

Summary
Since the structure determination of bacteriorhodopsin in 1990, much progress has been made in the further development and use of electron crystallography. In this review, we provide a concise overview of the new developments in electron crystallography concerning 2D crystallization, data collection and data processing. Based on electron crystallographic work on bacteriorhodopsin, the acetylcholine receptor and aquaporins, we highlight the unique advantages and future perspectives of electron crystallography for the structural study of membrane proteins. These advantages include the visualization of membrane proteins in their native environment without detergent-induced artifacts, the trapping of different states in a reaction pathway by time-resolved experiments, the study of non-specific protein-lipid interactions and the characterization of the charge state of individual residues in membrane proteins.
The structures
Electron crystallography began with Henderson and Unwin’s groundbreaking work on the structure of purple membrane. Introducing glucose embedding as a new specimen preparation method and electron crystallographic data processing, these pioneers could produce a density map of bacteriorhodopsin (bR) at 7Å resolution [1,2]. The map resolved for the first time membrane-spanning α-helices and provided the first view of the structural organization of a membrane protein. After improvements in specimen preparation, data collection and data processing, Henderson and co-workers could finally present an atomic model of bR, the first structure solved by electron crystallography [3]. Since then, a higher resolution map of bR was obtained [4] and the structures of six more membrane proteins were solved by electron crystallography: plant light-harvesting complex 2 [5], aquaporin-1 [6,7], torpedo ray nicotinic acetylcholine receptor [8,9], sheep aquaporin-0 [10,11●●], rat aquaporin-4 (solved using recombinant protein expressed in insect cells) [12●●] and rat microsomal glutathione transferase 1 [13●].
A game of numbers
The number of solved structures
Seven structures may appear a modest accomplishment compared to the many more membrane protein structures that have been determined by x-ray crystallography over the past few years. Considering, however, that less than two dozen groups are currently pursuing electron crystallography, compared to the hundreds of x-ray groups, the seven membrane protein structures determined by electron crystallography compare quite favorably to those determined by x-ray crystallography. More importantly, however, unlike x-ray crystallography, electron crystallography is not an “all or nothing” approach, and structural information can be extracted even from poorly ordered 2D crystals. Electron crystallography has thus contributed structural information of many membrane proteins in the 10Å range, providing valuable information on the arrangement of the transmembrane α-helices in these proteins (e.g., [14–16]).
The time it takes to solve a structure
Structure determination by electron crystallography used to be a tedious and time-consuming task as it took not only a long time to produce high quality 2D crystals but also years to collect and process the data. Over the past years the methodology has seen major advances. While the best 2D crystals are still being produced by dialysis [17], new ways of producing 2D crystals of membrane proteins have been introduced [18–20]. Still lacking are 2D crystallization screens as those available for 3D crystallization, but efforts to establish such screens are currently underway. Specimen preparation is one of the most crucial steps to obtain good images of 2D crystals, which has to be optimized for each specimen. Nevertheless, the development of the carbon sandwich specimen preparation technique [21] has dramatically increased the yield of good images taken at high tilt angles from a number of 2D crystals. The development of an electron microscope equipped with a helium-cooled top-entry specimen stage by Fujiyoshi and co-workers [22] has made it much easier not only to collect high quality images but also to quickly exchange EM grids until a suitable specimen has been obtained for data collection. The programs developed at the MRC in Cambridge (U.K.) have been the software of choice to process electron crystallographic data to date [23]. While versatile and powerful, the MRC programs are not nearly as user-friendly as the software packages used in x-ray crystallography. Recently the 2dx package has been released, which introduces a user-friendly graphical interface for the MRC programs and represents a further step towards automation of the data processing [24●]. In Europe, the Network of Excellence for three-dimensional electron microscopy is reevaluating and advancing the methodology in electron microscopy, and has also already contributed new concepts and software tools in electron crystallography (e.g., [25,26●,27]). In addition, established x-ray crystallography approaches are now being adapted to electron crystallography. Recent examples are the determination of the AQP0 and AQP4 structures by molecular replacement [10,11●●,12●●]. Phase extension by density modification calculations is another example of an x-ray technique that could be adapted to electron crystallography. The application of single particle averaging techniques to unit cells extracted from poorly ordered 2D crystals (e.g., [28–30]) is yet another under-developed approach that could enhance the use of 2D crystals.
