

Abstract
The signaling processes that maintain the homeostatic proliferation of peripheral T-cells and result in their self-renewal largely remain to be elucidated. Much focus has been placed on the anti-apoptotic function of the cytokine, interleukin-7 (IL-7), during T-cell development. But a more critical role has been ascribed to IL-7 as a mediator of peripheral T-cell maintenance. The biological effects responsive to IL-7 signaling are transduced through only a few well-known pathways. In this review we will focus on the signals transduced by IL-7 and similar cytokines, examining how proliferative signals originate from cytokine receptors, are amplified and eventually alter gene expression. In this regard we will highlight the crosstalk between pathways that promote survival, drive cell cycle progression and most importantly provide the needed energy to sustain these critical cellular activities. Though this review showcases much of what has been learned about IL-7 proliferative signaling, it also reveals the significant gaps in our knowledge about cytokine signaling in the very relevant context of peripheral T-cell homeostasis.
Keywords: cytokine, proliferation, lymphocytes, homeostasis, metabolism
Interleukin-7 (IL-7) is an essential survival factor for lymphocytes, but evidence is accumulating that IL-7 has indispensable functions that go beyond protection from cell death. IL-7 was originally discovered as a result of its proliferative activity on immature murine B-cells in vitro 61. Early work with IL-7 and IL-7 receptor knockout mice revealed the critical role of this cytokine in T-cell development57;71;95 and demonstrated that loss of IL-7 signaling is a major cause of severe combined immunodeficiency syndrome (SCID)73, establishing the importance of IL-7 in T-cell biology. More recently, IL-7 has been recognized as a mediator of the homeostatic mechanisms that maintain stable numbers of naïve and memory T-cells in the peripheral immune system 77;84;89. Much of what is currently known about the activity of IL-7 concerns its role preventing T-cell apoptosis and far less is known about its proliferative function. Notably, it is this proliferative capacity of IL-7 that has the greatest therapeutic implication in the treatment of clinical conditions such as lymphopenia. It is widely recognized that the survival activity of IL-7 is dependent upon the regulation of the BCL-2 family of anti-apoptotic and pro-apoptotic proteins. However, this survival activity is not sufficient to fully rescue the lymphopenic conditions that result upon ablation of the IL-7 signaling pathway 41. Sustained expression of the anti-apoptotic protein BCL-2 56 or deletion of the pro-apoptotic proteins BAX 41 or BIM 70, protected dependent cells from death, but failed, in the long term, to fully reconstitute a functional T-cell compartment. Clearly, in addition to its role as a survival factor, IL-7 also acts as a growth stimulus for T-cells. In this review, we will examine what is currently known about the biology of IL-7, specifically in the context of T-cell proliferation.
IL-7 Receptor and Signaling for Proliferation
IL-7 is not produced by lymphocytes but is generally considered a product of the stromal cells found in lymphoid tissues as well as the skin and intestinal epithelium (reviewed in 2;33;58). Binding of IL-7 to its cell surface receptor leads to the activation of a few signaling pathways that have the potential to drive T-cell proliferation. The IL-7 receptor consists of the IL-7Rα chain (IL-7R) and the common cytokine γ chain (γc) that is also shared by the receptors for IL-2, IL-4, IL-9, IL-15, and IL-21 46. Upon ligand binding, the two receptor chains heterodimerize 101, which leads to the activation of the receptor associated tyrosine Janus kinases, JAK1 (IL-7R) and JAK3 (γc) 85. The activated JAK proteins in turn phosphorylate specific residues on the IL-7R creating docking sites for signaling molecules that have Src homology 2 (SH2) domains such as STAT5 (signal transducers and activators of transcription 5) 48 and to a lesser extent STAT1 and STAT3 100. Disruption of any of the components of the IL-7 signaling pathway leads to a significant loss of both developing and mature T-cells 62;71;99. Work by several groups has been instrumental in identifying key regions in the IL-7R responsible for mediating the growth promoting activities of this cytokine.
Signals transduced by IL-7 initiate from the intracellular signaling region of the IL-7R. This region consists of an acidic rich domain, a serine rich domain, and a tyrosine rich domain (T-domain) which contains three tyrosine (Y) residues (Y401, Y449, and Y456) 72 that are conserved in mice and humans. An additional domain known as Box1 is located proximal to the membrane and is thought to be the binding site for JAK1 90. Studies with T-cell lines expressing mutant IL-4/IL-7 chimeric receptors revealed that loss of Box1 results in rapid cell death, deletion of the serine rich region has minimal effects, while the Y449F mutation causes cell cycle arrest that precedes cell death 34. Hence, signals originating from Box1 and Y449 are essential for IL-7 mediated T-cell survival and proliferation 34. This was reinforced in mice expressing a knock-in mutation (IL-7RαY449F) that displayed defective homeostatic proliferation of naïve CD4 and CD8 T-cells 66. The Y449 site is thus of particular interest because two critical IL-7 signaling pathways, the JAK/STAT pathway and the phosphatidylinositol 3-kinase (PI3K)/AKT pathway may originate from this site 68.
JAK/STAT
The current model for JAK/STAT signaling from the IL-7R is that upon binding to Y449, STAT5 proteins, specifically STAT5a and STAT5b, are phosphorylated by JAKs. The phosphorylated STATs dimerize and translocate to the nucleus where they modulate the expression of a number of genes involved in cell survival and proliferation such as BCL-XL, c-myc, and the D-type cyclins 3;22;99. There have also been reports that STAT5 has functions independent of its transcriptional activities. Within the cytoplasm, STAT5 can, by associating with the scaffolding protein, Gab2, activate two signaling pathways that are involved in cell proliferation, the PI3K/AKT and Ras/MAPK pathways63. This was shown in IL-3 dependent Ba/F3 cells which signal through JAK2. However, the Ras/MAPK pathway, which is activated by IL-2 and IL-15, does not appear to be activated by IL-7 18;42;74, so it remains to be determined whether STAT5 association with Gab2 occurs in response to IL-7 in peripheral T-cells and is a means for the activation of downstream proliferative signaling pathways.
Because JAKs and STATs are critical for transducing the IL-7 growth signal, mutations in these proteins can contribute to the development of immunodeficiencies as well as lymphomas. A deficiency in JAK3 is one of the major causes of severe combined immunodeficiency syndrome (SCID) in humans. Mice completely deficient in STAT5a and STAT5b also develop a SCID-like phenotype 64;99. The lymphopenic conditions resulting from loss of these signaling molecules could be due to defects in T-cell proliferation. However, this is difficult to prove given the critical role of IL-7 in early T-cell development. Hence, to study the role of these proteins in the context of T-cell proliferation, cell cycling effects on peripheral T-cells must be examined.
The role of STAT5 in cytokine-induced cell cycling has been examined in several murine models. Peripheral T-cells in STAT5a/b deficient mice fail to proliferate in response to IL-2 and this was correlated with the loss of expression of the cell cycle proteins, cyclin D2, cyclin D3, cyclin A, and cdk6 59. T-cells from an IL-7RαY449F knock-in mouse did not activate STAT5 66, suggesting that signals transduced through Y449 of the Il-7R regulate STAT5 activity. A link between IL-7 induced proliferation and STAT5 was shown in an elegant study utilizing STAT5a/b+/− compound heterozygous mice which express reduced copy numbers of STAT5a and STAT5b (haploinsufficiency). In this study, mice that expressed an IL-7 transgene rapidly developed lymphomas and died, while STAT5a/b+/− mice had improved survival rates and fewer signs of lymphoma development1. Hence STAT5 seems to enhance proliferation in mouse models in which multiple cell lineages are present, but findings differ when evaluating individual cell lines. A constitutively active STAT5a was able to protect an IL-7 dependent T-cell line from death due to cytokine-withdrawal but did not promote proliferation 34, while a constitutively active STAT5a or STAT5b could sustain proliferation in the absence of IL-3 in a dependent B-cell line but this may have been due to a gain of function mutation 65. It thus remains unclear whether STAT5 is required solely for survival signaling or also for the induction of proliferative activity.
