

Abstract
Bacterial tRNAs frequently have 4-thiouridine (s4U) modification at position 8, which is adjacent to the C13-G22-m7G46 base triple in the elbow region of the tRNA tertiary structure. Irradiation with light in the UVA range induces an efficient photocrosslink between s4U8 and C13. The temperature dependence of the rate constants for photocrosslinking between the s4U8 and C13 has been used to investigate the tRNA conformational energy and structure in Escherichia coli tRNAVal, tRNAPhe, and tRNAfMet under different conditions. Corrections have been made in the measured rate constants to compensate for differences in the excited state lifetimes due to tRNA identity, buffer conditions, and temperature. The resulting rate constants are related to the rate at which the s4U8 and C13 come into the alignment needed for photoreaction; this depends on an activation energy, attributable to the conformational potential energy that occurs during the photoreaction, and on the extent of the structural change. Different photocrosslinking rate constants and temperature dependencies occur in the three tRNAs, and these differences are due both to modest differences in the activation energies and in the apparent s4U8–C13 geometries. Analysis of tRNAVal in buffers without Mg2+ indicate a smaller activation energy (∼13 kJ mol−1) and a larger apparent s4U8–C13 distance (∼12 Å) compared to values for the same parameters in buffers with Mg2+ (∼26 kJ mol−1 and 0.36 Å, respectively). These measurements are a quantitative indication of the strong constraint that Mg2+ imposes on the tRNA flexibility and structure.
Keywords: RNA tertiary structure, photocrosslinking rates, activation energy, conformational energy, T1 excited state lifetimes
INTRODUCTION
The tRNA L-shaped tertiary structure is critical to many of its biological functions and there has been a continuing interest in the tRNA structure in translation (Yarus and Smith 1995; Yarus et al. 2003; Ogle and Ramakrishnan 2005). The connection between divalent ions and the stabilization of the tRNA tertiary structure has been addressed by several approaches. Binding analysis indicates that at least one Mg2+ in tRNAfMet (Cohn et al. 1969; Stein and Crothers 1976a,b) or two to three Mg2+ in tRNAPhe (Lynch and Schimmel 1974; Römer and Hach 1975) are bound at high affinity sites and are associated with formation of the tertiary structure. The locations for the strong binding sites for Mg2+ are suggested by the crystal structure; two likely locations are at a site between the D loop (at nucleotides 19–20) and the T loop (at nucleotides 59–60), and at a site in the sharp turn around nucleotides 8–12 between the 5′ strand of the acceptor stem and the 5′ strand of the D stem (Jack et al. 1977; Holbrook et al. 1977; Hingerty et al. 1978). Hydrodynamic experiments indicate an overall more extended shape in the tRNA at magnesium concentrations below 100 μM (Eisinger et al. 1970; Danchin and Gueron 1970; Römer et al. 1970; Willick and Kay 1971; Robison and Zimmerman 1971). Quantitative measurements made by a hydrodynamic method (Friederich and Hagerman 1997; Hagerman 1997) and by gel mobility shift (Friederich et al. 1998) show that above 3 mM Mg2+ the interstem angle between the acceptor stem and anticodon stem is nearly the same as that observed in the crystal structure, but at micromolar Mg2+ concentrations the interstem angle is ∼150°.
Favre and coworkers (1975) used UVA irradiation (λ>324 nm) of various Escherichia coli tRNAs to activate the 4-thiouridine at position 8 (s4U8) and showed that it specifically crosslinks to nucleotide C13. The rate for the photocrosslinking reaction at 25°C is different for different tRNAs and depends on the buffer conditions (Favre et al. 1975, 1979). It was also found that a mutation in the D stem of (sus+) tRNATrp caused a decrease the rate of photocrosslinking compared to value seen in the (wild-type) tRNATrp; this was taken as evidence for a conformational difference due to the alteration of the tRNA structure in the D stem (Favre et al. 1975; Favre and Thomas 1981). In addition, the photocrosslinking rate for tRNAPhe was reduced by more than twofold when it was irradiated in a complex with tRNA synthetase (Favre et al. 1979). These studies indicate that the photocrosslinking reaction is very sensitive to conformational differences in the tRNA (Favre et al. 1975).
A quantitative model for the photocrosslinking process will help to connect differences in photocrosslinking rates to differences in the molecular properties. Information relevant to the mechanism came from our analysis of RNA–RNA photocrosslinks in ribosomal RNA induced by UV irradiation (Wilms et al. 1997; Shapkina et al. 2004) or by UVA irradiation of ribosomes containing s4U (Nanda and Wollenzien 2004). These are both zero-length crosslinking reactions, like the s4U8–C13 crosslink in tRNA, and, for both of these reactions, formation of the photocrosslinks depends on the direct contact and correct orientation of the two participating nucleobases. The availability of the atomic structure of the 30S ribosomal subunit (Wimberly et al. 2000) allowed calculation of parameters that describe the internucleotide geometry at the sites where photocrosslinks had been observed. These measurements indicated a correlation between the reciprocal C1′–C1′ internucleotide distances and the photocrosslinking frequencies; the correlation coefficients have values between 0.39 to 0.7 using the Thermus thermophilus 30S structure (Wimberly et al. 2000) or two E. coli 30S structures (Schuwirth et al. 2005). These correlations and the comparison of the data for the UV- and UVA-s4U-induced crosslinks are consistent with reaction mechanisms that involve transient movements, which occur during the excited electronic state lifetime of the nucleobase, to bring the pairs of nucleobases from their equilibrium positions to the arrangement needed for photoreaction, rather than mechanisms in which there are stable alternate local conformations at the crosslinking sites (Huggins et al. 2005). An important feature of this mechanism is that the transition state for the photoreaction is reached without the system having to go through any higher potential energy. The tRNA tertiary structure at s4U8–C13 suggests that a similar situation exists in the tRNA, and the same mechanism is assumed in generating an empirical equation to describe the rate of photocrosslinking.
The correlation coefficients between crosslinking frequencies and internucleotide distances are less than unity, so another factor must also affect the crosslinking frequencies. For the 16S rRNA it is likely that local flexibility, determined by hydrogen bonding and packing density, is responsible for inhibition at many potential sites and the modulation of the frequency at the sites where photocrosslinks occur (Huggins et al. 2005; W. Huggins, K. Nanda, S.K. Ghosh, T. Shapkina, and P. Wollenzien, unpubl.). If this is the case, then the rate of photocrosslinking should increase as the temperature increases to reflect greater accessibility to the precise structures needed for photoreaction, which is determined by the conformational potential energy landscape. Such a response should allow the determination of the activation energy for the movement of the two nucleobases that participate in the photoreaction.
The temperature dependence of the rate of photocrosslinking for tRNAfMet, tRNAPhe, and tRNAVal under different conditions has been investigated in this report. The data of Favre et al. (1975) that the photocrosslinking reaction follows first order kinetics are confirmed, and we further show that the rate constants increase with temperature and follow Arrhenius behavior, so that an activation energy can be determined. The apparent geometry at the photocrosslinking site also can be calculated for tRNA under different conditions. The measurements show that differences in geometry and flexibility can be separately determined by this approach. The data quantitatively confirm the stabilization of the canonical form of the tRNA by Mg2+.