The achievable resolution
Compared to x-ray crystallography, electron crystallography used to be a “low-resolution” technique, as the resolution of density maps were typically limited to ~3.5Å. Recently, the structure of the aquaporin-0 mediated membrane junction was determined to 1.9Å resolution, revealing for the first time water molecules in an electron crystallographic density map [11●●]. Electron diffraction patterns showing a comparable resolution have now also been recorded from purple membranes and 2D crystals of two aquaporins (Yoshinori Fujiyoshi and Andreas Engel, personal communication), solidifying electron crystallography as a method that under favorable conditions can produce structures at resolutions comparable to those obtained by x-ray crystallography.
Advantages of electron crystallography for the study of membrane proteins
The structure of membrane proteins in their native environment
Detergent micelles are an appropriate but imperfect substitution for the lipids surrounding membrane proteins in situ. Crystal structures of membrane proteins in detergent micelles thus always carry a certain risk not to represent the native structure of the protein. This risk is avoided with 2D crystals, in which the protein is embedded in a lipid bilayer. Density maps obtained with 2D crystals, even at low resolution, can thus be used to critically assess differences in the structures that membrane proteins adopt in detergent micelles and lipid bilayers. In the case of the multidrug transporter EmrE, for example, the electron crystallographic structure, which is likely to represent a biologically relevant state, is different from two x-ray structures, which seem to represent non-native states of the protein [31]. It can thus be interesting to visualize membrane proteins by electron crystallography even if crystal structures already exist. The most intriguing example are voltage-gated ion channels [32–34], in which case it would be of great interest to visualize the orientation of the voltage sensors within the lipid bilayer. Another example are the rhomboid family of intramembrane proteases [35,36], in which case it would be interesting to see the disposition of the predominantly α-helical L1 loop in the context of the membrane, as the crystal structures show that it extends parallel to the membrane and is embedded only in the extracellular leaflet of the lipid bilayer.
The difference between a detergent micelle and a lipid bilayer can also affect the behavior of a membrane protein, as illustrated by the junction-forming interactions between AQP0 tetramers. In double-layered 2D crystals the two AQP0 tetramers in the adjoining membranes were exactly aligned (Fig. 1a) [10,11●●], whereas in loosely packed 3D crystals the paired AQP0 tetramers were rotated by 24° with respect to each other (Fig. 1b) [37]. The interaction observed in the 3D crystals ought to be the preferred interaction, in which unrestricted AQP0 tetramers engage, but since the thin junctions between fiber cells are formed by square arrays of AQP0 [38], the interaction observed in the double-layered 2D crystals is presumably the one used by AQP0 in vivo. Interestingly, two other aquaporins also formed double-layered 2D crystals, AQP4 (Fig. 1c) which was subsequently localized to both non-junctional and junctional membrane areas in thin sections through the hypothalamus [12●●] and plant aquaporin SoPIP2;1 [15].
Figure 1.

Membrane junctions formed by aquaporins-0 and 4. (a) Structure of the aquaporin-0 mediated membrane junction visualized by electron crystallography of double-layered 2D crystals (pdb accession code: 2B6O) [10,11●●]. The paired tetramers are exactly in register. (b) Paired aquaporin-0 tetramers as seen in loosely packed 3D crystals (pdb accession code: 2C32) [37]. The paired tetramers are rotated by 24° with respect to each other. (c) Structure of the aquaporin-4 mediated membrane junction visualized by electron crystallography of double-layered 2D crystals (pdb accession code: 2D57) [12●●].