One study tried to address a role for STAT5 in T-cell proliferation by assessing the phosphorylation state of STAT5. In this study it was reported that the level of IL-7 that induced STAT5 activation (as measured by tyrosine phosphorylation of STAT5) did not directly correspond to IL-7-driven T-cell proliferation. That is quiescent T-cells or those undergoing minimal cell cycle divisions had equivalent levels of STAT5 tyrosine phosphorylation as did actively proliferating cells87. Yet in another study that measured ex vivo expression of STAT5, increased levels of phosphorylated STAT5 were detected in freshly isolated CD8 T-cells recovered from bone marrow12. This was indicative of the increased proliferative capacity of cells derived from the bone marrow compared to spleen, but these same cells also displayed decreased expression of the IL-7R (CD127), suggesting that STAT5 phosphorylation was sustained even after the IL-7 signal was down-regulated. It thus remains to be determined whether the activation of STAT5 directly confers cell cycle progression or whether additional pathways are involved in regulating T-cell proliferation.
PI3K/AKT
The PI3K/AKT pathway is a potential T-cell survival pathway that in many cell types also regulates diverse cellular functions such as cell cycle progression, transcription, and metabolism. Mutations in the PI3K/AKT pathway and the tumor suppressor PTEN, an inhibitor of PI3K/AKT signaling, are frequent causes of cancer in humans 21;94. PI3K is a lipid kinase that when activated by growth factors, translocates to the plasma membrane where it phosphorylates the lipid, PIP2 92. Conversion of PIP2 into PIP3, recruits AKT, the major effector of PI3K signaling, to the membrane where it becomes activated. AKT is a serine/threonine kinase and is estimated to have nearly 900 cellular targets 52. In this section, we will examine several AKT targets that are involved in initiating cell cycle progression that could be mediators of IL-7’s proliferative activity.
In terms of cell proliferation, two major activities critical for cell cycle progression have been attributed to PI3K/AKT signaling: metabolism, which will be discussed later, and regulation of cyclin-dependent kinases (cdks) 26. Previous studies by us and others have shown that in the absence of growth signals like IL-7 or IL-3, dependent lymphocytes arrest in the G0/G1 phase of the cell cycle 39;44;45. To exit from this phase, cells must typically activate specific G1 cdks/cyclins that are required for cell cycle entry as well as down regulate cell cycle inhibitors such as p27kip1. PI3K through AKT promotes stability of the cell cycle proteins c-myc and cyclin D by inactivating glycogen synthase kinase-3β (GSK3β), a negative regulator of c-myc and cyclin D (reviewed in 94). Using cytokine-dependent cell lines or epithelial-derived cells, AKT was shown to inactivate the Forkhead family of transcription factors, preventing the transcription of genes that negatively regulate the cell cycle like p27kip1 83 as well as mediators of apoptosis19. In addition to transcriptional regulation, AKT has also been shown to directly interact with and phosphorylate p27kip1 in epithelial cells, targeting it for cytoplasmic localization and degradation 25.
While there is evidence to support a role for IL-7 in the activation of PI3K/AKT signaling, most of these studies were performed in transformed cell lines, T-acute lymphoblastic leukemia (T-ALL) cells 4, or thymocytes 68. Hence, the data obtained from these studies may not be truly representative of what is occurring in the peripheral T-cell compartment. Interestingly, one study which did use untransformed mature T-cells found that IL-7 did not activate the PI3K pathway (as measured by phosphorylation of AKT), even though these cells did proliferate in response to IL-7 47, and this was supported by a recent study in which IL-7 again did not induce the phosphorylation of AKT in primary murine T-cells 66. Hence the PI3K/AKT pathway may not be directly required for T-cell proliferation, at least in response of primary T-cells to IL-7. Results using cell lines expressing a mutant IL-2 receptor also suggested that PI3K signaling was not immediately activated in response to cytokine signaling 14;47. However, when the kinetics of PI3K signaling were examined, IL-2 was found to induce two waves of PI3K activity: one rapid wave occurring within minutes and a second later wave, occurring within hours, that contributed to T-cell growth 47. More importantly, IL-7 was found to also induce this later wave of PI3K activity that may be responsible for cell cycling, although differences in the response of thymocytes versus mature T-cells were noted 47. Hence, the immediate, early activation of PI3K signaling does not occur in response to IL-7, while the function of the later wave of activity remains unclear. This is in contrast to the more rapid induction of STAT5 activity. The PI3K pathway may thus play a lesser role in the maintenance of the peripheral T-cells by IL-7 and initiating T-cell proliferation through an IL-7 signal may in fact be dependent on recruiting other signaling pathways.
In an IL-3 dependent B-cell line, induction of DNA synthesis required activation of both the PI3K/AKT pathway and the Raf/Mek/Erk pathway. Expression of either activated Raf or AKT alone was unable to stimulate B-cell proliferation in the absence of the cytokine signal 81. However, the Erk pathway, does not appear to be activated by IL-7 signaling in T-cells 18. Furthermore, the PI3K signaling pathway in T-cells may be quite different from B-cells. PI3K signaling in T-cells may be associated with the γc receptor through JAK3 80 and not the IL-7R 34, impacting on the downstream consequences of this signaling pathway. In recent thymic emigrants (RTE), inhibition of PI3K did impede cell cycling 87, but this could have been due to the loss of survival or metabolic activities sustained by PI3K, with proliferation dependent on other signals. Hence, in T-cells, the proliferative signal mediated by IL-7 could involve the activation of PI3K/AKT in order to support pro-life signals or bioenergetics, but the essential mediators of cell division activated in response to IL-7 may not be dependent on this pathway and remain to be identified.
IL-7 Controls Cell Cycling
Studies of IL-7 dose responses can clearly distinguish the survival function of this cytokine from its proliferative activity by demonstrating that low doses of IL-7 (<1 ng/ml) sustain only survival, while high doses of IL-7 (>1 ng/ml) promote survival and cell cycling 44;87. Early studies suggesting that IL-7 is a proliferative factor 76 have been recently substantiated by the observation that administration of IL-7 leads to increased T-cell numbers 28. However, these findings are in contrast with other studies that describe the failure of IL-7 to induce the cell division of T-cells 75 along with the decrease in thymic numbers observed upon transgenic expression of IL-7R 60. In fact, one primate study showed that the proliferative response of T-cells to IL-7 treatment was transient and that the responding T-cells eventually returned to a quiescent state even in the continued presence of IL-7 7.
