

SUMMARY
The periplasmic molecular chaperone protein SurA facilitates correct folding and maturation of outer membrane proteins in gram-negative bacteria. It preferentially binds peptides that have a high fraction of aromatic amino acids. Phage display selections, isothermal titration calorimetry and crystallographic structure determination have been used to elucidate the basis of the binding specificity. The peptide recognition is imparted by the first peptidyl-prolyl isomerase (PPIase) domain of SurA. Crystal structures of complexes between peptides of sequence WEYIPNV and NFTLKFWDIFRK with the first PPIase domain of the Escherichia coli SurA protein at 1.3 Å resolution, and of a complex between the dodecapeptide and a SurA fragment lacking the second PPIase domain at 3.4 Å resolution, have been solved. SurA binds as a monomer to the heptapeptide in an extended conformation. It binds as a dimer to the dodecapeptide in an alpha helical conformation, predicated on a substantial structural rearrangement of the SurA protein. In both cases, side chains of aromatic residues contribute a large fraction of the binding interactions. SurA therefore asserts a recognition preference for aromatic amino acids in a variety of sequence configurations by adopting alternative tertiary and quaternary structures to bind peptides in different conformations.
Keywords: molecular chaperone, membrane protein folding, crystallography, phage display, isothermal titration calorimetry
INTRODUCTION
The Eschericia coli SurA protein is a periplasmic molecular chaperone whose absence, when implemented through genetic deletion, results in reduced levels of integral outer membrane proteins (OMPs)1–5. Studies on the folding and maturation of the trimeric LamB protein of E. coli suggested that SurA specifically facilitates the correct folding of LamB protomers prior to their assembly into trimers3,5. By extrapolation, SurA may suppress misfolding of OMP protomers in general. SurA was first identified as a gene necessary for stationary phase survival6, but is not necessary for growth under normal conditions4.
The amino acid sequence of the mature SurA protein reveals four separate domains: an amino-terminal domain (“N-domain”) of ~150 amino acids, followed by two peptidyl-prolyl isomerase domains of the parvulin class7 (PPIase P1 and P2) of ~100 residues each, and finally, a carboxy-terminal domain (“C-domain”) of ~40 amino acids. The x-ray crystallographic structure of SurA has been solved8. In the tertiary structure, the N-domain, P1 and C-domain form a globular “core fragment”, while the P2 domain is a satellite tethered to the core fragment by polypeptide linkers approximately 30Å in length. A stable core fragment in which the P2 domain and linkers are deleted (“SurA(ΔP2)”), constituted of residues 21-281 plus 390-428, has been expressed9 and crystallized (data not shown). The core fragment has an extended crevice ~50 Å in length that could readily accommodate extended polypeptides; it has been noted that this crevice is an attractive candidate for a site of interaction with peptides9.
Phage display selection experiments, using an M13 library with a randomized amino-terminal 7-residue peptide on the minor coat protein pIII, identified a consensus SurA binding motif of Ar-X1-Ar-X2-P, where Ar is an aromatic residue, X1 is usually polar, X2 is usually aliphatic nonpolar9. In the phage clones that were sequenced, the first aromatic residue of the consensus was either phenylalanine or tryptophan. Parallel, independent selection experiments with SurA and SurA(ΔP2) produced the same consensus binding motif for both protein constructs. The peptide showing the highest affinity for SurA, with sequence WEYIPNV, was synthesized; isothermal calorimetry (ITC) measurements determined a peptide binding constant in the micromolar range for both SurA and SurA(ΔP2). Competition binding experiments with phage-presented SurA-binding peptides and unfolded outer membrane proteins showed that binding of phage-selected peptides competed with, and by implication, mimicked, binding of unfolded OMPs10. Ar-X-Ar tripeptide motifs are common in OMPs, the membrane-embedded fragments of which are antiparallel β-barrel structures with exterior bands of aromatic residues at the extremities of the hydrophobic membrane interior; the Ar-X-Ar motif would place two aromatic residues consecutively on one side of a beta strand.