RESULTS
Determination of apparent photocrosslinking rate constants by gel electrophoresis analysis of irradiated tRNA
UVA irradiation was done on [32P] 3′ end-labeled E. coli tRNAfMet and tRNAVal for 1–20 min to induce the s4U8–C13 photocrosslink (Fig. 1) previously characterized by Favre et al. (1975) and Favre (1990). The separation of the tRNAs by electrophoresis on 8% polyacrylamide gels resulted in the appearance of a faster migrating band in the irradiated samples (Fig. 2A), rather than a slower mobility band, which would be expected for a molecule with a covalent loop (Lemaigre-Debreuil et al. 1991; Behlen et al. 1992; Wilms et al. 1997). To confirm that this faster band contained the s4U8–C13 photocrosslink, RNA from both bands was isolated and subjected to partial alkaline hydrolysis and RNA sequencing (Supplemental Material). In the RNA from the faster moving band, C13 was involved in intramolecular crosslink; its partner must be s4U8, since that is the only nucleotide that absorbs the wavelengths used. The sequencing experiments ruled out other crosslinks involving s4U8 for the tRNA in solution. In addition, the gel electrophoresis analysis showed that there is some background radioactivity present at the position of the crosslinked tRNA even without irradiation and that not all of the original tRNA is photoreactive (Fig. 2A). The maximum amount of crosslinking in different samples is 70%–90% of the total. The background and maximum reaction need to be taken into account when determining the extent of reaction (see Materials and Methods).
FIGURE 1.

Location of the s4U8–C13 photocrosslinking site in the tRNA secondary and tertiary structures. (A) Cloverleaf structure and sequence of E. coli tRNAVal with the s4U8 and C13 nucleotides indicated. (B) Location of U8 and C13 in the yeast tRNAPhe tertiary structure. U8 is not thiolated in yeast tRNAPhe.
FIGURE 2.

Analysis of kinetics of crosslink formation. (A) Gel electrophoresis separation of irradiated tRNA. tRNAfMet and tRNAVal were 3′ end exchange-labeled and irradiated in HiFi buffer with wavelengths >320 nm for the indicated times at 0°C and 45°C. Bands 1 and 2 separate after electrophoresis on 8% polyacrylamide urea gel and contain the uncrosslinked and crosslinked tRNA as confirmed by RNA sequencing (Supplemental Material). (B) Time course of photocrosslinking for tRNAfMet and tRNAVal. Plot of fraction crosslinked versus irradiation time (in seconds) for tRNAfMet and tRNAVal at 0°C and 45°C as indicated in the figure. The irradiations were done in a buffer containing 20 mM Tris (pH 7.5), 100 mM NH4Cl, and 20 mM Mg2+. The curves were fitted using parameters for the first order rate constants and for the total amount reactive. The total extent of crosslinking for tRNAfMet and tRNAVal is 0.80 ± 0.01 and 0.72 ± 0.02, respectively. (C) First order plot of rate of photocrosslinking. The logarithm of the fraction of tRNA unreacted for the s4U8×C13 photocrosslink in tRNAfMet and tRNAVal is plotted versus time for the irradiations at 0°C and 45°C.
The progress of the s4U×C13 crosslink formation was followed by plotting the fraction of crosslinked tRNAfMet and tRNAVal at 0°C or 45°C versus time (Fig. 2B). The s4U×C13 crosslinking reaction is faster for tRNAfMet, and the maximum amount is reached by 5 min of irradiation (Fig. 2B). In comparison, for the tRNAVal photoreaction the maximum amount of crosslink formation is reached after longer times and has a greater response to temperature than that seen for tRNAfMet (Fig. 2B). For both tRNAs, the reactions go to the same total extent at all temperatures studied. The fit of these curves with a single rate indicates that the entire photoreactive sample behaves with the same kinetics. The tRNAs need to be renatured for reproducible results, indicating that the rates are attributable to a property of the native structure.
Plots of natural logarithm of the fraction of unreacted tRNA versus time were done to determine if the reactions were first order and to determine the apparent rate constants (Fig. 2C). For both tRNAs, irradiations at both temperatures produced straight lines confirming a first order reaction. These data rule out temperature-dependent photoreactions other than crosslink formation that would prevent calculation of the photocrosslinking rate constant.
Correction of measured photocrosslinking rate constants for differences in excited state T1 lifetimes between tRNAs and at different temperatures
Different tRNAs have different s4U excited state lifetimes (Favre 1990), and corrections have been made to the measured photocrosslinking rate constants to compensate for the longer or shorter times for reaction for the s4U in different tRNAs. The factors for these corrections are listed in the Supplemental Materials. In addition, for a particular tRNA, there may be differences in the excited state lifetimes due to other temperature-dependent or buffer effects. For s4U, the photoreactive state is the T1 state derived from the S1 state by intersystem crossing. Intersystem crossing is complete for s4U (Favre 1990, Favre et al. 1998), and the formation of neither the S1 nor T1 states is expected to be temperature dependent. However, there are several possible fates for the T1 state besides photochemistry, including nonradiative energy transfer, radiative deexcitation (phosphorescence), collisional quenching, and nonradiative deexcitation, which would alter the excited state lifetime. Measurements made in buffers containing chloride ion, which quenches the T1 excited state (Favre 1990), or acetate ion indicate that collisional quenching does not occur as long as the tRNA is in the native conformation (data not shown). Of the remaining processes, nonradiative deexcitation must account for the majority of the deexcitation (Favre 1990) and this could be temperature dependent (Turro 1991). To determine if this were the case, the phosphorescence emission intensity, which reflects the excited state lifetime, was measured at different temperatures; it was seen that all of the tRNAs exhibit decreases in phosphorescence emission intensity as the temperature increases. Factors to correct the measured photocrosslinking rate constants were calculated from the emission intensities using emission at 25°C as the reference temperature. The correction factors for the temperature effect are listed in the Supplemental Material.
Arrhenius plots to determine activation energies for photocrosslinking
Rate constants for a photochemical reaction depend on the maximum rate of the photochemistry and, through the Boltzmann relationship, on an activation energy (E a) associated with bringing the reactants into the transition state (Turro 1991). For the tRNA photocrosslinking reaction, the activation energy is attributable to the conformational potential energy change needed to move the photoreactive bonds of s4U8 and C13 away from their relaxed equilibrium positions and into van der Waals contact and correct alignment for reaction. The rate constant could also depend on the entropy change associated with reaching the transition state, although for a unimolecular reaction this is usually negligible (Jencks 1969). This would be part of the preexponential term in the rate constant equation. In tRNA this could result from changes in the interaction with solvent or ions or changes in the conformational entropy. It will be seen that the preexponential term varies by >20-fold under different conditions, and it is very unlikely that this difference is due to differences in the entropy change (see Discussion). On the other hand, we had already observed that photocrosslinking frequencies tend to be larger for sites in the rRNA where the internucleotide distance is smaller (Huggins et al. 2005). Therefore the expression for the crosslinking rate constant should contain the inverse of the s4U8–C13 internucleotide distance to incorporate this dependence. This gives the empirical equation
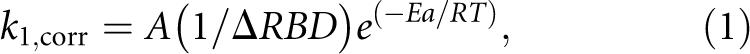 |
where k 1, corr is the measured first-order rate constant corrected for differences in the excited state lifetime values; A is the factor that incorporates the rate of the photochemical reaction as well as instrumentation factors; ΔRBD is the distance between the centers of the C4–S4 bond of s4U8 and the center of the C5–C6 bond of C13 minus the distance between the bonds at direct contact, 3.4 Å; E a is the activation energy; and R and T are the gas constant and temperature.