One theory that could explain such disparate findings is that transcription and expression of the IL-7R is negatively regulated by its ligand, IL-7 60;69, perhaps through activation of transcriptional repressors 69, and that this “altruistic” behavior could modulate the proliferative response to the cytokine. Down-regulation of the IL-7R was also observed in response to diverse growth stimuli such as engagement of the T-cell receptor (TCR) 24 and other cytokines 69. Though the significance of this differential expression of the IL-7R remains elusive, it may play a role in the differentiation of memory T-cells from effector T-cells 37 through mechanisms poorly understood. IL-7R levels do appear to reflect the IL-7 dose and duration of signal, with detection of surface IL-7R decreased at higher doses of the cytokine. Thus down-regulation of the IL-7R occurs not at the lower doses of IL-7 that maintain survival, but at the higher doses that support cell cycling 87;88. This may suggest that a strong IL-7 signal is needed to provide an initial proliferative stimulus that is dispensable once a cell begins the DNA replicative phase. The complex nature of the IL-7 growth signal remains unknown, but it likely involves regulation of the activity of key components of the cell cycle machinery.
One mechanism by which IL-7 could promote cell proliferation is through the regulation of proteins which are involved in the G1/S cell cycle transition, for example through substrates activated by the PI3K/AKT pathway as previously discussed. Indeed the activity of cyclin-dependent kinase 2 (cdk2), which is required for progression through G1/S, is increased in lymphocytes isolated from mice that were treated with IL-7 compared to those treated with a control28. Furthermore, protein levels of cyclin E and phosphorylated retinoblastoma (Rb) protein were elevated following IL-7 treatment 28. However, in cytokine-dependent lymphocyte cell lines, although mRNA levels for cdk4 and cyclin D3 were observed to decrease following cytokine withdrawal, their protein levels were still detectable at a time point when the cells had already undergone growth arrest 39. This suggest that while a cytokine signal is required for the transcription of cdks and cyclins 39;43, the presence of the proteins themselves is not sufficient to sustain cell cycling in the absence of a growth factor signal. Instead, it may be the control of the activity of two regulatory factors: the cdk inhibitor p27kip1 and the cdk activating phosphatase, Cdc25A, which are the primary mechanisms by which IL-7 promotes T-cell proliferation.
p27kip1
p27kip1 is a member of the CIP/KIP family of cyclin-dependent cell cycle inhibitors (CKIs) that negatively regulates the G1/S transition 93 but may also promote the assembly of G1 cdk/cyclin D complexes in a manner not well understood (reviewed in 16). p27kip1 binds to and inhibits cdk2 and its cyclin partner by occupying the kinase’s catalytic cleft, causing conformational changes that inactivate the kinase/cyclin complex, leading to G1 cell cycle arrest 82. Levels of p27kip1 are highest in quiescent cells and must be down-regulated in order for cells to enter cell cycling 38. Recently, the discovery that p27kip1 is a phosphoprotein containing 6-8 phosphorylatable residues suggests a mechanism to explain how growth signals can modulate the CKI’s activity. Phosphorylation of p27kip1 by tyrosine kinases, such as Src, inhibited its association with cdk2, leading to the activation of cdk2, which in turn further phosphorylated p27kip1, targeting the CKI for degradation 31. Other kinases that could be involved in the phosphorylation of p27kip1 include PKCθ, shown to regulate p27kip1 stability in an IL-7 dependent T-cell line 49, and the PI3K/AKT pathway, shown to regulate phosphorylation of the transcription factor FoxO3, which induces p27kip1 expression in IL-2 dependent T-cells 83. In IL-7 dependent T-cells, the expression of p27kip1 was sufficient to cause G1 arrest in the presence of IL-7. Furthermore, IL-7 withdrawal induced the upregulation of p27kip1 and arrested cells in G1 while siRNA knockout of p27kip1 enhanced cell cycle progression compared to control cells 49. However, adoptive transfer of p27kip1 deficient lymphocytes into IL-7 deficient mice demonstrated that loss of p27kip1 could only partially compensate for the IL-7 signal needed by T-cells to expand in a lymphopenic environment 49. Hence p27kip1 may be needed to negatively regulate cdk2 activity, but its absence is not sufficient to fully induce cell cycling under lymphopenic conditions.
Cdc25A
The positive activation of cdk/cyclin complexes needed to promote cell cycle progression is mediated by the dephosphorylating activity of a family of dual-function protein phosphatases collectively known as Cdc25. There are three isoforms of Cdc25 in mammalian cells: Cdc25A, Cdc25B, and Cdc25C. Few studies have examined the activities of these proteins in T-cells. What is known from examining epithelial-derived cell lines is that Cdc25A has a critical role in the G1/S transition by activating cdk2, and can also act on cdk1 to promote mitosis 53. Overexpression of Cdc25A is correlated with accelerated entry into S-phase and a shortened G110. Hence Cdc25A is the rate limiting factor for S-phase progression 10;79. In contrast, Cdc25B and Cdc25C have more limited roles which are restricted to the G2/M phases. The importance of Cdc25B and Cdc25C in cell cycling has been challenged by the finding that cells from Cdc25B and Cdc25C knockout mice, have normal cell cycling activities 23. Hence, it has been proposed that Cdc25A may be able to compensate for the activities of both Cdc25B and Cdc25C. Cdc25A is of particular interest in terms of IL-7 proliferative signaling because IL-7 withdrawal results in G1/S cell cycle arrest and is correlated with loss of cdk2 activity 28;39, two events regulated by the dephosphorylating activity of Cdc25A.
Cdk activity is negatively regulated through inhibitory phosphorylation mediated by Wee1/Mik1/Myt1 kinases 9. This leads to the accumulation of an inactive pool of cdks. In order to re-activate cdks, like cdk2, the dephosphorylating activity of Cdc25A is required 35. Cdc25A is in turn negatively regulated by phosphorylation which triggers its degradation through a ubiquitin-proteasome dependent pathway 8. Two kinase families known to phosphorylate and regulate the stability of Cdc25A are the MAP kinases (MAPK), specifically p38 MAPK, and the checkpoint kinases 1 and 2 (CHK1 and CHK2) 29;32.
In cytokine-dependent lympocytes, p38 MAPK activity has been linked to both proliferative effects 17 and the induction of apoptosis 42. Such conflicting results, may be explained by examining the kinetics of p38 MAPK activity, which indicate that cytokine signaling maintains a basal level of activity that is greatly increased upon cytokine withdrawal 42. Localization and availability of substrates may also influence the outcome of p38 MAPK activity. Using IL-3 dependent B-cells, the increase in activity of p38 MAPK, but not CHK1, following cytokine withdrawal in dependent lymphocytes correlated with a rapid decline in the protein levels of Cdc25A 39. In epithelial cells, p38 MAPK was found to phosphorylate Cdc25A and this phosphorylation in turn triggered degradation of the phosphatase 29, resulting in G1 arrest. To determine whether Cdc25A could have a role in IL-7 induced proliferation of T-cells, a p38 MAPK-resistant Cdc25A mutant was generated by mutating the two p38 MAPK target sites on Cdc25A. Once expressed, this Cdc25A mutant protein remained stable during IL-7 withdrawal and was not targeted for degradation by p38 MAPK phosphorylation. Expression of the Cdc25A mutant in an IL-7-dependent T-cell line as well as in peripheral, primary T-cells was sufficient to sustain cell survival and promote cell cycling for several days in the absence of IL-7. This was accompanied by increased phosphorylation of cdk2 and Rb39;44. Hence, Cdc25A, like p27kip1, is responsive to IL-7 proliferative signaling. Interestingly, when p27kip1 levels were examined in cytokine-deprived lymphocytes expressing the Cdc25A mutant protein, these were found to be elevated and yet did not interfere with the cell cycling activity conferred by Cdc25A expression 44. The positive effects of Cdc25A upon cdk activity can thus overcome the inhibitory action of p27kip1. Though little is known of the IL-7 receptor proximal events that control p38 MAPK activity and the stability of Cdc25A, these likely involve mechanisms that control transcriptional activity (perhaps through STAT5), cycling controls (perhaps through PI3K) and stress responses through pathways that link survival signaling with the MAPK pathway. What can be surmised from the current findings is that the controller of cell cycle progression, Cdc25A, can provide signals that ensure not only successful DNA replication 44 but may also sustain the necessary metabolic activity required for mitotic cell division in response to an IL-7 proliferative signal.