Of related interest, it has been shown that the carboxy-terminal Ar-X-Ar tripeptide of OMPs is a signal for the periplasmic unfolded protein response11; this raises the additional question of whether SurA would also bind a carboxy-terminal Ar-X-Ar motif, and in doing so, suppress the unfolded protein response.
Binding of SurA and SurA(ΔP2) to cellulose-tethered libraries of 13-residue peptides which scanned the sequences of E. coli LamB, OmpF, and OmpA has also been reported12. These experiments indicate a binding preference for peptides with a high content of aromatic residues in specific configurations, and little preference for proline. Analysis of the peptide sequences of the libraries with the secondary structure prediction program PsiPred suggested that the majority of “good” and “weak” binding peptides would favor a beta strand or random coil structure, while 5% of the good binders and 15% of the weak binders were predicted to favor an alpha helical conformation.
To further characterize the peptide binding specificity of SurA, we have carried out phage display selections using an M13 library presenting a peptide that is both longer and on the opposite terminus of the protein than used previously, a randomized 12-residue peptide on the carboxy terminus of the P8 major coat protein13,14. This has identified peptides that bind SurA with significant affinity, but whose sequences are not consonant with the Ar-X-Ar-X-P consensus derived previously9 or with the patterns derived from peptide arrays12. We have synthesized one of these peptides and characterized its interactions with SurA and subfragments thereof. Additionally, we have crystallized peptide complexes with SurA fragments and have solved structures that show (a) SurA can bind peptides in both extended and helical conformations while asserting a binding preference for aromatic residues in both cases, (b) the specific peptide binding activity is in the P1 PPIase domain, and (c) binding helical peptides induces dimerization of the protein. And, we have determined that SurA fails to bind a representative carboxy-terminal OMP peptide ending with the sequence YQF.
RESULTS
In this work, phage display selection experiments identified a carboxy-terminal peptide of sequence NFTLKFWDIFRK (“C-peptide”) which bound SurA and SurA(ΔP2) nearly as tightly as a relatively high-affinity 7-residue peptide selected previously from a phage library displaying randomized amino-terminal peptides (sequence WEYIPNV, “N-peptide”)9. Competition experiments demonstrated that the binding sites of the C-peptide and N-peptide overlapped. However, the C-peptide did not conform to even the minimal consensus tripeptide pattern of aromatic-polar-aromatic identified with the phage library presenting amino-terminal peptides. We crystallized a complex between the C-peptide and SurA(ΔP2) and solved its structure to 3.4 Å resolution. This revealed a single peptide bound in a helical conformation between two prolyl isomerase domains of a dimeric SurA(ΔP2). Mutation of residues in the binding cleft impaired binding of both the C-peptide and the N-peptide. Consequently, we expressed the first prolyl isomerase domain alone, “SurA-P1”, and crystallized and solved structures of complexes with both the C-peptide and the N-peptide, both at 1.3 Å resolution, revealing the detailed interactions of the peptides with the SurA-P1 domain. The results are described below in logical, rather than chronological, order.
Phage Selection Identifies a Peptide Not Fitting the Previous Consensus
Selection experiments with a library displaying carboxy-terminal 12-residue peptides of random sequence14 identified several peptides with significant affinities for SurA, one of which, denoted hereafter as the C-peptide, was chosen for further characterization. Comparative ELISA assays showed that phage displaying the C-peptide bound SurA with an affinity of the same order as phage displaying the N-peptide (Figure 1A). A competition assay showed that the N-peptide displaced phage presenting the C-peptide from SurA in a concentration-dependent manner, indicating that the two peptides probably have overlapping binding sites (Figure 1B).
Figure 1.