The activation energy can be determined by the Arrhenius method by finding the slope of the dependence of ln (k 1,corr) on 1/T:
Favre and coworkers (1975) previously characterized three different classes of E. coli tRNAs depending on whether their rates for photoreaction at 25°C were faster, slower, or equal to bulk unpurified E. coli tRNA. Three tRNAs were used to investigate the temperature dependence of the photocrosslinking rate: tRNAfMet (faster rate than bulk tRNA), tRNAPhe (slower rate than bulk tRNA), and tRNAVal (rate equal to that of bulk tRNA). Each tRNA was [32P] labeled and renatured in HiFi buffer (50 mM Tris at pH 7.5, 30 mM KCl, 70 mM NH4Cl, 3.5 mM MgCl2, 8 mM putrescine, 0.5 mM spermidine) before irradiation as described in the Materials and Methods. Samples were irradiated for 1 min at temperatures from 0°C to 50°C to obtain apparent k 1 values. The k 1 values were corrected by multiplying by the factors to compensate for differences in the excited state lifetime intrinsic to each tRNA and due to temperature (Supplemental Material). Plots of corrected rate constants versus temperature for the three tRNAs are shown in Figure 3A.
FIGURE 3.

Temperature dependence of rate constants for formation of the s4U×C13 crosslink in tRNAfMet, tRNAVal, and tRNAPhe. (A) First order rate constants calculated from 1-min irradiations at different temperatures and corrected for changes in excited state lifetimes. The fitted lines are exponential functions. Uncertainty bars are standard deviations of measurements at each temperature. (B) Arrhenius plots for the corrected first order rate constants. The fitted lines are linear regression lines.
The photocrosslinking rate constants increase with temperature for all three tRNAs. At 25°C the largest rate constant occurs in tRNAfMet, 0.022 sec−1, which is nearly three times the rate constant for tRNAVal, 0.0077 sec−1, and six times the rate constant for tRNAPhe, 0.0036 sec−1 (Fig. 3A; Table 1). This trend is in good agreement with the results of Favre et al. (1975), although the absolute values between the tRNAs are different than in their report, but this can be attributed to differences in irradiation and buffer conditions and to the fact that the rate constants have been corrected for excited state lifetime differences.
TABLE 1.
Summary of experiments and calculation of apparent geometry and force constants

Arrhenius plots were done with the data (Fig. 3B). The appearance of the plots for the three tRNAs are very similar, but there is a significantly smaller slope in the regression line for tRNAfMet compared to the two other tRNAs, which corresponds to an activation energy ∼5 kJ mol−1 less than for either tRNAVal or tRNAPhe.
Calculation of internucleotide distances for s4U8–C13 for the different tRNAs
Rearrangement of Equation 1 allows calculation of the apparent extent of movement of the photoreactive bonds (C4-S4 of s4U8 and C5-C6 of C13) during the photocrosslinking reaction:
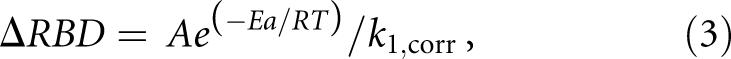 |
where all terms are the same as in Equation 1. Since k 1, corr and E a are experimentally determined, this provides a method to determine the distance between the reactive bonds at 25°C under different conditions if the value of the preexponential factor A is known. Since the reactive bonds have to be in van der Waals contact at the time of photoaddition, the calculation is an indication of what their distance separation must be in the equilibrium structure. A was determined by postulating that there is a single value for it for all three tRNAs. High resolution structures for the E. coli tRNAs are not available, so the atomic structures of yeast tRNAfMet (Basavappa and Sigler 1991), tRNAPhe (Shi and Moore 2000), and tRNAAsp (Comarmond et al. 1986) were used to calculate values of ΔRBD for initiator tRNA and for elongator tRNA. With these values, A was calculated from the experimentally determined k 1, corr and E a values for each tRNA. These values were averaged and then used to recalculate ΔRBD for each tRNA.
The data and calculations of the distances between the reactive bonds at 25°C for the three tRNAs in HiFi are summarized in Table 1. The rate constants are partly determined by the activation energy term of the rate equation. However the activation energies for tRNAPhe and tRNAVal are nearly the same, but there is a twofold difference in the rate constant. The correction made for the difference in the excited state lifetimes contributes to the rate difference, but, even without that correction, a difference exists in the rate constants that can be attributed to a difference in the geometries of the molecules. For tRNAfMet, the much higher rate constant is correlated to the lower activation energy; however, the rate constant would be even greater if tRNAfMet had an s4U8–C13 internucleotide distance similar to either of the other tRNAs. Therefore in this case the discrepancy between the rate constant and the activation energy again can be attributed in a reasonable way to a difference in the internucleotide distance.
Calculation of force constant for the conformational flexibility in the tRNA
The activation energy for the photocrosslinking reaction must originate in the tRNA tertiary structure. Nucleotide s4U8 is the first nucleotide following the acceptor stem, and its arrangement must be tied closely to the position of the acceptor stem. Nucleotide C13 is one half of the terminal base pair of the D stem with G22, and G22 in turn is also hydrogen bonded with m7G46 to form a base triple. In addition, s4U8 is hydrogen bonded to A14, which positions it in close proximity to the C13–G21–G46 base triple. As a consequence, changes in the orientation of the s4U8 and C13 nucleobases will require changes in the geometry of the tRNA core.
Values of the apparent change in the internucleotide distance, ΔRBD, and the activation energy (E a) can be combined to calculate an apparent force constant for the tRNA movement. This can be done by assuming quadratic behavior for the potential energy with respect to the energy present in the structure when the s4U8 and C13 are in their equilibrium positions (see Materials and Methods). Rearrangement of the standard expression for the potential energy gives the force constant:
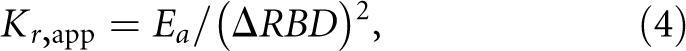 |
where E a is the activation energy, K r, app is the apparent force constant for a virtual bond between the centers of the photoreactive bonds, and ΔRBD is the distance displacement of the reactive bonds away from their equilibrium positions toward each other. The force constant is called apparent since it is based on the assumption that all of the geometry dependence of the rate constant is due to a distance change between the reactive bonds. The apparent force constants for tRNAfMet, tRNAPhe, and tRNAVal in HiFi buffer vary from 21 ± 8 to 190 ± 100 kJ mol−1 Å−2 (Table 1). The large uncertainties in the force constant values come from amplification of the uncertainties in the E a values when the exponential function of E a is calculated.
Dependence of activation energy of crosslink formation on buffer conditions
The stability and flexibility of RNA is known to depend on the concentration of both monovalent (K+, NH4 +, Cl−) and divalent ions, such as magnesium and putrescine, and polyamines, such as spermidine, in solution (Herman and Westhof 1998; Misra et al. 2003; Koculi et al. 2004). Therefore alterations in the monovalent ion or Mg2+ concentrations should produce differences in the photocrosslinking rates that would reflect changes in either, or both, the flexibility and structure of the tRNA. To measure this effect, the temperature dependence experiments were repeated on tRNAVal using buffers differing in monovalent and divalent ion concentration and identity. The first series of buffers was varied in the Mg2+ concentration (0–20 mM Mg2+) and the second series was varied in the monovalent ion concentration (100–1000 mM NH4Cl). The last buffer was HiFi buffer without Mg2+ to contrast the effects of the polyamines and magnesium.