IL-7 Controls Metabolism (and cell size)
Maintenance of cell survival and proliferation is dependent upon the ability of cells to acquire sufficient nutrients to support cellular metabolism. Failure to maintain energy production, specifically through glucose metabolism, is especially detrimental to actively cycling cells which have increased energetic demands. Indeed one of the cellular requirements for progressing from G1 into S-phase is to acquire the appropriate cell size, which is indicative of cell growth. This was elegantly shown in yeast in which cell cycling stops when cell growth is delayed 36. However, in mammalian cells, it has been argued that pro-growth extracellular signals are the main requirement for G1/S progression 15. Yet, as found with yeast, evidence for cell size controlling mammalian cell division has also been reported. IL-3 dependent cells from mice deficient in the apoptotic proteins, BAX and BAK, survived in the absence of IL-3 but decreased in size. Re-addition of IL-3 restored cell proliferation but only after the cells had reached the size of normal cycling cells, suggesting that size control was linked to cytokine-driven cycling 51;97. As with IL-3 dependent cells, IL-7 dependent cells undergo atrophy or decrease in cell size when the cytokine is withdrawn 39;75. The control of cell size mediated by IL-7 was linked to the maintenance of glucose metabolism through signals transduced by PI3K/AKT 5 and the mammalian target of rapamycin (mTOR) 78, resulting in the regulation of the expression and activity of glucose transporters.
GLUT1
A function attributed to growth factors like IL-7 is the maintenance of metabolic activity through the uptake of glucose from the extracellular milieu. How this process occurs in peripheral T-cells is poorly understood. Mitogen-stimulated thymocytes, for example, are dependent upon glucose uptake for proliferation, switching from oxidative to glycolytic energy production during the G1/S transition 30, a trend also observed in cancerous cells (Warburg effect) 96. We and others have found that IL-7 deprivation caused dependent cells to rapidly cease glucose transport 40. The main facilitative glucose transporter in peripheral T-cells is GLUT1 13, which is also the receptor for the human T-cell leukemia virus (HTLV) 54. Increased GLUT1 expression occurs upon T-cell activation, for example, through engagement of the TCR55. GLUT1 expression is also upregulated by cytokine signaling. In IL-3 dependent B cells, GLUT1 translocation to the surface and stimulation of glucose transport was controlled by the presence or absence of IL-3, a process inhibited by PI3K inhibitors 6 as well as inhibitors of mTOR 98. Similar findings were made in IL-7 dependent T-ALL cells in which PI3K signaling was required for maintenance of cell size and GLUT1 expression 4. In recent thymic emigrants, the PI3K pathway was also responsible for the up-regulation of GLUT1 and increased glucose uptake that correlated with IL-7 induced proliferation 87. In addition to GLUT1, cytokine signaling also maintains expression of other glycolytic enzymes including hexokinase 2 and phosphofructokinase 1 – enzymes whose activity rapidly declined upon growth factor withdrawal 91. In the absence of such growth factor signaling, metabolism ceases in dependent cells, causing progressive atrophy, as measured by a decrease in cell size, and ultimately death.
In addition to regulating GLUT1, IL-7 is also linked to the expression of another nutrient transporter, the transferrin receptor (CD71). High levels of CD71 were observed on cells that also expressed GLUT1 transporters and in fact characterized a highly proliferative subset of thymocytes 86;87. Like GLUT1, surface expression of CD71 was blocked by inhibition of PI3K in the IL-7 responsive T-ALL cells 4. Expression of both GLUT1 and CD71 is thus associated with high metabolic activity, characteristic of actively cycling cells. Hence, maintenance of glucose uptake and metabolism is a requirement for active cell cycling as well as the prevention of apoptosis. But given that very little is known about glucose metabolism in peripheral T-cells and that the inhibition of PI3K signaling in an IL-7 dependent T-cell line did not cause cell death in the presence of the cytokine but only accelerated death during IL-7 withdrawal 50, it is possible that in IL-7 dependent mature T-cells, sustained glucose metabolism and its associated effects upon proliferation and survival also rely upon PI3K-independent mechanisms that are yet to be discovered.
Conclusions
IL-7 is a multi-potent growth factor known to provide survival signals for T-cells. More importantly IL-7 sustains lymphocyte development and provides proliferative signals that maintain the peripheral T-cell compartment. As shown in Figure 1, the current model for the IL-7 growth signal involves only a small number of known signaling pathways. The JAK/STAT pathway provides the essential survival and growth signaling, the PI3K/AKT pathway provides additional survival signals and metabolic activity, and the MAPK pathway initiates apoptosis and growth arrest. Though much has been learned, many critical questions remained unresolved. In response to IL-7, does STAT5 directly initiate proliferative signaling and are there non-transcriptional activities that promote cell survival or does STAT5 indirectly induce the expression of novel proliferative (X) or survival (Y) factors? What is the importance of PI3K signaling and how is it initiated – directly by binding the IL-7 receptor or is there an intermediate step (X), perhaps linked to the JAK/STAT pathway? Are there unknown IL-7-induced secondary messengers that activate transcription factors (Z) needed for inducing survival gene products? How is the MAPK pathway regulated – directly from the IL-7 receptor (X) or via feedback from the JAK/STAT or PI3K/AKT pathways? Lastly, how are these pathways connected to provide the complex signals that mediate proliferation, survival and metabolism?
Figure 1. IL-7 Signaling Pathways that Drive Proliferation, Survival and Metabolism.
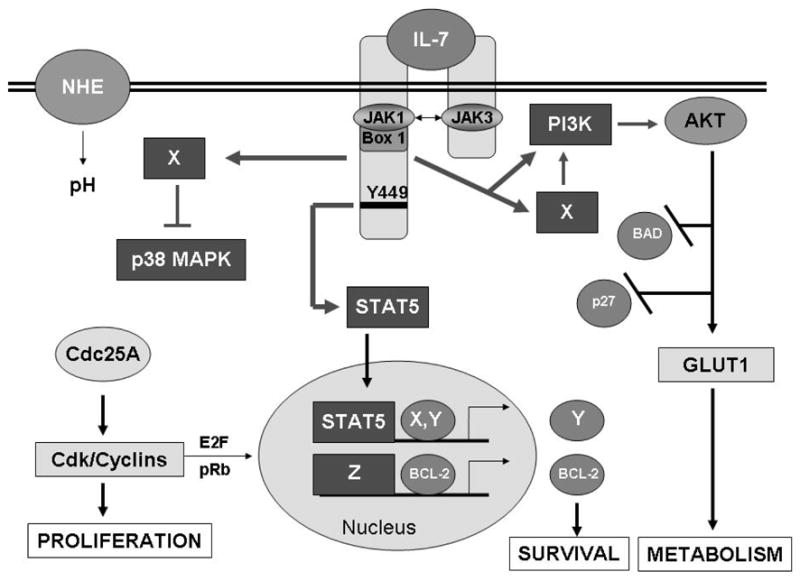
Binding of IL-7 causes dimerization of its receptor chains and activation of the receptor–associated JAK1 and JAK3. JAKs in turn phosphorylate receptor docking sites for STAT5, leading to the transcription factor’s phosphorylation, dimerization and nuclear translocation, perhaps inducing novel gene products that promote survival (X, Y). Other signaling pathways that may stem directly or indirectly (X) from the IL-7 receptor include the PI3K/AKT pathway which inhibits apoptotic proteins and the cell cycle inhibitor, p27kip1, and promotes glucose uptake through the glucose transporter, GLUT1. Unknown signaling pathways (X) also regulate the activity of the stress kinase, p38 MAPK. Down-regulation of p38 MAPK leads to the stable expression of Cdc25A, the activator of cdk/cyclin complexes that promotes proliferation and gene expression likely through activation of the E2F transcription factors. Expression of the survival protein, BCL-2, is up-regulated by IL-7, perhaps through STAT5 activity, but more likely through unknown transcriptional activity (Z). T-cell proliferation in response to IL-7 thus requires signals that drive cell cycle progression but also provide survival signals that prevent apoptosis and produce energy to support cell division.