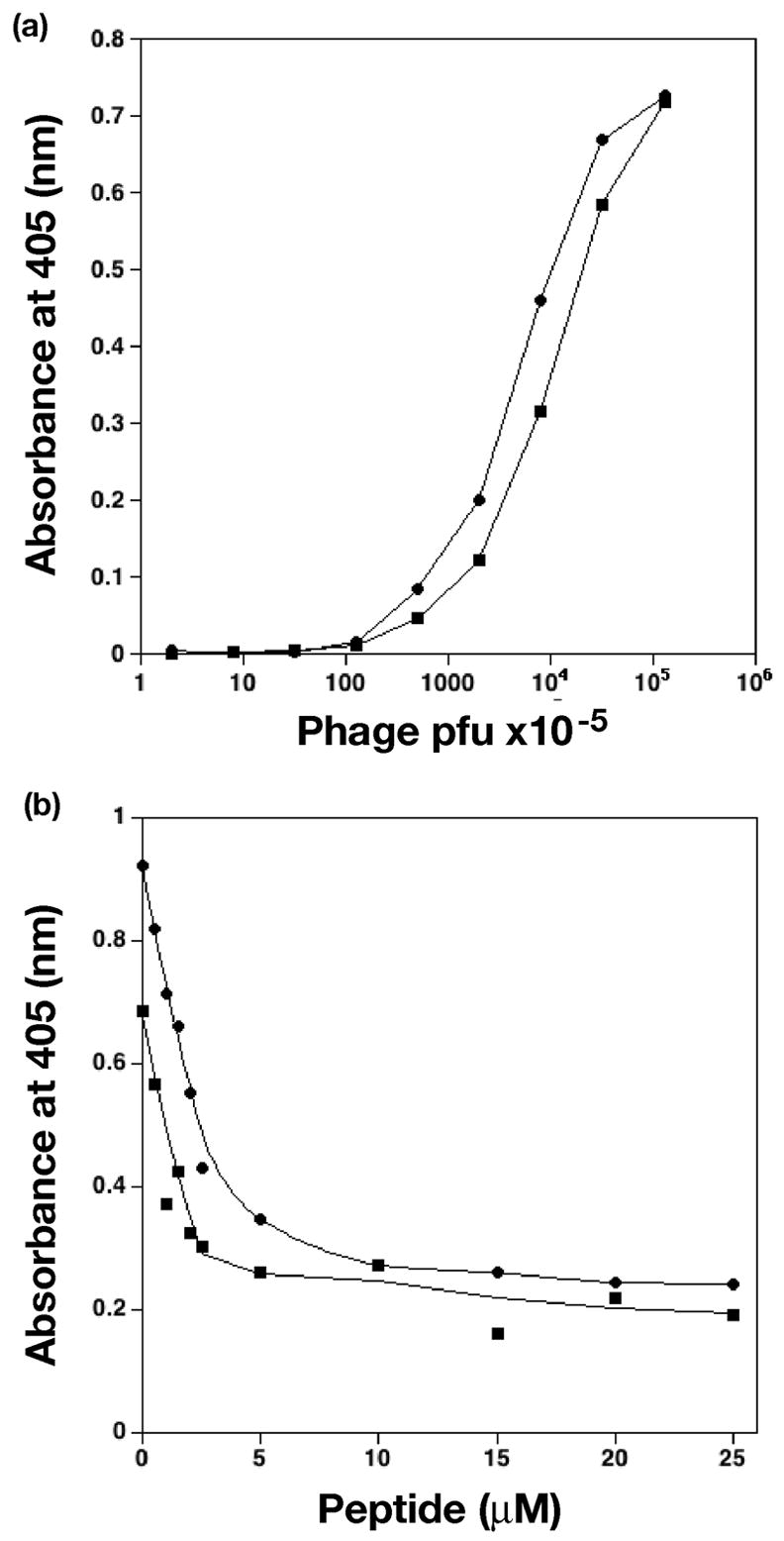
ELISA assays of peptide binding. (a) Comparative ELISA of phage presenting N-peptide (circles) and C-peptide (squares) binding SurA. Consecutive data points are connected by lines. (b) Competitive ELISA. C-peptide presenting phage were 1.0 × 1011 pfu (circles) or 2.5 × 1010 pfu (squares). Concentration of N-peptide is as shown. Smooth curves were fit to experimental data for visual clarity. Graphs were made with the program KaleidaGraph v. 3.6 (Synergy Software).