Functional and time-resolved studies on 2D crystals
To test for their activity, membrane proteins are often reconstituted into liposomes. Since vesicular 2D crystals are essentially liposomes that contain regularly packed protein, they can be used to perform functional assays. This was done, for example, to establish that AQP1 in 2D crystals retained its biological activity [39]. Time-resolved electron crystallography studies take functional analyses of membrane proteins much further. The basis of such experiments is to induce a conformational change and to trap the protein in its new state by quick-freezing the 2D crystals in liquid ethane for structural analysis by electron crystallography. By carefully timing the freezing after induction of the reaction, various intermediate states in a reaction pathway can be trapped. An impressive example is the time-resolved electron crystallographic investigation of the bR photocycle by Henderson and co-workers [40]. The conformational changes in the membrane-spanning α-helices upon light-induced unbending of the retinal were, however, eventually elucidated using mutant bR, in which the full extent of the light-driven conformational change is present with or without illumination (Fig. 2) [41]. Elegant time-resolved studies by Unwin and co-workers on tubular crystals of the acetylcholine receptor provided unique insights into the mechanism of gate opening upon binding of acetycholine to the receptor (Fig. 3) [8]. The open state of the channel was captured by spraying acetylcholine on the tubular crystals and quick-freezing the samples within less than 5 ms [42]. By increasing the time between spraying and freezing, the same approach should also eventually yield the structure of the desensitized state. In contrast to tubular crystals such as those formed by the acetylcholine receptor, which only provide access to the outside of the tube but not to its lumen, sheet-like 2D crystals provide access to both sides of the crystallized protein. The most recent example of functional studies on 2D crystals, although much more preliminary than those on the acetylcholine receptor, is the application of electron crystallography to visualize the pH-dependent conformational change that occurs in the Na+/H+ antiporter from Methanococcus jannaschii [43]. The projection maps of the protein at low and high pH presumably represent the closed and open states of the antiporter.
Figure 2.
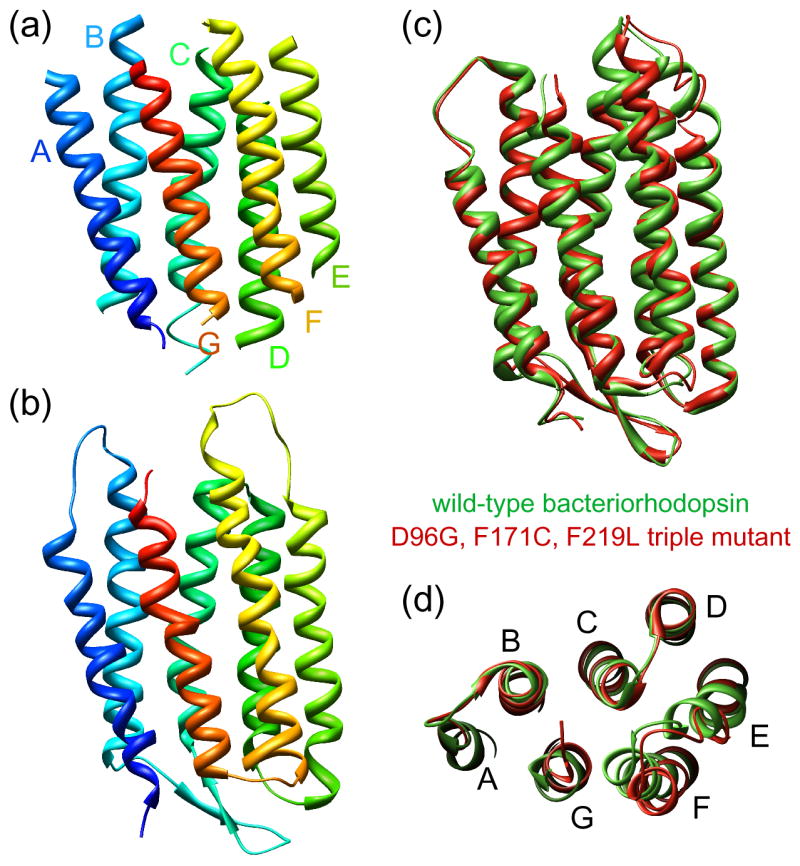
Structural basis of vectorial proton translocation by bacteriorhodopsin. (a) The bR structure determined by Henderson and co-workers, which was the first atomic model produced by electron crystallography (pdb accession code: 1BRD) [3]. (b) 3Å resolution structure of bR determined by Fujiyoshi and co-workers (pdb accession code: 2AT9). (c,d) Superposition of wild-type bacteriorhodopsin (green; pdb accession code: 1FBB) and the D96G, F171C, F219L triple mutant (red; pdb accession code: 1FBK), which always shows the full extent of the light-driven conformational change, illustrating differences in the cytoplasmic portions of helices F and G [41].