Answers for these questions will require evaluating the IL-7 signaling pathway for novel interactions beyond those already known. A few new signal transducers that cooperate with or substitute for IL-7 have already been discovered. For example the Notch pathway synergizes with IL-7 signaling to drive the proliferation of thymocytes 27 and the TNF receptor family member, CD27, has been shown to maintain expression of the IL-7R leading to clonal expansion of T-cells 11. Glutamate receptors and pH regulators like the sodium hydrogen exchanger (NHE) (Figure 1) have known roles in T-cell growth 20;67 and links to IL-7 signaling remain to be established. Clues to novel downstream substrates of the IL-7 signaling pathway have also come from gene array studies in which cell cycle regulators, like cdc20, nuclear factors, like ALY, and cell adhesion molecules and intracellular signal transducers were upregulated by IL-7 43. Future studies may continue to unravel the connections between the survival, metabolic and proliferative signals mediated by IL-7 in dependent T-cells by extrapolating from parallel findings in other cell types, such as neurons whose growth factor dependency in many ways resembles that of immune cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Abraham N, Ma MC, Snow JW, Miners MJ, Herndier BG, Goldsmith MA. Haploinsufficiency identifies STAT5 as a modifier of IL-7-induced lymphomas. Oncogene. 2005;24:5252–5257. doi: 10.1038/sj.onc.1208726. [DOI] [PubMed] [Google Scholar]
- 2.Barata JT, Cardoso AA, Boussiotis VA. Interleukin-7 in T-cell acute lymphoblastic leukemia: an extrinsic factor supporting leukemogenesis? Leuk Lymphoma. 2005;46:483–495. doi: 10.1080/10428190400027852. [DOI] [PubMed] [Google Scholar]
- 3.Barata JT, Cardoso AA, Nadler LM, Boussiotis VA. Interleukin-7 promotes survival and cell cycle progression of T-cell acute lymphoblastic leukemia cells by down-regulating the cyclin-dependent kinase inhibitor p27(kip1) Blood. 2001;98:1524–1531. doi: 10.1182/blood.v98.5.1524. [DOI] [PubMed] [Google Scholar]
- 4.Barata JT, Silva A, Brandao JG, Nadler LM, Cardoso AA, Boussiotis VA. Activation of PI3K is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J Exp Med. 2004;200:659–669. doi: 10.1084/jem.20040789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP, Jr, Roth RA. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem. 1999;274:20281–20286. doi: 10.1074/jbc.274.29.20281. [DOI] [PubMed] [Google Scholar]
- 6.Bentley J, Itchayanan D, Barnes K, McIntosh E, Tang X, Downes CP, Holman GD, Whetton AD, Owen-Lynch PJ, Baldwin SA. Interleukin-3-mediated cell survival signals include phosphatidylinositol 3-kinase-dependent translocation of the glucose transporter GLUT1 to the cell surface. J Biol Chem. 2003;278:39337–39348. doi: 10.1074/jbc.M305689200. [DOI] [PubMed] [Google Scholar]
- 7.Beq S, Nugeyre MT, Ho Tsong FR, Gautier D, Legrand R, Schmitt N, Estaquier J, Barre-Sinoussi F, Hurtrel B, Cheynier R, Israel N. IL-7 induces immunological improvement in SIV-infected rhesus macaques under antiviral therapy. J Immunol. 2006;176:914–922. doi: 10.4049/jimmunol.176.2.914. [DOI] [PubMed] [Google Scholar]
- 8.Bernardi R, Liebermann DA, Hoffman B. Cdc25A stability is controlled by the ubiquitin-proteasome pathway during cell cycle progression and terminal differentiation. Oncogene. 2000;19:2447–2454. doi: 10.1038/sj.onc.1203564. [DOI] [PubMed] [Google Scholar]
- 9.Berry LD, Gould KL. Regulation of Cdc2 activity by phosphorylation at T14/Y15. Prog Cell Cycle Res. 1996;2:99–105. doi: 10.1007/978-1-4615-5873-6_10. [DOI] [PubMed] [Google Scholar]
- 10.Blomberg I, Hoffmann I. Ectopic expression of Cdc25A accelerates the G(1)/S transition and leads to premature activation of cyclin E- and cyclin A-dependent kinases. Mol Cell Biol. 1999;19:6183–6194. doi: 10.1128/mcb.19.9.6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carr JM, Carrasco MJ, Thaventhiran JE, Bambrough PJ, Kraman M, Edwards AD, Al Shamkhani A, Fearon DT. CD27 mediates interleukin-2-independent clonal expansion of the CD8+ T cell without effector differentiation. Proc Natl Acad Sci USA. 2006;103:19454–19459. doi: 10.1073/pnas.0609706104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cassese G, Parretta E, Pisapia L, Santoni A, Guardiola J, Di Rosa F. Bone marrow CD8 cells down-modulate membrane IL-7R{alpha} expression and exhibit increased STAT-5 and p38 MAPK phosphorylation in the organ environment. Blood. 2007 doi: 10.1182/blood-2006-09-045807. [DOI] [PubMed] [Google Scholar]
- 13.Chakrabarti R, Jung CY, Lee TP, Liu H, Mookerjee BK. Changes in glucose transport and transporter isoforms during the activation of human peripheral blood lymphocytes by phytohemagglutinin. J Immunol. 1994;152:2660–2668. [PubMed] [Google Scholar]
- 14.Cipres A, Carrasco S, Merida I. Deletion of the acidic-rich domain of the IL-2Rbeta chain increases receptor-associated PI3K activity. FEBS Lett. 2001;500:99–104. doi: 10.1016/s0014-5793(01)02594-7. [DOI] [PubMed] [Google Scholar]
- 15.Conlon IJ, Dunn GA, Mudge AW, Raff MC. Extracellular control of cell size. Nat Cell Biol. 2001;3:918–921. doi: 10.1038/ncb1001-918. [DOI] [PubMed] [Google Scholar]
- 16.Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 2003;13:65–70. doi: 10.1016/s0962-8924(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 17.Crawley JB, Rawlinson L, Lali FV, Page TH, Saklatvala J, Foxwell BM. T cell proliferation in response to interleukins 2 and 7 requires p38MAP kinase activation. J Biol Chem. 1997;272:15023–15027. doi: 10.1074/jbc.272.23.15023. [DOI] [PubMed] [Google Scholar]
- 18.Crawley JB, Willcocks J, Foxwell BM. Interleukin-7 induces T cell proliferation in the absence of Erk/MAP kinase activity. Eur J Immunol. 1996;26:2717–2723. doi: 10.1002/eji.1830261125. [DOI] [PubMed] [Google Scholar]
- 19.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 20.De Vito P. The sodium/hydrogen exchanger: a possible mediator of immunity. Cell Immunol. 2006;240:69–85. doi: 10.1016/j.cellimm.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000;100:387–390. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- 22.Dumon S, Santos SC, Debierre-Grockiego F, Gouilleux-Gruart V, Cocault L, Boucheron C, Mollat P, Gisselbrecht S, Gouilleux F. IL-3 dependent regulation of Bcl-xL gene expression by STAT5 in a bone marrow derived cell line. Oncogene. 1999;18:4191–4199. doi: 10.1038/sj.onc.1202796. [DOI] [PubMed] [Google Scholar]
