google Scholar]
- Tran ND, Correale J, Schreiber SS, Fisher M. Transforming growth factor-beta mediates astrocyte-specific regulation of brain endothelial anticoagulant factors. Stroke. 1999;30:1671–1678. doi: 10.1161/01.str.30.8.1671. [DOI] [PubMed] [Google Scholar]
- Utsumi H, Chiba H, Kamimura Y, Osanai M, Igarashi Y, Tobioka H, Mori M, Sawada N. Expression of GFRalpha-1, receptor for GDNF, in rat brain capillary during postnatal development of the BBB. Am J Physiol Cell Physiol. 2000;279:C361–C368. doi: 10.1152/ajpcell.2000.279.2.C361. [DOI] [PubMed] [Google Scholar]
- Vaudry D, Gonzalez BJ, Basille M, Fournier A, Vaudry H. Neurotrophic activity of pituitary adenylate cyclase-activating polypeptide on rat cerebellar cortex during development. Proc Natl Acad Sci U S A. 1999;96:9415–9420. doi: 10.1073/pnas.96.16.9415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Morisaki N, Tezuka M, Fukuda K, Ueda S, Koyama N, Yokote K, Kanzaki T, Yoshida S, Saito Y. Cultured retinal pericytes stimulate in vitro angiogenesis of endothelial cells through secretion of a fibroblast growth factor-like molecule. Atherosclerosis. 1997;130:101–107. doi: 10.1016/s0021-9150(96)06050-9. [DOI] [PubMed] [Google Scholar]
- Yamano T, Paldino AM, Suzuki K. Ultrastructural and morphometric studies of Purkinje cells of brindled mouse after administration of cupric chloride. J Neuropathol Exp Neurol. 1985;44:97–107. doi: 10.1097/00005072-198501000-00008. [DOI] [PubMed] [Google Scholar]
- Yamano T, Shimada M, Onaga A, Kawasaki H, Iwane S, Ono K, Nishimura M. Electron microscopic study on brain of macular mutant mouse after copper therapy. Acta Neuropathol (Berl) 1988;76:574–580. doi: 10.1007/BF00689595. [DOI] [PubMed] [Google Scholar]
- Yamano T, Suzuki K. Abnormalities of Purkinje cell arborization in brindled mouse cerebellum. A Golgi study. J Neuropathol Exp Neurol. 1985;44:85–96. doi: 10.1097/00005072-198501000-00007. [DOI] [PubMed] [Google Scholar]
- Yoshimura N. Histochemical localization of copper in various organs of brindled mice. Pathol Int. 1994;44:14–19. doi: 10.1111/j.1440-1827.1994.tb02580.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SOD1 and SOD3 levels are unchanged in MoBr/y brain. A. SOD1 immunoblot of SDS-solubilized homogenates from the indicated P8 wild-type and MoBr/y tissues. B. Quantification of SOD1 signal intensity; error bars, range of duplicates. C. SOD3 immunoblot of the indicated homogenates; lung and kidney (Kdy) were included as positive controls (Folz et al., 1997; Ookawara et al., 1998). Mouse SOD3 (NP_035565) has a predicted molecular mass of 27.3 kDa; 35 and 33 kDa bands are detected in tissue, indicative of glycosylation (Ookawara et al., 1997; Ookawara et al., 1998)