Corrections were made to the measured rate constants to account for differences in the excited state lifetimes at 25°C and to account for the temperature dependence of the lifetimes as described in the previous section. Plots of the rate constants versus temperature for tRNAVal irradiated in these buffers show that the rate constants increase with increasing temperature in all buffers, and Arrhenius plots are linear and show that the activation energies depend strongly on the conditions (Supplemental Material).
Trends in the rate constants and activation energies are present in the two series of experiments that used increasing NH4 + or increasing Mg2+ concentrations (Fig. 4). For tRNAVal in 100 mM NH4Cl, the activation energy is less than half its value in HiFi buffer, and the rate constant is such that the apparent distance between s4U8 and C13 is ∼15 Å. As a consequence of these values, the force constant is over three orders smaller in the 100 mM NH4 + buffer compared to its value in the HiFi buffer. These parameters indicate that in the absence of Mg2+ there is a much larger flexibility in the molecule and that the structure must be significantly different than the structure in the presence of Mg2+. As the NH4Cl concentration is increased from 100 to 1000 mM, the activation energy increases by ∼70% to nearly 18 kJ mol−1. However, the photocrosslinking rate constant is such that the apparent s4U8–C13 distance is still ∼7 Å in 1000 mM NH4Cl. As a consequence, the apparent force constant is only ∼7 times the value it has in the 100 mM NH4 + buffer. This is still over 500 times less than the value in HiFi buffer or in the buffer containing 20 mM Mg2+.
FIGURE 4.

Dependence of the corrected rate constants, energies of activation, and calculated parameters on NH4OAc and Mg2+ concentrations for the tRNAVal s4U8×C13 crosslink. (A) Dependence of the corrected first order rate constants on NH4 + and Mg2+ concentrations. All data in this and subsequent panels are from Table 1. (B) Dependence of activation energies on NH4 + and Mg2+ concentrations. (C) Dependence of the s4U8–C13 apparent distance on NH4 + and Mg2+ concentrations. (D) Dependence of force constants on NH4 + and Mg2+ concentrations. Note that there is a 100-fold change in scale in the left and right parts of panel D. For all experiments, the dependence on different NH4 + concentrations was determined in the presence of 20 mM Tris (pH 7.5) and the dependence on different Mg2+ concentrations was determined in the presence of 20 mM Tris (pH 7.5), 100 mM NH4Cl.
In the buffer containing 100 mM NH4 + plus 0.5 mM Mg2+, the activation energy is nearly 22 kJ mol−1 and the rate constant is such that the apparent s4U8–C13 distance is 1.5 Å. These values correspond to an apparent force constant of 9.7 kJ mol−1 Å−2. When the Mg2+ concentration is increased to 1 mM there is a significant increase in activation energy that results in a significant decrease in the apparent s4U8–C13 distance and an increase in the apparent force constant. Additional increases in the Mg2+ concentration up to 20 mM result in increases in the activation energy together with decreases in the photocrosslinking rate constants. The combination of values in the buffer containing 20 mM Mg2+ results in an apparent internucleotide distance that is nearly the same as it is in HiFi buffer.
tRNAVal was also measured in a buffer containing the components of HiFi buffer but without Mg2+. Under these conditions there is a slight reduction in the rate constant and a little larger reduction in the activation energy. These values indicate a little larger distance separation between s4U8 and C13 and a sixfold reduction in the apparent force constant. This result indicates that the putrescine and spermidine that are in the HiFi buffer do not replace the role of Mg2+ in the tRNA structure.
DISCUSSION
The tRNA photocrosslinking reactions show a single first order behavior indicating a single structural conformer at the s4U8–C13 photocrosslinking site. In addition, Arrhenius behavior is seen in all of the samples with activation energies that depend on the identity of the tRNA and on the buffer conditions. These results support the model for the photocrosslinking mechanism in which the transition state for the reaction is reached during conformational excursions from an equilibrium lowest free energy structure. As a consequence the rate of reaction is determined by the conformational potential energy of the transition state rather than by the potential energy of any barrier that might occur between the lowest potential energy state and the transition state. Differences in the observed rate constants are not accounted for by the activation energies alone. From a study of the connections between internucleotide geometry and photocrosslinking frequency in ribosomal RNA, we expected that photocrosslinking rates would depend on the internucleotide distance (Huggins et al. 2005). This dependence is probably due to the fact that the excited state lifetime of the s4U limits the extent of the conformational excursions. The expression for the rate constant therefore contains terms for both the activation energy and the apparent displacement of the photoreaction bonds during the reaction.
The preexponential part of the equation for the rate constant could depend on the entropy change associated with reaching the transition state (Jencks 1969). In the case of the unimolecular reaction in tRNA, entropy changes could occur because of changes in the interactions with water or ions or due to changes in the conformational entropy. Since the same photochemistry occurs under all of the buffer conditions and temperatures, the reactions must go through the same transition state, so any differences in the entropy change under different conditions would have to be due to entropy differences in the equilibrium structures. The preexponential term in the equation describing the rate constant is larger by >20-fold for the reaction when the tRNA is in the more extended form in the absence of Mg2+ compared to the reaction when the tRNA is in the canonical form in the presence of Mg2+. If this difference were due to interactions between tRNA and water or ions, the extent of the interactions would have to be less in the extended form, which seems unlikely. On the other hand, an entropy change could be due to differences in the conformational entropy. In this case the extended tRNA form would have to possess larger conformational entropy than the canonical form and this would be consistent with a greater flexibility. However since the entropy is determined by the logarithm of the conformational space, this would require over a 20-fold greater conformational space for the extended form compared to the canonical form. It is impossible to rule this out, but it is unlikely, since only a small fraction of the nucleotides in the tRNA seem to be involved with its conformational change (Tung et al. 1984; Nakamura and Doi 1994; Matsumoto et al. 1999). In addition, in-line probing experiments, which measure the spontaneous hydrolysis at the phosphodiester bonds and depend on the intrinsic flexibility of the nucleotide unit (Dock-Bregeon and Moras 1987; Soukup and Breaker 1999), have been done on tRNA with buffers with and without Mg2+ and show that a limited number of nucleotides have differences in reactivity, and these changes involve both increases and decreases in reactivity (data not shown). As a consequence, differences in the conformational entropy of the tRNA under different conditions could probably at best account for a small part of the variation in the preexponential term.
The corrected rate constants for photocrosslinking for tRNAVal, tRNAPhe, and tRNAfMet in HiFi buffer are all different in spite of the fact that the activation energies are similar for tRNAVal and tRNAPhe, 24.6 kJ mol−1, but are smaller for tRNAfMet, 19.8 kJ mol−1. The rate constant differences can be accounted for by modest differences in the s4U8–C13 geometry in each tRNA. In addition, in tRNAVal, which has been studied as a representative, the activation energies increase as the monovalent ion concentration or the Mg2+ concentration increase—the range is 12.1 kJmol−1 to 26.6 kJmol−1—and, under the same conditions, the values calculated for the s4U8–C13 distances decrease—the range is from ∼15 Å to 0.36 Å. These changes in the properties of the tRNA under different conditions show that the rate equation links the rate constants to the conformational energy and the geometry in a self-consistent way.