- 23.Ferguson AM, White LS, Donovan PJ, Piwnica-Worms H. Normal cell cycle and checkpoint responses in mice and cells lacking Cdc25B and Cdc25C protein phosphatases. Mol Cell Biol. 2005;25:2853–2860. doi: 10.1128/MCB.25.7.2853-2860.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franchimont D, Galon J, Vacchio MS, Fan S, Visconti R, Frucht DM, Geenen V, Chrousos GP, Ashwell JD, O’Shea JJ. Positive effects of glucocorticoids on T cell function by up-regulation of IL-7 receptor alpha. J Immunol. 2002;168:2212–2218. doi: 10.4049/jimmunol.168.5.2212. [DOI] [PubMed] [Google Scholar]
- 25.Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2002;277:28706–28713. doi: 10.1074/jbc.M203668200. [DOI] [PubMed] [Google Scholar]
- 26.Garcia Z, Silio V, Marques M, Cortes I, Kumar A, Hernandez C, Checa AI, Serrano A, Carrera AC. A PI3K activity-independent function of p85 regulatory subunit in control of mammalian cytokinesis. EMBO J. 2006;25:4740–4751. doi: 10.1038/sj.emboj.7601324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Peydro M, de Yebenes VG, Toribio ML. Notch1 and IL-7 receptor interplay maintains proliferation of human thymic progenitors while suppressing non-T cell fates. J Immunol. 2006;177:3711–3720. doi: 10.4049/jimmunol.177.6.3711. [DOI] [PubMed] [Google Scholar]
- 28.Geiselhart LA, Humphries CA, Gregorio TA, Mou S, Subleski J, Komschlies KL. IL-7 administration alters the CD4:CD8 ratio, increases T cell numbers, and increases T cell function in the absence of activation. J Immunol. 2001;166:3019–3027. doi: 10.4049/jimmunol.166.5.3019. [DOI] [PubMed] [Google Scholar]
- 29.Goloudina A, Yamaguchi H, Chervyakova DB, Appella E, Fornace AJ, Jr, Bulavin DV. Regulation of Human Cdc25A Stability by Serine 75 Phosphorylation Is Not Sufficient to Activate a S Phase Checkpoint. Cell Cycle. 2003;2:473–478. [PubMed] [Google Scholar]
- 30.Greiner EF, Guppy M, Brand K. Glucose is essential for proliferation and the glycolytic enzyme induction that provokes a transition to glycolytic energy production. J Biol Chem. 1994;269:31484–31490. [PubMed] [Google Scholar]
- 31.Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, Jakel H, Kullmann M, Kriwacki RW, Hengst L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128:269–280. doi: 10.1016/j.cell.2006.11.047. [DOI] [PubMed] [Google Scholar]
- 32.Hassepass I, Voit R, Hoffmann I. Phosphorylation at serine 75 is required for UV-mediated degradation of human Cdc25A phosphatase at the S-phase checkpoint. J Biol Chem. 2003;278:29824–29829. doi: 10.1074/jbc.M302704200. [DOI] [PubMed] [Google Scholar]
- 33.Jiang Q, Li WQ, Aiello FB, Mazzucchelli R, Asefa B, Khaled AR, Durum SK. Cell biology of IL-7, a key lymphotrophin. Cytokine Growth Factor Rev. 2005;16:513–533. doi: 10.1016/j.cytogfr.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 34.Jiang Q, Li WQ, Hofmeister RR, Young HA, Hodge DR, Keller JR, Khaled AR, Durum SK. Distinct regions of the interleukin-7 receptor regulate different Bcl2 family members. Mol Cell Biol. 2004;24:6501–6513. doi: 10.1128/MCB.24.14.6501-6513.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jinno S, Suto K, Nagata A, Igarashi M, Kanaoka Y, Nojima H, Okayama H. Cdc25A is a novel phosphatase functioning early in the cell cycle. EMBO J. 1994;13:1549–1556. doi: 10.1002/j.1460-2075.1994.tb06417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnston GC, Pringle JR, Hartwell LH. Coordination of growth with cell division in the yeast Saccharomyces cerevisiae. Exp Cell Res. 1977;105:79–98. doi: 10.1016/0014-4827(77)90154-9. [DOI] [PubMed] [Google Scholar]
- 37.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 38.Kaldis P. Another piece of the p27Kip1 puzzle. Cell. 2007;128:241–244. doi: 10.1016/j.cell.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 39.Khaled AR, Bulavin DV, Kittipatarin C, Li WQ, Alvarez M, Kim K, Young HA, Fornace AJ, Durum SK. Cytokine-driven cell cycling is mediated through Cdc25A. J Cell Biol. 2005;169:755–763. doi: 10.1083/jcb.200409099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khaled AR, Durum SK. Death and Baxes: mechanisms of lymphotrophic cytokines. Immunol Rev. 2003;193:48–57. doi: 10.1034/j.1600-065x.2003.00050.x. [DOI] [PubMed] [Google Scholar]
- 41.Khaled AR, Li WQ, Huang J, Fry TJ, Khaled AS, Mackall CL, Muegge K, Young HA, Durum SK. Bax deficiency partially corrects IL-7 receptor alpha deficiency. Immunity. 2002;17:561–573. doi: 10.1016/s1074-7613(02)00450-8. [DOI] [PubMed] [Google Scholar]
- 42.Khaled AR, Moor AN, Li A, Kim K, Ferris DK, Muegge K, Fisher RJ, Fliegel L, Durum SK. Trophic Factor Withdrawal: p38 Mitogen-Activated Protein Kinase Activates NHE1, Which Induces Intracellular Alkalinization. Mol Cell Biol. 2001;21:7545–7557. doi: 10.1128/MCB.21.22.7545-7557.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim K, Khaled AR, Reynolds D, Young HA, Lee CK, Durum SK. Characterization of an interleukin-7-dependent thymic cell line derived from a p53(−/−) mouse. J Immunol Methods. 2003;274:177–184. doi: 10.1016/s0022-1759(02)00513-6. [DOI] [PubMed] [Google Scholar]
- 44.Kittipatarin C, Li WQ, Bulavin DV, Durum SK, Khaled AR. Cell Cycling Through Cdc25A: Transducer of Cytokine Proliferative Signals. Cell Cycle. 2006:5. doi: 10.4161/cc.5.9.2693. [DOI] [PubMed] [Google Scholar]
- 45.Komschlies KL, Gregorio TA, Gruys ME, Back TC, Faltynek CR, Wiltrout RH. Administration of recombinant human IL-7 to mice alters the composition of B-lineage cells and T cell subsets, enhances T cell function, and induces regression of established metastases. J Immunol. 1994;152:5776–5784. [PubMed] [Google Scholar]
- 46.Kovanen PE, Leonard WJ. Cytokines and immunodeficiency diseases: critical roles of the gamma(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol Rev. 2004;202:67–83. doi: 10.1111/j.0105-2896.2004.00203.x. [DOI] [PubMed] [Google Scholar]