Figure 3.
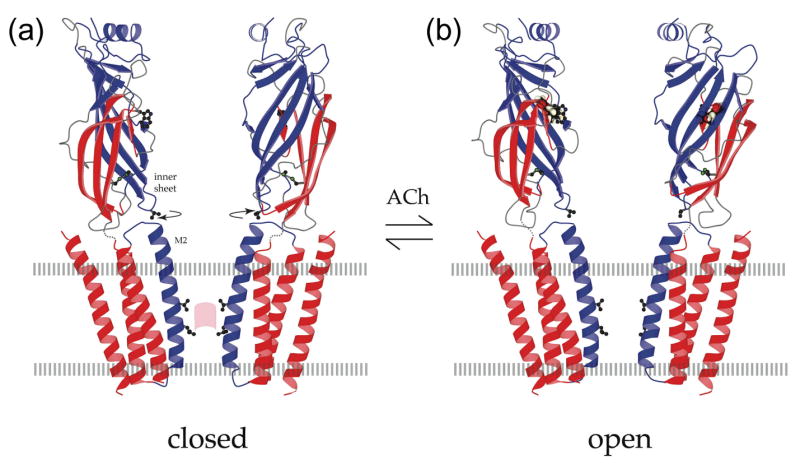
Open and closed states of the acetylcholine receptor. (a) Atomic model of the acetylcholine receptor in the closed conformation based on a 4Å density map [9]. (b) Binding of acetylcholine opens the channel by initiating rotational movements (arrows) of the inner β-sheets of the α subunits in the ligand-binding domain. These movements are communicated to the inner (M2) helices lining the pore and break apart the gate – a hydrophobic girdle in the middle of the membrane – so that ions can flow through. A tryptophan side chain in the ligand-binding domain identifies the acetylcholine-binding region; a valine side chain links the inner sheet to the inner helix; leucine and valine side chains on the inner helices make the gate (pink patch); the locations of the membrane surfaces are indicated by broken lines; the relevant moving parts are in blue. Figure reprinted by permission of Federation of the European Biochemical Societies from “Structure and action of the nicotinic acetylcholine receptor explored by electron microscopy” by Nigel Unwin, FEBS Letters, 555, 91–95, 2003.
Lipid-protein interactions
X-ray structures of membrane proteins occasionally contain lipid molecules (reviewed in [44–46]). Most notably, the structure of bR from lipid cubic phase crystallization revealed 13 phytanyl lipids, seven of which formed a bilayer structure, and a squalene [47]. 10 lipids were also included in the refinement of the electron crystallographic structure of bR [48]. These, and all lipids resolved in crystal structures so far, originate from the native membrane. The electron crystallographic structure of the AQP0 membrane junction revealed nine lipid molecules per monomer [11●●]. These lipids were not co-purified from the native membrane but dimyristoyl phosphatidyl choline (DMPC) molecules, the artificial lipid used for 2D crystallization. Since DMPC is not present in native lens membranes, the structure revealed the non-specific interactions of a membrane protein with the surrounding lipids in the bilayer (Fig. 4a). In addition to the seven annular lipids that form a sheath protecting the hydrophobic surfaces of the protein, the density map also revealed partial density for two lipids not in contact with the protein, thus representing bulk lipids (Fig. 4b). The density map showed branched densities for several of the annular lipids’ acyl chains (unpublished data). The data set was only sufficient, however, to model the predominant conformation of the individual lipids. By including more electron diffraction patterns in the data set, it should be possible in the future to also model the alternative conformations of the lipids and to determine the relative occupancies of the different conformations. It may thus be possible to directly visualize the conformational dynamics of bilayer lipids in the presence of a membrane protein. Furthermore, 2D crystals of aquaporins can form with a variety of different lipids. Highly ordered 2D crystals of aquaporin-1 formed with E. coli polar lipids [6], DMPC [49] and dioleoyl phosphatidyl choline [7]. By reconstituting AQP0 into 2D crystals composed of lipids with different acyl chains and different headgroups, it may thus be possible to study how different lipids interact non-specifically with a membrane protein and to understand the influence of a membrane protein on the structure and dynamics of lipids in the surrounding bilayer.