The temperature dependence of the s4U emission intensity at 510 nm provides additional evidence for flexibility changes in the tRNA. The plots of emission intensity show exponential decrease with temperature (see Supplemental Material). Emission intensity must be inversely related to the rate of the nonradiative deexcitation, which is the predominant mode for deexcitation. Arrhenius plots of the reciprocal emission intensity provide estimates of the activation energy for the deexcitation process, which are ∼15 kJ mol−1 in HiFi buffer and 10 kJ mol−1 in buffers without Mg2+, irrespective of the tRNA identity. The reason for the temperature dependence of the deexcitation rate is not clear, but it must come from an internal process somehow connected to the structure because the s4U8 in the tRNA is not subject to collisional quenching or energy transfer. The activation energies for deexcitation are different than for photocrosslinking, but they are larger for tRNA in buffers with higher concentrations of Mg2+ and smaller in buffers containing only monovalent cations; this difference is consistent with the idea that the internal flexibility is changing in response to the Mg2+ content.
The photocrosslinking data indicate progressive conformational differences in the tRNAVal as the monovalent ion concentration is increased from 100 to 1000 mM and a large change when even submillimolar Mg2+ is present. Overall the changes in the apparent distance between s4U8 and C13 decrease from a value of ∼15 Å in the buffer with 100 mM NH4 + to values of ∼0.4 Å in HiFi buffer or in the buffer with 20 mM Mg2+; this last value is consistent with the structure seen in the crystal. The values of the apparent force constant for the relative movement of s4U8 and C13 are 0.05 kJ mol−1Å−2 for the tRNA in 100 mM NH4Cl and ∼200 kJ mol−1Å−2 for the tRNA in the HiFi buffer or the buffer with 20 mM Mg2+. These values indicate an exceedingly flexible structure under the lowest ionic strength. However, even the largest of these force constants is small compared to the C–C covalent bond stretching force constant, 1200–2000 kJ mol−1 Å−2 (Weiner et al. 1986). The apparent force constants determined here are in keeping with the energy change associated with bending the tRNA in small angles.
The shape of yeast tRNAPhe determined by transient electric birefringence in a buffer containing 15 mM NaPO4 (pH 7) and 0.01–10 μM Mg2+ indicated an interstem angle of 154° (Friederich and Hagerman 1997). There was a large change in the tRNA behavior at Mg2+ concentrations between 10 and 200 μM. The terminal decay time decreased to a minimum at 200 μM Mg2+, corresponding to an interstem angle of 70°, and increased a little as the Mg2+ concentration was increased to 10 mM, corresponding to an angle of 80°–90°. Changes in the tRNA interstem angle and flexibility were also measured by the gel electrophoretic mobility of RNA molecules with the yeast tRNAPhe sequence (Friederich et al. 1998). Mobility differences confirmed the Mg2+ dependence of both the tRNA bend angle and the flexibility. The flexibility change could not be unambiguously calculated. However, one model predicted a 50-fold decrease in the torsional elastic constant, a measure of the flexibility, although the values in low Mg2+ concentrations should also be affected by changes in the persistence length of the RNA (Friederich et al. 1998).
It was not possible for us to use the same ionic conditions of Friederich and Hagerman (1997) and Friederich et al. (1998) due to the instability of the tRNA at higher temperatures. However, it is likely that we are observing the same overall conformational transition in the tRNA from the extended flexible structure that it has in 0–10 μM Mg2+ to the canonical structure that it has in buffers containing at least 200 μM Mg2+. The photocrosslinking indicates an apparent distance between s4U8 and C13 and although this does not directly provide information about the overall extension of the tRNA structure, the changes in its value are correlated to the changes in the interstem angle determined by Friederich et al. (1998).
The fold difference in the apparent force constant calculated from photocrosslinking in buffers with high Mg2+ compared to buffers with no Mg2+ is ∼4000, much larger than the fold difference estimated from the gel electrophoresis mobility measurements. This could be due to the different ionic conditions that have been used in the experiments. Alternatively, this could be due to an overestimate of the apparent s4U8–C13 distance, especially under conditions of 0 mM Mg2+, which would strongly affect the calculation of the apparent force constant. The reason for a possible overestimate of the distance change is that we have used the dependence of the rate constant on internucleotide geometry as being due entirely to the distance between the photoreactive bonds. It would be reasonable that the rate constant will actually depend on a combination of changes in the distance and angle between the photoreactive bonds. Dependence on a combination of distance and angle was not used here because, in the statistical analysis of the factors for the crosslinking frequency in the rRNA, the internucleotide angle did not show a correlation to the frequency (Huggins et al. 2005).
There have been several descriptions of the flexibility of the tRNA from normal mode analysis and from a computational simulation of conformational changes. Two studies that employed normal mode analysis identified very similar lowest frequency modes that corresponded to movements of the acceptor stem and anticodon stem in the same direction and in the opposite direction of the elbow region (Nakamura and Doi 1994; Matsumoto et al. 1999). A maximum value of the bend angle due to thermal fluctuations of ∼5° was described by Nakamura and Doi (1994), and they concluded that this bend originated from a hinge centered in the elbow region. Matsumoto et al. (1999) concluded that the movements originated at deformations around the base pair U7–A66, at the interior end of the acceptor stem, and at deformations around the base pair C28–G42 in the anticodon stem, so that the tRNA could be thought of as three blocks. The calculation of the RMS displacement values for the lowest mode and the mode frequencies are very similar in these two studies, so it is likely that there are different interpretations because of the way in which the nucleotide movements were classified.
A molecular mechanics analysis was done by Tung et al. (1984) to determine how the acceptor end–anticodon end distance could lengthen or shorten with as small as possible an energy increase. The conformational change was treated as an angular bending movement of the arms of the tRNA. A series of bending centers were identified that resulted in the low energy increases as the tRNA extended or bent acutely. For increase or decrease in the angle of bending up to ∼6°, the center of bending was located at a point halfway between the phosphates of nucleotides 8 and 49 (P8 and P49), and the energy change was ≲5 kcal. For additional extension up to 36°, the bending center was switched to the N1 atom of nucleotide 59. The total energy change for the maximum angle of bending in the core of 118° was 34 kcal mol−1. Additional extension was centered progressively around the Robertus/Klug hinge at the acceptor stem/T stem junction, at a point between P26 and P46, and then at a point between P27 and P44. The total increase in the angle was 54° at an energy increase of 52 kcal mol−1. Since the angle in the canonical form is 82°, the total maximum angle between the acceptor stem and anticodon stem is ∼136°.
The activation energy for tRNAVal in HiFi buffer associated with the s4U8–C13 movement, in which ΔRBD is equal to 0.36 Å, is ∼26 kJ mol−1 (6.2 kcal mol−1). The bending angle associated with this distance displacement, if the bend center is at a point between P8 and P49, would be a little <3°. This is in good agreement with the extent of movement under thermal energy determined by normal mode analysis (Nakamura and Doi 1994; Matsumoto et al. 1999) as well as with the energy associated with small deformations calculated by molecular mechanics (Tung et al. 1984).