- 47.Lali FV, Crawley J, McCulloch DA, Foxwell BM. A late, prolonged activation of the phosphatidylinositol 3-kinase pathway is required for T cell proliferation. J Immunol. 2004;172:3527–3534. doi: 10.4049/jimmunol.172.6.3527. [DOI] [PubMed] [Google Scholar]
- 48.Leonard WJ. Role of Jak kinases and STATs in cytokine signal transduction. Int J Hematol. 2001;73:271–277. doi: 10.1007/BF02981951. [DOI] [PubMed] [Google Scholar]
- 49.Li WQ, Jiang Q, Aleem E, Kaldis P, Khaled AR, Durum SK. IL-7 promotes T cell proliferation through destabilization of p27Kip1. J Exp Med. 2006;203:573–582. doi: 10.1084/jem.20051520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li WQ, Jiang Q, Khaled AR, Keller JR, Durum SK. Interleukin-7 inactivates the pro-apoptotic protein bad promoting T cell survival. J Biol Chem. 2004;279:29160–29166. doi: 10.1074/jbc.M401656200. [DOI] [PubMed] [Google Scholar]
- 51.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 52.Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, Zuse A, Eshraghi M, Manda KD, Wiechec E, Los M. Cell survival, cell death and cell cycle pathways are interconnected: Implications for cancer therapy. Drug Resist Updat. 2007;10:13–29. doi: 10.1016/j.drup.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 53.Mailand N, Podtelejnikov AV, Groth A, Mann M, Bartek J, Lukas J. Regulation of G(2)/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J. 2002;21:5911–5920. doi: 10.1093/emboj/cdf567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manel N, Kim FJ, Kinet S, Taylor N, Sitbon M, Battini JL. The ubiquitous glucose transporter GLUT-1 is a receptor for HTLV. Cell. 2003;115:449–459. doi: 10.1016/s0092-8674(03)00881-x. [DOI] [PubMed] [Google Scholar]
- 55.Manel N, Kinet S, Battini JL, Kim FJ, Taylor N, Sitbon M. The HTLV receptor is an early T-cell activation marker whose expression requires de novo protein synthesis. Blood. 2003;101:1913–1918. doi: 10.1182/blood-2002-09-2681. [DOI] [PubMed] [Google Scholar]
- 56.Maraskovsky E, O’Reilly LA, Teepe M, Corcoran LM, Peschon JJ, Strasser A. Bcl-2 can rescue T lymphocyte development in interleukin-7 receptor-deficient mice but not in mutant rag-1−/− mice. Cell. 1997;89:1011–1019. doi: 10.1016/s0092-8674(00)80289-5. [DOI] [PubMed] [Google Scholar]
- 57.Maraskovsky E, Teepe M, Morrissey PJ, Braddy S, Miller RE, Lynch DH, Peschon JJ. Impaired survival and proliferation in IL-7 receptor-deficient peripheral T cells. J Immunol. 1996;157:5315–5323. [PubMed] [Google Scholar]
- 58.Mazzucchelli R, Durum SK. Interleukin-7 receptor expression: intelligent design. Nat Rev Immunol. 2007;7:144–154. doi: 10.1038/nri2023. [DOI] [PubMed] [Google Scholar]
- 59.Moriggl R, Sexl V, Piekorz R, Topham D, Ihle JN. Stat5 activation is uniquely associated with cytokine signaling in peripheral T cells. Immunity. 1999;11:225–230. doi: 10.1016/s1074-7613(00)80097-7. [DOI] [PubMed] [Google Scholar]
- 60.Munitic I, Williams JA, Yang Y, Dong B, Lucas PJ, El Kassar N, Gress RE, Ashwell JD. Dynamic regulation of IL-7 receptor expression is required for normal thymopoiesis. Blood. 2004;104:4165–4172. doi: 10.1182/blood-2004-06-2484. [DOI] [PubMed] [Google Scholar]
- 61.Namen AE, Lupton S, Hjerrild K, Wignall J, Mochizuki DY, Schmierer A, Mosley B, March CJ, Urdal D, Gillis S. Stimulation of B-cell progenitors by cloned murine interleukin-7. Nature. 1988;333:571–573. doi: 10.1038/333571a0. [DOI] [PubMed] [Google Scholar]
- 62.Nosaka T, van Deursen JM, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, Doherty PC, Grosveld GC, Ihle JN. Defective lymphoid development in mice lacking Jak3. Science. 1995;270:800–802. doi: 10.1126/science.270.5237.800. [DOI] [PubMed] [Google Scholar]
- 63.Nyga R, Pecquet C, Harir N, Gu H, Dhennin-Duthille I, Regnier A, Gouilleux-Gruart V, Lassoued K, Gouilleux F. Activated STAT5 proteins induce activation of the PI 3-kinase/Akt and Ras/MAPK pathways via the Gab2 scaffolding adapter. Biochem J. 2005;390:359–366. doi: 10.1042/BJ20041523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Shea JJ, Husa M, Li D, Hofmann SR, Watford W, Roberts JL, Buckley RH, Changelian P, Candotti F. Jak3 and the pathogenesis of severe combined immunodeficiency. Mol Immunol. 2004;41:727–737. doi: 10.1016/j.molimm.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 65.Onishi M, Nosaka T, Misawa K, Mui AL, Gorman D, McMahon M, Miyajima A, Kitamura T. Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol Cell Biol. 1998;18:3871–3879. doi: 10.1128/mcb.18.7.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Osborne LC, Dhanji S, Snow JW, Priatel JJ, Ma MC, Miners MJ, Teh HS, Goldsmith MA, Abraham N. Impaired CD8 T cell memory and CD4 T cell primary responses in IL-7R alpha mutant mice. J Exp Med. 2007;204:619–631. doi: 10.1084/jem.20061871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pacheco R, Gallart T, Lluis C, Franco R. Role of glutamate on T-cell mediated immunity. J Neuroimmunol. 2007;185:9–19. doi: 10.1016/j.jneuroim.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 68.Pallard C, Stegmann AP, van Kleffens T, Smart F, Venkitaraman A, Spits H. Distinct roles of the phosphatidylinositol 3-kinase and STAT5 pathways in IL-7-mediated development of human thymocyte precursors. Immunity. 1999;10:525–535. doi: 10.1016/s1074-7613(00)80052-7. [DOI] [PubMed] [Google Scholar]
- 69.Park JH, Yu Q, Erman B, Appelbaum JS, Montoya-Durango D, Grimes HL, Singer A. Suppression of IL7Ralpha transcription by IL-7 and other prosurvival cytokines: a novel mechanism for maximizing IL-7-dependent T cell survival. Immunity. 2004;21:289–302. doi: 10.1016/j.immuni.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 70.Pellegrini M, Bouillet P, Robati M, Belz GT, Davey GM, Strasser A. Loss of Bim increases T cell production and function in interleukin 7 receptor-deficient mice. J Exp Med. 2004;200:1189–1195. doi: 10.1084/jem.20041328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peschon JJ, Morrissey PJ, Grabstein KH, Ramsdell FJ, Maraskovsky E, Gliniak BC, Park LS, Ziegler SF, Williams DE, Ware CB, et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J Exp Med. 1994;180:1955–1960. doi: 10.1084/jem.180.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Porter BO, Scibelli P, Malek TR. Control of T cell development in vivo by subdomains within the IL-7 receptor alpha-chain cytoplasmic tail. J Immunol. 2001;166:262–269. doi: 10.4049/jimmunol.166.1.262. [DOI] [PubMed] [Google Scholar]