Figure 4.
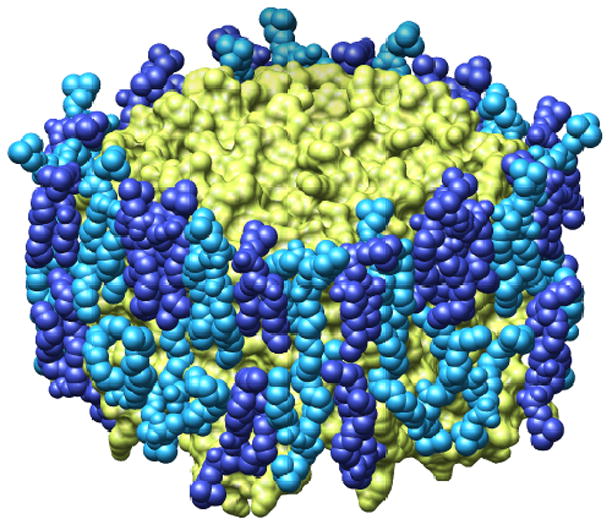
Lipids surrounding an aquaporin-0. (a) Space-filling model of the lipid bilayer surrounding an aquaporin-0 tetramer; the protein is shown in yellow and the lipids in two shades of blue. (b) The nine lipids surrounding an aquaporin-0 monomer; the protein is shown as ribbon diagram and the lipids as ball-and-stick models (adapted from [11●●]).
Visualizing the charge of atoms
Unlike x-rays, electrons carry a negative charge and electron scattering is thus affected by the charge state of the scattering atoms in the specimen. Especially in the resolution range below 5Å, atomic scattering factors for electrons are very different for neutral and charged atoms. By comparing differences in density maps calculated with and without the low-resolution information, Fujiyoshi and co-workers saw effects in the 3Å density map of bR that may reflect the charge state of individual residues in the protein [4,50]. While more work will be needed to fully understand the true potential, the initial results with bR illustrate that it might be possible to use electron crystallography to visualize the charge state of individual residues in a protein. In combination with time-resolved studies, it might thus be possible one day to directly follow the path of the proton in bR as it is pumped through the membrane.
Conclusions
Structures of membrane proteins in different functional states, structures at resolutions comparable to those of x-ray crystal structures, unresolved questions concerning membrane protein structures determined by x-ray crystallography, and the potential to study non-specific protein-lipid interactions and the charge state of individual amino acid residues in membrane proteins all contribute to the renewed interest in electron crystallography. The community continues to advance the methodology, and the First Electron Crystallography Workshop at the University of California Davis (http://2dx.org/workshop) and the upcoming Special Issue of the Journal of Structural Biology on electron crystallography are signs of the new momentum gained by electron crystallography.