Our data are also consistent with a change in the bending center as the tRNA becomes extended. As described, the s4U8–C13 distance of 0.36 Å is easy to reconcile with a bend center at P8–P49. However, for the s4U8–C13 distance of 7 Å that is measured when the tRNA is in the T20A1000 buffer, the bend center must be at a location farther away from s4U8–C13. The reason for this is that if the bend were at P8–P49, a 7 Å s4U8–C13 distance would predict an increase in the bend angle of 42°, which is larger than possible for the core structure. On the other hand, if the bend center is at N1 of A59, an increase in the angle of bending would be 30 Å, closer to the measurements of the tRNA bend from the hydrodynamic measurements and the molecular mechanics prediction. However, for the measured s4U8–C13 distance of 15.8 Å, even with the bend center at N1 of A59, an angular increase of ∼60° is predicted that is larger than the angle calculated from the gel electrophoresis measurements or the molecular mechanics study. This suggests that part of the dependence of the photocrosslinking rate on geometry is on the angle between the bonds, which would reduce the apparent distance between s4U8 and C13.
Beyond small degrees of bending, there must be a significant change in the organization of the tRNA core region that involves the loss of the Mg2+ binding site. The apparent force constant for s4U8–C13 movement in HiFi buffer or in T20A100M20 buffer is 200 kJ mol−1 Å−2. If that apparent force constant described the energy when the s4U8–C13 distance increased to 15.8 Å, it would predict an energy increase of ∼47,000 kJ mol−1 (11,000 kcal mol−1), far more than the 90 kcal mol−1 estimated from the molecular mechanics calculation or the 12.1 kJ mol−1 (2.8 kcal mol−1) measured in the photocrosslinking experiments when the tRNA is without Mg2+. These results point to the strong effect of Mg2+ in maintaining the structure in the tRNA core that is resistant to deformations. This is a very specific effect because there is quite a large difference in the properties of the tRNA in T20A100M7 versus HiFi M0, which have nearly the same divalent cation concentration but have 7 mM Mg2+ replaced with 8 mM putrescine and 0.5 mM spermidine. There is a fourfold larger apparent force constant in the T20A100M7 buffer than in the HiFi M0 buffer, so the putrescine and spermidine do not functionally replace Mg2+. The apparent force constant is much smaller when the divalent ion is replaced by even large concentrations of monovalent ions. The apparent force constant is 30-fold larger in the T20A100M0.5 buffer compared to the T20A1000 buffer.
The experimental strategy described here provides the two measurements, k 1 and E a, to characterize the photochemical reaction. The quantitative analysis shows that the photocrosslinking rate constant can be accounted for in terms of the geometry at the photoreactive site together with the activation energy, which can be associated with the conformational energy change during the photocrosslinking reaction. These give self-consistent values for the changes in the tRNA, particularly with respect to the properties it has under conditions with different Mg2+ concentration. The experiments demonstrate a high sensitivity to the environmental conditions.
MATERIALS AND METHODS
3′-end labeling of tRNAs
E. coli tRNAfMet, tRNAVal, and tRNAPhe (Sigma or Chemical Block) were 3′-end labeled by the PPi exchange reaction using the Bacillus stearothermophilus CCA enzyme as previously described (Yue et al. 1998; Wolfson and Uhlenbeck 2002; Huggins and Wollenzien 2004). To ensure that the tRNA had fully restored ends, after incubation with [α-32P]ATP under exchange conditions, ATP and CTP were added to a final concentration of 1 μM and the mixture was again incubated at 55°C for 3 min. The samples were then phenol and ether extracted and ethanol precipitated twice with 4 μg glycogen.
Irradiation procedures
tRNAs were irradiated with light in the UVA region as described (Nanda and Wollenzien 2004). Aliquots of 3′ [32P] exchange-labeled tRNAPhe, tRNAVal, or tRNAfMet in microfuge tubes were submerged in a water filled sample well for irradiation with a 400 W mercury vapor lamp. The sample well was surrounded by a glass jacket containing a circulating solution of Co(NO3)2 to filter out wavelengths <320 nm and to regulate the temperature (Isaacs et al. 1977). The light intensity in this device impinging on the microfuge tubes is ∼200 mW cm−2.
For time courses, 50 pmol of either 3′ [32P] end-labeled tRNAfMet or tRNAVal were dissolved in 200 μL HiFi buffer (50 mM Tris at pH 7.5, 30 mM KCl, 70 mM NH4Cl, 8 mM putrescine, 3.5 mM MgCl2, 0.5 mM spermidine), incubated at 10 min at 45°C followed by slow cooling, aliquoted into microfuge tubes in 100 μL portions, and stored on ice until needed. The samples were preincubated at least 3 min at 0°C or 45°C before irradiation. Samples of 10 μL were removed from each portion at 1–5, 10, 15, and 20 min. The irradiated tRNA was then precipitated with 0.5 M NH4OAc (pH 5.5), 4 μg glycogen, and 2.5 volumes of ethanol. After precipitation, samples were separated by electrophoresis on 8% PAGE sequencing gels, with 8.3 M urea in TBE buffer, before quantitation by ImageQuant software.
For the temperature studies, 100 pmol of 3′ [32P] end-labeled tRNAfMet, tRNAPhe, or tRNAVal were dissolved in 1000 μL HiFi buffer and renatured for 10 min at 45°C, aliquoted into 50 μL portions, and stored on ice. The tRNAs were equilibrated at different temperatures (every 5°C between 0°C and 50°C) at least 3 min in a separate water bath and irradiated for 1 min at the same temperature. After irradiation, RNA was ethanol precipitated with 4 μg of glycogen as a carrier before electrophoreses.
For the experiments in the different buffers, samples were 3′ [32P] end-labeled and renatured in HiFi buffer as described above. The samples were then dialyzed overnight against the appropriate buffer at 4°C, changing the buffer once. After dialysis, the samples were aliquoted, irradiated, and analyzed as described above.
Calculation of the rate constant for the s4U8×C13 photocrosslinking
The amount of reaction at each time was determined by first subtracting the amount of background radioactivity present in the control (unirradiated) lane, and then dividing the amount of radioactivity in the crosslinked band by the total amount of radioactivity in the uncrosslinked and crosslinked bands RNA (Fig. 1). This number was then subtracted from the total fraction of crosslinking possible (the fractional extent of crosslinking in the samples irradiated 20 min at 0°C or 45°C), and finally the difference was divided by the total fraction of crosslinking possible. This calculation yields the normalized fraction of uncrosslinked tRNA at each temperature. The natural logarithm of this value was plotted against time in the first order graphs (Fig. 2C). The first order rate constants for the photocrosslinking reaction of s4U8×C13 in both tRNAVal and tRNAfMet were calculated from the negative slope of these graphs.
Calculation of first order rate constants and activation energies
The first order rate constants for the photocrosslinking reactions at each temperature were determined from the fractional amount of crosslink product at 1 min. The correction and normalization described above yielded a quantity that was the fraction of noncrosslinked tRNA after 1 min irradiation. This was converted into the rate constant by taking the natural logarithm and dividing by 60 sec.
The activation energy was then calculated by plotting the natural logarithm of the rate constants versus the inverse of temperature in degrees Kelvin as described by Equation 2. The slopes of the regression lines to those graphs are −E a/R from which E a values were calculated.