Isothermal Titration Calorimetry: the C-peptide Binds SurA with Micromolar Affinity
The C-peptide was synthesized for further characterization. Isothermal titration calorimetry (ITC) experiments showed that the C-peptide binds SurA and SurA(ΔP2) with affinities in the micromolar range, and that the affinities are only slightly weaker (approximately twofold) than those of the N-peptide (Table 1). The experiments also indicated a binding stoichiometry of 1:2, peptide:protein, for the C-peptide, in contrast to the 1:1 stoichiometry computed for complexes with the N-peptide. Gel filtration experiments confirmed that the complexes of SurA and SurA(ΔP2) with the C-peptide migrate with an apparent molecular size consistent with two protein protomers in each of the complexes.
Table 1.
Dissociation constants and binding stoichiometry computed from isothermal titration calorimetry data for binding of peptides to SurA and its subfragments.
| Target | N-Peptide | C-Peptide | ||
|---|---|---|---|---|
| na | Kd (μM) | na | Kd (μM) | |
| SurA | 0.88±0.01b | 3.58±0.08b | 0.66±0.03 | 6.62±1.03 |
| SurA(ΔP2) | 1.17±0.01b | 2.23±0.07b | 0.50±0.02 | 5.57±0.64 |
| SurA-P1 | 0.96±0.01 | 0.073±0.003 | 0.65±0.01 | 1.23±0.15 |
computed number of peptide molecules per protein molecule in the complex.
data from Bitto & McKay, 20039 measured in 20 mM phosphate buffer, pH7.3 at 25 °C. Other data measured in 50mM sodium acetate buffer, pH 5.0 at 25 °C as described under “Experimental Procedures”.
Subsequently, ITC and gel filtration experiments were carried out on the isolated prolyl isomerase domain, SurA-P1. The N-peptide binds the P1 domain approximately fifty-fold more tightly than it binds SurA, while the C-peptide binds about fivefold more tightly (Figure 2 and Table 1). As with SurA and SurA(ΔP2), the binding stoichiometry of peptide to protein was 1:1 for the N-peptide and 1:2 for the C-peptide. Gel filtration confirmed that the complex of SurA-P1 with the C-peptide migrates as a protein dimer while the complex with the N-peptide migrates as a protein monomer.
Figure 2.

Isothermal calorimetry of N-peptide and C-peptide binding to SurA-P1. (a) The binding between N-peptide and SurA-P1; (b) The binding between C-peptide and SurA-P1. Data were obtained in 50 mM sodium acetate, pH 5.0 at 25 °C described under “Experimental Procedures”
Structure of the Complex of the N-peptide with SurA-P1
The crystallographic structure of the complex between the N-peptide of sequence WEYIPNV and the SurA P1 domain, determined at 1.3 Å resolution, shows the peptide binds a single protomer of the protein (Figure 3). The protein presents a concave, predominantly nonpolar surface that is approximately 20 Å long and 10 Å wide to which the peptide binds. The binding surface on the protein includes the exposed face of the beta sheet and the loops and short beta strand of the sheet (β2) between helices α2 and α3, specifically residues 223–240. The side chain of Trp1 of the peptide binds in a deep cavity; notably, the base of the cavity is nonpolar, consistent with phage display results that allowed tryptophan or phenylalanine, but not tyrosine, at this position in selected peptides. The planar aromatic ring of Tyr3 of the peptide is sandwiched between a “wall” formed primarily by the aliphatic side chains of Gln223 and Gln224 of the protein on one side, and Pro5 of the peptide on the other. Nonpolar residues Ile4, Pro5 and Val7 of the peptide interact with the extended nonpolar binding surface of the protein. The side chains of polar residues Glu2 and Asn6 of the peptide face the solvent and have little interaction with the protein. The total accessible surface area of the peptide that interfaces with the protein is 648 Å2 and is 72% nonpolar. One measure of contributions of residues to the interface, the percentage of interface accessible surface area that individual residues contribute, is summarized in Table 2. Trp1 of the peptide accounts for 32% of the interface area, while Tyr3 and Val7 each account for approximately 20%. The interactions observed in the structure are consistent with the consensus sequence of (Trp/Phe)-polar-aromatic-nonpolar-Pro derived from phage display for the first five residues of the peptide. From the protein side, Met231 contributes 17% of the interface area, while other residues contribute less than 10% each.