Acknowledgments
The authors wish to thank Andreas Engel, Yoshinori Fujiyoshi, Henning Stahlberg and members of the Walz group for stimulating discussions and critical reading of the manuscript. Electron crystallographic work on membrane proteins in the Walz laboratory is supported by NIH grants P01 GM062580 and R01 EY015107.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
● of special interest
●● of outstanding interest
- 1.Unwin PN, Henderson R. Molecular structure determination by electron microscopy of unstained crystalline specimens. J Mol Biol. 1975;94:425–440. doi: 10.1016/0022-2836(75)90212-0. [DOI] [PubMed] [Google Scholar]
- 2.Henderson R, Unwin PN. Three-dimensional model of purple membrane obtained by electron microscopy. Nature. 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- 3.Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J Mol Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- 4.Kimura Y, Vassylyev DG, Miyazawa A, Kidera A, Matsushima M, Mitsuoka K, Murata K, Hirai T, Fujiyoshi Y. Surface of bacteriorhodopsin revealed by high-resolution electron crystallography. Nature. 1997;389:206–211. doi: 10.1038/38323. [DOI] [PubMed] [Google Scholar]
- 5.Kuhlbrandt W, Wang DN, Fujiyoshi Y. Atomic model of plant light-harvesting complex by electron crystallography. Nature. 1994;367:614–621. doi: 10.1038/367614a0. [DOI] [PubMed] [Google Scholar]
- 6.Murata K, Mitsuoka K, Hirai T, Walz T, Agre P, Heymann JB, Engel A, Fujiyoshi Y. Structural determinants of water permeation through aquaporin-1. Nature. 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- 7.Ren G, Reddy VS, Cheng A, Melnyk P, Mitra AK. Visualization of a water-selective pore by electron crystallography in vitreous ice. Proc Natl Acad Sci USA. 2001;98:1398–1403. doi: 10.1073/pnas.041489198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyazawa A, Fujiyoshi Y, Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- 9.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4Å resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 10.Gonen T, Sliz P, Kistler J, Cheng Y, Walz T. Aquaporin-0 membrane junctions reveal the structure of a closed water pore. Nature. 2004a;429:193–197. doi: 10.1038/nature02503. [DOI] [PubMed] [Google Scholar]
- 11.Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. Lipid-protein interactions in double-layered two-dimensional crystals of aquaporin-0. Nature. 2005;438:633–638. doi: 10.1038/nature04321. ●● The 1.9Å resolution structure of aquaporin-0 is the highest resolution density map determined by electron crystallography and revealed water molecules in the channel as well as nine lipid molecules per monomer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hiroaki Y, Kazutoshi T, Kamegawa A, Gyobo N, Nishikawa K, Suzuki H, Walz T, Sasaki S, Mitsuoka K, Kimura K, et al. Implications of the aquaporin-4 structure on array formation and cell adhesion. J Mol Biol. 2006;355:628–639. doi: 10.1016/j.jmb.2005.10.081. ●● The aquaporin-4 structure is the first structure of a multi-span mammalian membrane protein determined using recombinant protein and showed that aquaporin-4 can also form membrane junctions. [DOI] [PubMed] [Google Scholar]
- 13.Holm PJ, Bhakat P, Jegerschold C, Gyobu N, Mitsuoka K, Fujiyoshi Y, Morgenstern R, Hebert H. Structural basis for detoxification and oxidative stress protection in membranes. J Mol Biol. 2006;360:934–945. doi: 10.1016/j.jmb.2006.05.056. ●The structure of rat microsomal glutathione transferase is the most recent structure determined by electron crystallography and showed that the glutathione binding site is different frrom that of the canonical soluble glutathione transferases. [DOI] [PubMed] [Google Scholar]
- 14.Breyton C, Haase W, Rapoport TA, Kuhlbrandt W, Collinson I. Three-dimensional structure of the bacterial protein-translocation complex SecYEG. Nature. 2002;418:662–665. doi: 10.1038/nature00827. [DOI] [PubMed] [Google Scholar]