Calculation of the preexponential A factor and ΔRBD
The rate constant for photocrosslinking, k 1, and the activation energy, E a, are explicitly determined from the experiments. For reasons described in the text, the preexponential term has been separated into a term that depends on the internucleotide distance, 1/ΔRBD, and a constant factor, A. The term A was calculated using measured values for k 1 and E a and values for ΔRBD calculated from the tRNA crystal structures. Then to calculate A, Equation 1 was rearranged to yield
Distances between the reactive bonds (between the midpoint of the C4–O4 bond of U8 and the midpoint of the C5–C6 bond) in the X-ray structures of yeast tRNAPhe (Shi and Moore 2000), yeast tRNAfMet (Basavappa and Sigler 1991), and yeast tRNAAsp (Comarmond et al. 1986) were calculated and 3.4 Å, for the distance between aliphatic carbon atoms at van der Waals contact, was subtracted to obtain ΔRBD. The calculated ΔRBD value from yeast tRNAfMet was used with k 1 and E a for E. coli tRNAfMet to calculate A and the calculated ΔRBD values from yeast tRNAPhe and tRNAAsp were averaged and used with k 1 and E a values for E. coli tRNAVal and tRNAPhe to calculate A values. The three A values, 50.0, 98.7, and 20.0 Ås−1 for tRNAVal, tRNAPhe, and tRNAfMet, were averaged to give 56 Å sec−1, and this was used for the rest of the calculations.
Calculation of the apparent force constant associated with s4U8 and C13 movement
Values for ΔRBD and E a for a tRNA under a specific condition allows calculation of an apparent force constant associated with the energy involved in the s4U8–C13 movement. The quadratic force equation, E = Kr (X−X 0)2, was used for the energy associated with the movement of the reactive bonds away from the separation they have in the equilibrium structure. ΔRBD has already been defined as the distance displacement between the equilibrium distance and the distance needed for photoreaction, so it is equivalent to the displacement distance in the force law. Equation 4 follows immediately.
SUPPLEMENTAL DATA
Supplemental Material can be found at http://biochem.ncsu.edu/faculty/wollenzien/wollenzien.htm.
ACKNOWLEDGMENTS
This work was supported by funds from the College of Agriculture and Life Sciences, NC State University. Paul Agris and Keith Gagnon are thanked for help with the tRNA melting determination. Clay Clark is thanked for instructions and help with using the PTI spectrofluorimeter.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.656907.
REFERENCES
- Basavappa, R., Sigler, P.B. The 3 Å crystal structure of yeast initiator tRNA: Functional implications in initiator/elongator discrimination. EMBO J. 1991;10:3105–3111. doi: 10.1002/j.1460-2075.1991.tb07864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behlen, L.S., Sampson, J.R., Uhlenbeck, O.C. An ultraviolet light-induced crosslink in yeast tRNAPhe . Nucleic Acids Res. 1992;20:4055–4059. doi: 10.1093/nar/20.15.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn, M., Danchin, A., Grunberg-Manago, M. Proton magnetic relaxation studies of manganous complexes of transfer RNA and related compounds. J. Mol. Biol. 1969;39:199–217. doi: 10.1016/0022-2836(69)90342-8. [DOI] [PubMed] [Google Scholar]
- Comarmond, M.B., Giege, R., Thierry, J.C., Moras, D., Fischer, J. Three-dimensional structure of yeast tRNA-ASP. I. Structure determination. Acta Crystallogr., Sect. B. 1986;42:272–280. [Google Scholar]
- Danchin, A., Gueron, M. Cooperative binding of manganese (II) to transfer RNA. Eur. J. Biochem. 1970;16:532–536. doi: 10.1111/j.1432-1033.1970.tb01113.x. [DOI] [PubMed] [Google Scholar]
- Dock-Bregeon, A.C., Moras, D. Conformational changes and dynamics of tRNAs: Evidence from hydrolysis patterns. Cold Spring Harb. Symp. Quant. Biol. 1987;52:113–121. doi: 10.1101/sqb.1987.052.01.016. [DOI] [PubMed] [Google Scholar]
- Eisinger, J., Feuer, B., Yamane, T. Luminescence and binding studies on tRNA-Phe. Proc. Natl. Acad. Sci. 1970;65:638–644. doi: 10.1073/pnas.65.3.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre, A. 4-thiouridine as an intrinsic photoaffinity probe of nucleic acid structure and interactions. In: Morrison H., editor. Bioorganic photochemistry: Photochemistry and the nucleic acids. Vol. 1. Wiley; New York: 1990. pp. 379–425. [Google Scholar]
- Favre, A., Thomas, G. Transfer RNA: From photophysics to photobiology. Annu. Rev. Biophys. Bioeng. 1981;10:174–195. doi: 10.1146/annurev.bb.10.060181.001135. [DOI] [PubMed] [Google Scholar]
- Favre, A., Buckingham, R., Thomas, G. tRNA tertiary structure in solution as probed by the photochemically induced 8-13 crosslink. Nucleic Acids Res. 1975;2:1421–1431. doi: 10.1093/nar/2.8.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre, A., Ballini, J.P., Holler, E. Phenylalanyl-tRNA synthetase induced conformational change of Escherichia coli tRNAPhe . Biochemistry. 1979;18:2887–2895. doi: 10.1021/bi00580a033. [DOI] [PubMed] [Google Scholar]
- Favre, A., Saintome, C., Fourrey, J.-L., Clivio, P., Laugaa, P. Thionucleobases as intrinsic photoaffinity probes of nucleic acid structure and nucleic acid–protein interactions. J. Photochem. Photobiol. B. 1998;42:109–124. doi: 10.1016/s1011-1344(97)00116-4. [DOI] [PubMed] [Google Scholar]
- Friederich, M.W., Hagerman, P.J. The angle between the anticodon and aminoacyl acceptor stems of yeast tRNAPhe is strongly modulated by magnesium ions. Biochemistry. 1997;36:6090–6099. doi: 10.1021/bi970066f. [DOI] [PubMed] [Google Scholar]
- Friederich, M.W., Vacano, E., Hagerman, P.J. Global flexibility of tertiary structure in RNA: Yeast tRNAPhe as a model system. Proc. Natl. Acad. Sci. 1998;95:3572–3577. doi: 10.1073/pnas.95.7.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman, P.J. Flexibility of RNA. Annu. Rev. Biophys. Biomol. Struct. 1997;26:139–156. doi: 10.1146/annurev.biophys.26.1.139. [DOI] [PubMed] [Google Scholar]
- Herman, T., Westhof, E. Exploration of metal ion binding sites in RNA folds by Brownian-dynamics simulations. Structure. 1998;6:1303–1314. doi: 10.1016/s0969-2126(98)00130-0. [DOI] [PubMed] [Google Scholar]
- Hingerty, B., Brown, R.S., Jack, A. Further refinement of the structure of yeast tRNAPhe . J. Mol. Biol. 1978;124:523–534. doi: 10.1016/0022-2836(78)90185-7. [DOI] [PubMed] [Google Scholar]