Figure 3.
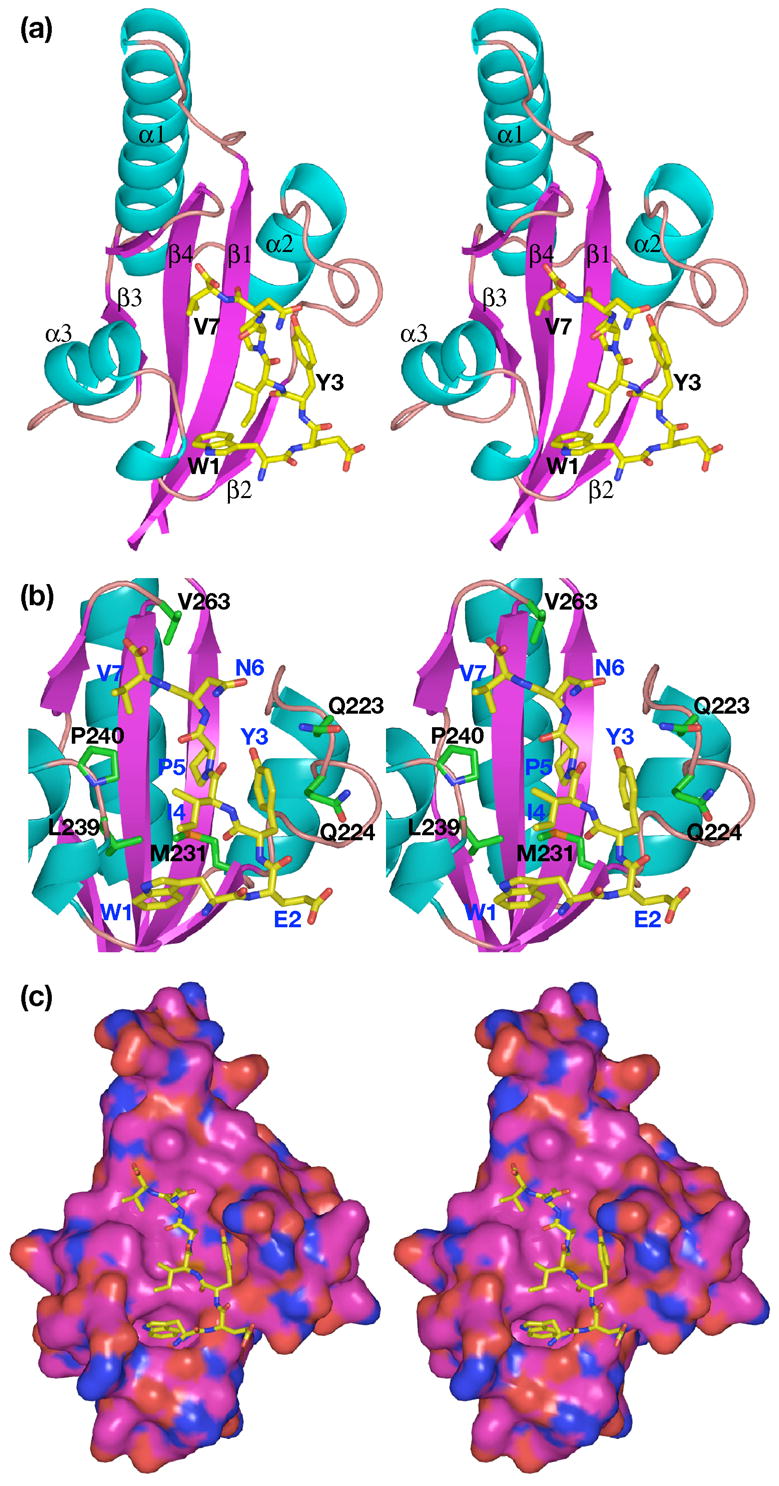
Stereo views of the complex of the N-peptide with SurA-P1. (a) Ribbon drawing of the complex. α-helices and strands of β-sheet are numbered sequentially; note that Pro258 introduces a break in β3. Helices are cyan; β-strands, magenta; oxygen atoms, red; nitrogen atoms, blue; carbon atoms, green on protein, yellow on peptide. (b) View showing interactions between residues on peptide and protein. Protein residues are labeled in black; peptide residues labeled in blue. Colors are same as in (a); additionally, sulfur atoms are orange. (c) Space-filling surface of protein. Colors as above, except carbon atoms of protein are magenta.