- 73.Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R expression in T(−)B(+)NK(+) severe combined immunodeficiency. Nat Genet. 1998;20:394–397. doi: 10.1038/3877. [DOI] [PubMed] [Google Scholar]
- 74.Rajnavolgyi E, Benbernou N, Rethi B, Reynolds D, Young HA, Magocsi M, Muegge K, Durum SK. IL-7 withdrawal induces a stress pathway activating p38 and Jun N-terminal kinases. Cell Signal. 2002;14:761–769. doi: 10.1016/s0898-6568(02)00026-8. [DOI] [PubMed] [Google Scholar]
- 75.Rathmell JC, Farkash EA, Gao W, Thompson CB. IL-7 enhances the survival and maintains the size of naive T cells. J Immunol. 2001;167:6869–6876. doi: 10.4049/jimmunol.167.12.6869. [DOI] [PubMed] [Google Scholar]
- 76.Rich BE, Leder P. Transgenic expression of interleukin 7 restores T cell populations in nude mice. J Exp Med. 1995;181:1223–1228. doi: 10.1084/jem.181.3.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schluns KS, Kieper WC, Jameson SC, Lefrancois L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol. 2000;1:426–432. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 78.Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–262. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 79.Sexl V, Diehl JA, Sherr CJ, Ashmun R, Beach D, Roussel MF. A rate limiting function of cdc25A for S phase entry inversely correlates with tyrosine dephosphorylation of Cdk2. Oncogene. 1999;18:573–582. doi: 10.1038/sj.onc.1202362. [DOI] [PubMed] [Google Scholar]
- 80.Sharfe N, Dadi HK, Roifman CM. JAK3 protein tyrosine kinase mediates interleukin-7-induced activation of phosphatidylinositol-3′ kinase. Blood. 1995;86:2077–2085. [PubMed] [Google Scholar]
- 81.Shelton JG, Steelman LS, Lee JT, Knapp SL, Blalock WL, Moye PW, Franklin RA, Pohnert SC, Mirza AM, McMahon M, McCubrey JA. Effects of the RAF/MEK/ERK and PI3K/AKT signal transduction pathways on the abrogation of cytokine-dependence and prevention of apoptosis in hematopoietic cells. Oncogene. 2003;22:2478–2492. doi: 10.1038/sj.onc.1206321. [DOI] [PubMed] [Google Scholar]
- 82.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 83.Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, Medema RH. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. 2002;168:5024–5031. doi: 10.4049/jimmunol.168.10.5024. [DOI] [PubMed] [Google Scholar]
- 84.Surh CD, Boyman O, Purton JF, Sprent J. Homeostasis of memory T cells. Immunol Rev. 2006;211:154–163. doi: 10.1111/j.0105-2896.2006.00401.x. [DOI] [PubMed] [Google Scholar]
- 85.Suzuki K, Nakajima H, Saito Y, Saito T, Leonard WJ, Iwamoto I. Janus kinase 3 (Jak3) is essential for common cytokine receptor gamma chain (gamma(c))-dependent signaling: comparative analysis of gamma(c), Jak3, and gamma(c) and Jak3 double-deficient mice. Int Immunol. 2000;12:123–132. doi: 10.1093/intimm/12.2.123. [DOI] [PubMed] [Google Scholar]
- 86.Swainson L, Kinet S, Manel N, Battini JL, Sitbon M, Taylor N. Glucose transporter 1 expression identifies a population of cycling CD4+ CD8+ human thymocytes with high CXCR4-induced chemotaxis. Proc Natl Acad Sci USA. 2005;102:12867–12872. doi: 10.1073/pnas.0503603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Swainson L, Kinet S, Mongellaz C, Sourisseau M, Henriques T, Taylor N. IL-7-induced proliferation of recent thymic emigrants requires activation of the PI3K pathway. Blood. 2007;109:1034–1042. doi: 10.1182/blood-2006-06-027912. [DOI] [PubMed] [Google Scholar]
- 88.Swainson L, Verhoeyen E, Cosset FL, Taylor N. IL-7R alpha gene expression is inversely correlated with cell cycle progression in IL-7-stimulated T lymphocytes. J Immunol. 2006;176:6702–6708. doi: 10.4049/jimmunol.176.11.6702. [DOI] [PubMed] [Google Scholar]
- 89.Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, Surh CD. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci U S A. 2001;98:8732–8737. doi: 10.1073/pnas.161126098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tanner JW, Chen W, Young RL, Longmore GD, Shaw AS. The conserved box 1 motif of cytokine receptors is required for association with JAK kinases. J Biol Chem. 1995;270:6523–6530. doi: 10.1074/jbc.270.12.6523. [DOI] [PubMed] [Google Scholar]
- 91.Vander HM, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol Cell Biol. 2001;21:5899–5912. doi: 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res. 1999;253:239–254. doi: 10.1006/excr.1999.4701. [DOI] [PubMed] [Google Scholar]
- 93.Vidal A, Koff A. Cell-cycle inhibitors: three families united by a common cause. Gene. 2000;247:1–15. doi: 10.1016/s0378-1119(00)00092-5. [DOI] [PubMed] [Google Scholar]
- 94.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 95.von-Freeden-Jeffry U, Vieira P, Lucian LA, McNeil T, Burdach SE, Murray R. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J Exp Med. 1995;181:1519–1526. doi: 10.1084/jem.181.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wallace DC. Mitochondria and cancer: Warburg addressed. Cold Spring Harb Symp Quant Biol. 2005;70:363–374. doi: 10.1101/sqb.2005.70.035. [DOI] [PubMed] [Google Scholar]
- 97.Wells WA. Does size matter? J Cell Biol. 2002;158:1156–1159. doi: 10.1083/jcb.200209042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. 2007;18:1437–1446. doi: 10.1091/mbc.E06-07-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, Li D, Durum SK, Jiang Q, Bhandoola A, Hennighausen L, O’Shea JJ. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci USA. 2006;103:1000–1005. doi: 10.1073/pnas.0507350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yu CR, Young HA, Ortaldo JR. Characterization of cytokine differential induction of STAT complexes in primary human T and NK cells. J Leukoc Biol. 1998;64:245–258. doi: 10.1002/jlb.64.2.245. [DOI] [PubMed] [Google Scholar]
- 101.Ziegler SE, Morella KK, Anderson D, Kumaki N, Leonard WJ, Cosman D, Baumann H. Reconstitution of a functional interleukin (IL)-7 receptor demonstrates that the IL-2 receptor gamma chain is required for IL-7 signal transduction. Eur J Immunol. 1995;25:399–404. doi: 10.1002/eji.1830250214. [DOI] [PubMed] [Google Scholar]