- Holbrook, S.R., Sussman, J.L., Warrant, R.W., Church, G.M., Kim, S.-H. RNA-ligand interactions. (I) Magnesium binding sites in yeast tRNAPhe . Nucleic Acids Res. 1977;4:2811–2820. doi: 10.1093/nar/4.8.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins, W., Wollenzien, P. A 16 S rRNA–tRNA product containing a nucleotide phototrimer and specific for tRNA in the P/E hybrid state in the Eschericia coli ribosome. Nucleic Acids Res. 2004;32:6548–6556. doi: 10.1093/nar/gkh1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins, W., Ghosh, S.K., Nanda, K., Wollenzien, P. Internucleotide movements during formation of 16S rRNA–rRNA photocrosslinks and their connection to the 30 S subunit conformational dynamics. J. Mol. Biol. 2005;354:358–374. doi: 10.1016/j.jmb.2005.09.060. [DOI] [PubMed] [Google Scholar]
- Isaacs, S.T., Shen, C.-K.J., Hearst, J.E., Rapoport, H. Synthesis and characterization of new psoralen derivatives with superior photoreactivity with DNA and RNA. Biochemistry. 1977;16:1058–1064. doi: 10.1021/bi00625a005. [DOI] [PubMed] [Google Scholar]
- Jack, A., Ladner, J.E., Rhodes, D., Brown, R.S., Klug, A. A crystallographic study of metal-binding to yeast phenylalanine transfer RNA. J. Mol. Biol. 1977;111:315–329. doi: 10.1016/s0022-2836(77)80054-5. [DOI] [PubMed] [Google Scholar]
- Jencks, W.P. Catalysis in chemistry and enzymology. McGraw-Hill; New York: 1969. [Google Scholar]
- Koculi, E., Lee, N.K., Thirumalai, D., Woodson, S.A. Folding of the Tetrahymena ribozyme by polyamines: Importance of counterion valence and size. J. Mol. Biol. 2004;341:27–36. doi: 10.1016/j.jmb.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Lemaigre-Debreuil, Y., Expert-Bezancon, A., Favre, A. Conformation and structural fluctuations of a 218 nucleotides long rRNA fragment: 4-thiouridine as an intrinsic photolabeling probe. Nucleic Acids Res. 1991;19:3653–3660. doi: 10.1093/nar/19.13.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, D.C., Schimmel, P.R. Cooperative binding of magnesium to transfer ribonucleic acid studied by a fluorescent probe. Biochemistry. 1974;13:1841–1852. doi: 10.1021/bi00706a012. [DOI] [PubMed] [Google Scholar]
- Matsumoto, A., Tomimoto, M., Go, N. Dynamical structure of transfer RNA studied by normal mode analysis. Eur. Biophys. J. 1999;28:369–379. doi: 10.1007/s002490050221. [DOI] [PubMed] [Google Scholar]
- Misra, V.K., Shiman, R., Draper, D.E. A thermodynamic framework for the magnesium-dependent folding of RNA. Biopolymers. 2003;69:118–136. doi: 10.1002/bip.10353. [DOI] [PubMed] [Google Scholar]
- Nakamura, S., Doi, J. Dynamics of transfer RNAs analyzed by normal mode calculation. Nucleic Acids Res. 1994;22:514–521. doi: 10.1093/nar/22.3.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda, K., Wollenzien, P. Pattern of 4-thiouridine-induced crosslinking in 16 S ribosomal RNA in the Escherichia coli 30S subunit. Biochemistry. 2004;43:8923–8934. doi: 10.1021/bi049702h. [DOI] [PubMed] [Google Scholar]
- Ogle, J.M., Ramakrishnan, V. Structural insights into translational fidelity. Annu. Rev. Biochem. 2005;74:129–177. doi: 10.1146/annurev.biochem.74.061903.155440. [DOI] [PubMed] [Google Scholar]
- Robison, B., Zimmerman, T.P. A conformational study of yeast phenylalanine transfer ribonucleic acid. J. Biol. Chem. 1971;246:110–117. [PubMed] [Google Scholar]
- Römer, R., Hach, R. tRNA conformation and magnesium binding. A study of a yeast phenylalanine-specific tRNA by a fluorescent indicator and differential melting curves. Eur. J. Biochem. 1975;55:271–284. doi: 10.1111/j.1432-1033.1975.tb02160.x. [DOI] [PubMed] [Google Scholar]
- Römer, R., Riesner, D., Maass, G. Resolution of five conformational transitions in phenylalanine-specific tRNA from yeast. FEBS Lett. 1970;10:352–357. doi: 10.1016/0014-5793(70)80471-9. [DOI] [PubMed] [Google Scholar]
- Schuwirth, B.S., Borovinskaya, M.A., Hau, W.W., Zhang, W., Vila-Sanjurjo, A., Holton, J.M., Cate, J.H.D. Structures of the bacterial ribosome at 3.5 Å resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- Shapkina, T., Lappi, S., Franzen, S., Wollenzien, P. Efficiency and pattern of UV pulse laser induced RNA–RNA crosslinking in the ribosome. Nucleic Acids Res. 2004;32:1518–1526. doi: 10.1093/nar/gkh320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, H., Moore, P.B. The crystal structure of yeast phenylalanine tRNA at 1.93 Å resolution: A classic structure revisited. RNA. 2000;6:1091–1105. doi: 10.1017/s1355838200000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukup, G.A., Breaker, R.R. Relationship between internucletide linkage geometry and the stability of RNA. RNA. 1999;5:1308–1325. doi: 10.1017/s1355838299990891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, A., Crothers, D.M. Equilibrium binding of magnesium (II) by Escherichia coli tRNAfMet. Biochemistry. 1976a;15:157–160. doi: 10.1021/bi00646a024. [DOI] [PubMed] [Google Scholar]
- Stein, A., Crothers, D.M. Conformational changes of transfer RNA. The role of magnesium (II) Biochemistry. 1976b;15:160–168. doi: 10.1021/bi00646a025. [DOI] [PubMed] [Google Scholar]
- Tung, C.-H., Harvey, S.C., McCammon, J.A. Large-amplitude bending motions in phenylalanine transfer RNA. Biopolymers. 1984;23:2173–2193. doi: 10.1002/bip.360231106. [DOI] [PubMed] [Google Scholar]
- Turro, N.J. Modern molecular photochemistry. University Science Books; Sausalito, CA: 1991. [Google Scholar]
- Weiner, S.J., Kollman, P.A., Nguyen, D.T., Case, D.A. An all atom force field for simulations of proteins and nucleic acids. J. Comput. Chem. 1986;7:230–252. doi: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- Willick, G.E., Kay, C.M. Magnesium-induced conformational change in transfer ribonucleic acid as measured by circular dichroism. Biochemistry. 1971;10:2216–2222. doi: 10.1021/bi00788a005. [DOI] [PubMed] [Google Scholar]
- Wilms, C., Noah, J.W., Zhong, D., Wollenzien, P. Exact determination of UV-induced crosslinks in 16S ribosomal RNA in 30S ribosomal subunits. RNA. 1997;3:602–612. [PMC free article] [PubMed] [Google Scholar]
- Wimberly, B.T., Brodersen, D.E., Clemons, W.M., Morgan-Warren, R.J., Carter, A.P., Vonrhein, C., Hartsch, T., Ramakrishnan, V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- Wolfson, A.D., Uhlenbeck, O.C. Modulation of tRNAAla identity by inorganic pyrophosphatase. Proc. Natl. Acad. Sci. 2002;99:5965–5970. doi: 10.1073/pnas.092152799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarus, M., Smith, D. tRNA on the ribosome: A waggle theory. In: Soll D., RajBhandary U., editors. tRNA: Structure, biosynthesis and function. American Society for Microbiology; Washington, DC: 1995. pp. 443–468. [Google Scholar]
- Yarus, M., Valle, M., Frank, J. A twisted tRNA intermediate sets the threshold for decoding. RNA. 2003;9:384–385. doi: 10.1261/rna.2184703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue, D., Weiner, A.M., Maizels, N. The CCA-adding enzyme has a single active site. J. Biol. Chem. 1998;273:29693–29700. doi: 10.1074/jbc.273.45.29693. [DOI] [PubMed] [Google Scholar]