- 15.Kukulski W, Schenk AD, Johanson U, Braun T, de Groot BL, Fotiadis D, Kjellbom P, Engel A. The 5Å structure of heterologously expressed plant aquaporin SoPIP2;1. J Mol Biol. 2005;350:611–616. doi: 10.1016/j.jmb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Min G, Wang H, Sun TT, Kong XP. Structural basis for tetraspanin functions as revealed by the cryo-EM structure of uroplakin complexes at 6-Å resolution. J Cell Biol. 2006;173:975–983. doi: 10.1083/jcb.200602086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuhlbrandt W. Two-dimensional crystallization of membrane proteins. Q Rev Biophys. 1992;25:1–49. doi: 10.1017/s0033583500004716. [DOI] [PubMed] [Google Scholar]
- 18.Levy D, Chami M, Rigaud JL. Two-dimensional crystallization of membrane proteins: the lipid layer strategy. FEBS Lett. 2001;504:187–193. doi: 10.1016/s0014-5793(01)02748-x. [DOI] [PubMed] [Google Scholar]
- 19.Remigy HW, Caujolle-Bert D, Suda K, Schenk A, Chami M, Engel A. Membrane protein reconstitution and crystallization by controlled dilution. FEBS Lett. 2003;555:160–169. doi: 10.1016/s0014-5793(03)01105-0. [DOI] [PubMed] [Google Scholar]
- 20.Signorell GA, Kaufmann TC, Kukulski W, Engel A, Remigy HW. Controlled 2D crystallization of membrane proteins using methyl-beta-cyclodextrin. J Struct Biol. 2007 doi: 10.1016/j.jsb.2006.07.011. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 21.Gyobu N, Tani K, Hiroaki Y, Kamegawa A, Mitsuoka K, Fujiyoshi Y. Improved specimen preparation for cryo-electron microscopy using a symmetric carbon sandwich technique. J Struct Biol. 2004;146:325–333. doi: 10.1016/j.jsb.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 22.Fujiyoshi Y, Mizusaki T, Morikawa K, Yamagishi H, Aoki Y, Kihara H, Harada Y. Development of a superfluid helium stage for high-resolution electron microscopy. Ultramicroscopy. 1991;38:241–251. [Google Scholar]
- 23.Crowther RA, Henderson R, Smith JM. MRC image processing programs. J Struct Biol. 1996;116:9–16. doi: 10.1006/jsbi.1996.0003. [DOI] [PubMed] [Google Scholar]
- 24.Gipson B, Zeng X, Zhang ZY, Stahlberg H. 2dx - user-friendly image processing for 2D crystals. J Struct Biol. 2007;157:64–72. doi: 10.1016/j.jsb.2006.07.020. ● This paper introduces 2dx, a new user-friendly and platform-independent software package for electron crystallographic data processing. [DOI] [PubMed] [Google Scholar]
- 25.Gil D, Carazo JM, Marabini R. On the nature of 2D crystal unbending. J Struct Biol. 2006;156:546–555. doi: 10.1016/j.jsb.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 26.Philippsen A, Schenk AD, Signorell GA, Mariani V, Berneche S, Engel A. Collaborative EM image processing with the IPLT image processing library and toolbox. J Struct Biol. 2007;157:28–37. doi: 10.1016/j.jsb.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 27.Philippsen A, Engel HA, Engel A. The contrast-imaging function for tilted specimens. Ultramicroscopy. 2007;107:202–212. doi: 10.1016/j.ultramic.2006.07.010. ● This paper introduces the Image Processing Library and Toolbox (IPLT), a flexible software architecture that offers an array of functions designed for electron crystallography image processing, and supports users to implement their own algorithms as processing code. [DOI] [PubMed] [Google Scholar]
- 28.Stahlberg H, Dubochet J, Vogel H, Ghosh R. Are the light-harvesting I complexes from Rhodospirillum rubrum arranged around the reaction centre in a square geometry? J Mol Biol. 1998;282:819–831. doi: 10.1006/jmbi.1998.1975. [DOI] [PubMed] [Google Scholar]
- 29.Walz T, Jamieson SJ, Bowers CM, Bullough PA, Hunter CN. Projection structures of three photosynthetic complexes from Rhodobacter sphaeroides: LH2 at 6 Å, LH1 and RC-LH1 at 25 Å. J Mol Biol. 1998;282:833–845. doi: 10.1006/jmbi.1998.2050. [DOI] [PubMed] [Google Scholar]
- 30.Tahara Y, Oshima A, Hirai T, Mitsuoka K, Fujiyoshi Y, Hayashi Y. The 11 A resolution projection map of Na+/K+-ATPase calculated by application of single particle analysis to two-dimensional crystal images. J Electron Microsc. 2000;49:583–587. doi: 10.1093/oxfordjournals.jmicro.a023847. [DOI] [PubMed] [Google Scholar]
- 31.Tate CG. Comparison of three structures of the multidrug transporter EmrE. Curr Opin Struct Biol. 2006;16:457–464. doi: 10.1016/j.sbi.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 32.Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 33.Long SB, Campbell EB, MacKinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005a;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 34.Long SB, Campbell EB, MacKinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005b;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444:179–180. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- 36.Wu Z, Yan N, Feng L, Oberstein A, Yan H, Baker RP, Gu L, Jeffrey PD, Urban S, Shi Y. Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat Struct Mol Biol. 2006;13:1084–1091. doi: 10.1038/nsmb1179. [DOI] [PubMed] [Google Scholar]
- 37.Palanivelu DVEKD, Engel A, Suda K, Lustig A, Agre P, Schirmer T. Co-axial association of recombinant eye lens aquaporin-0 observed in loosely packed 3D crystals. J Mol Biol. 2006;355:605–611. doi: 10.1016/j.jmb.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 38.Zampighi G, Simon SA, Robertson JD, McIntosh TJ, Costello MJ. On the structural organization of isolated bovine lens fiber junctions. J Cell Biol. 1982;93:175–189. doi: 10.1083/jcb.93.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walz T, Smith BL, Zeidel ML, Engel A, Agre P. Biologically active two-dimensional crystals of aquaporin CHIP. J Biol Chem. 1994;269:1583–1586. [PubMed] [Google Scholar]
- 40.Subramaniam S, Lindahl M, Bullough P, Faruqi AR, Tittor J, Oesterhelt D, Brown L, Lanyi J, Henderson R. Protein conformational changes in the bacteriorhodopsin photocycle. J Mol Biol. 1999;287:145–161. doi: 10.1006/jmbi.1999.2589. [DOI] [PubMed] [Google Scholar]
- 41.Subramaniam S, Henderson R. Molecular mechanism of vectorial proton translocation by bacteriorhodopsin. Nature. 2000;406:653–657. doi: 10.1038/35020614. [DOI] [PubMed] [Google Scholar]
- 42.Unwin N. Acetylcholine receptor channel imaged in the open state. Nature. 1995;373:37–43. doi: 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- 43.Vinothkumar KR, Smits SH, Kuhlbrandt W. pH-induced structural change in a sodium/proton antiporter from Methanococcus jannaschii. EMBO J. 2005;24:2720–2729. doi: 10.1038/sj.emboj.7600727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palsdottir H, Hunte C. Lipids in membrane protein structures. Biochim Biophys Acta. 2004;1666:2–18. doi: 10.1016/j.bbamem.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 45.Hunte C. Specific protein-lipid interactions in membrane proteins. Biochem Soc Trans. 2005;33:938–942. doi: 10.1042/BST20050938. [DOI] [PubMed] [Google Scholar]
- 46.Wiener M. How proteins shape lipids. In: Tamm LK, editor. Protein-lipid interactions: from membrane domains to cellular networks. WILEY-VCH: Verlag GmbH & Co; 2005. [Google Scholar]
- 47.Luecke H, Schobert B, Richter HT, Cartailler JP, Lanyi JK. Structure of bacteriorhodopsin at 1.55 Å resolution. J Mol Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 48.Grigorieff N, Ceska TA, Downing KH, Baldwin JM, Henderson R. Electron-crystallographic refinement of the structure of bacteriorhodopsin. J Mol Biol. 1996;259:393–421. doi: 10.1006/jmbi.1996.0328. [DOI] [PubMed] [Google Scholar]
- 49.Jap BK, Li H. Structure of the osmo-regulated H2O-channel, AQP-CHIP, in projection at 3.5 Å resolution. J Mol Biol. 1995;251:413–420. doi: 10.1006/jmbi.1995.0444. [DOI] [PubMed] [Google Scholar]
- 50.Mitsuoka K, Hirai T, Murata K, Miyazawa A, Kidera A, Kimura Y, Fujiyoshi Y. The structure of bacteriorhodopsin at 3.0 Å resolution based on electron crystallography: implication of the charge distribution. J Mol Biol. 1999;286:861–882. doi: 10.1006/jmbi.1998.2529. [DOI] [PubMed] [Google Scholar]
- 51.Unwin N. Structure and action of the nicotinic acetylcholine receptor explored by electron microscopy. FEBS Lett. 2003;555:91–95. doi: 10.1016/s0014-5793(03)01084-6. [DOI] [PubMed] [Google Scholar]