Table 2.
Interface accessible surface area (ASA) of complex between N-peptide and monomeric SurA-P1. For SurA-P1, only values >5% are shown. Parameters were computed using the Protein-Protein Interaction Server (http://www.biochem.ucl.ac.uk/bsm/PP/server/).
| SurA-P1 residue | % interface ASA in complex | Interface ASA (A2) |
|---|---|---|
| His178 | 5.0 | 22.7 |
| Gln223 | 6.6 | 29.7 |
| Gln224 | 8.3 | 37.6 |
| Met231 | 17.1 | 77.0 |
| Gly234 | 5.7 | 25.8 |
| Glu238 | 6.2 | 27.9 |
| Leu239 | 7.2 | 32.4 |
| Pro240 | 9.7 | 43.7 |
| Val263 | 6.6 | 29.8 |
|
| ||
| N-peptide | % interface ASA in complex | Interface ASA (A2) |
|
| ||
| Trp1 | 32.1 | 205.8 |
| Glu2 | 3.9 | 24.8 |
| Tyr3 | 19.0 | 122.2 |
| Ile4 | 7.0 | 44.6 |
| Pro5 | 12.6 | 80.8 |
| Asn6 | 5.2 | 33.0 |
| Val7 | 20.3 | 130.3 |
A structure of a different protein of the parvulin class, Pin1, complexed with the dipeptide Ala-Pro is available (PDB 1PIN)15. Pin1 is an active peptidyl-prolyl isomerase, and in the structure, the peptide bond of the bound Ala-Pro dipeptide is in the cis conformation. In the SurA P1-N-peptide structure, the Ile-Pro peptide bond remains in the trans conformation, but the proline is bound at approximately the same position on the protein as in the Pin1-Ala-Pro structure. Many of the residues that line the binding surface are similar in the two proteins; superposition of the domains reveals His178, His266, Phe243 and Leu268 of SurA superimpose on His59, His157, Phe134 and Ile159 of Pin1. Two residues that interact extensively with the peptide in SurA, Met231 and Leu239, are “reversed” in Pin1; Met231 aligns with Leu122 of Pin1 and Leu239 aligns with Met130 of Pin1. A major difference between the two proteins is that Cys113 of Pin1, which is thought to be essential for catalyzing peptide bond isomerization, corresponds to Asp222 of SurA, consistent with the observation that the SurA P1 domain shows no peptide bond isomerization activity16.
Structure of the Complex of the C-peptide with SurA-P1
The crystal structure at 1.3 Å resolution reveals the C-peptide of sequence NFTLKFWDIFRK binds in an α-helical conformation to a dimer of SurA-P1 (Figures 4–5), interfacing to the binding surfaces of both protomers with approximately equal accessible surface area; the peptide surface area involved in binding the first protomer (subunits A and C of the two dimers in the asymmetric unit respectively, using the chain definitions of PDB file 2PV2) is ~660 Å2, and the second protomer (subunits B and D), ~630 Å2. The interface of the peptide to the first subunit of the protein is predominantly through the side chains of Phe2, Phe6, Ile9 and Phe10 (Table 3), with Phe2 bound in the same deep pocket as Trp1 of the N-peptide. Phe2 accounts for approximately 20% of the peptide interface surface area, while the other three residues each account for approximately 15%, and other residues in the peptide each contribute less than 10%. The three aromatic residues therefore contribute half of the interface area.
Figure 4.
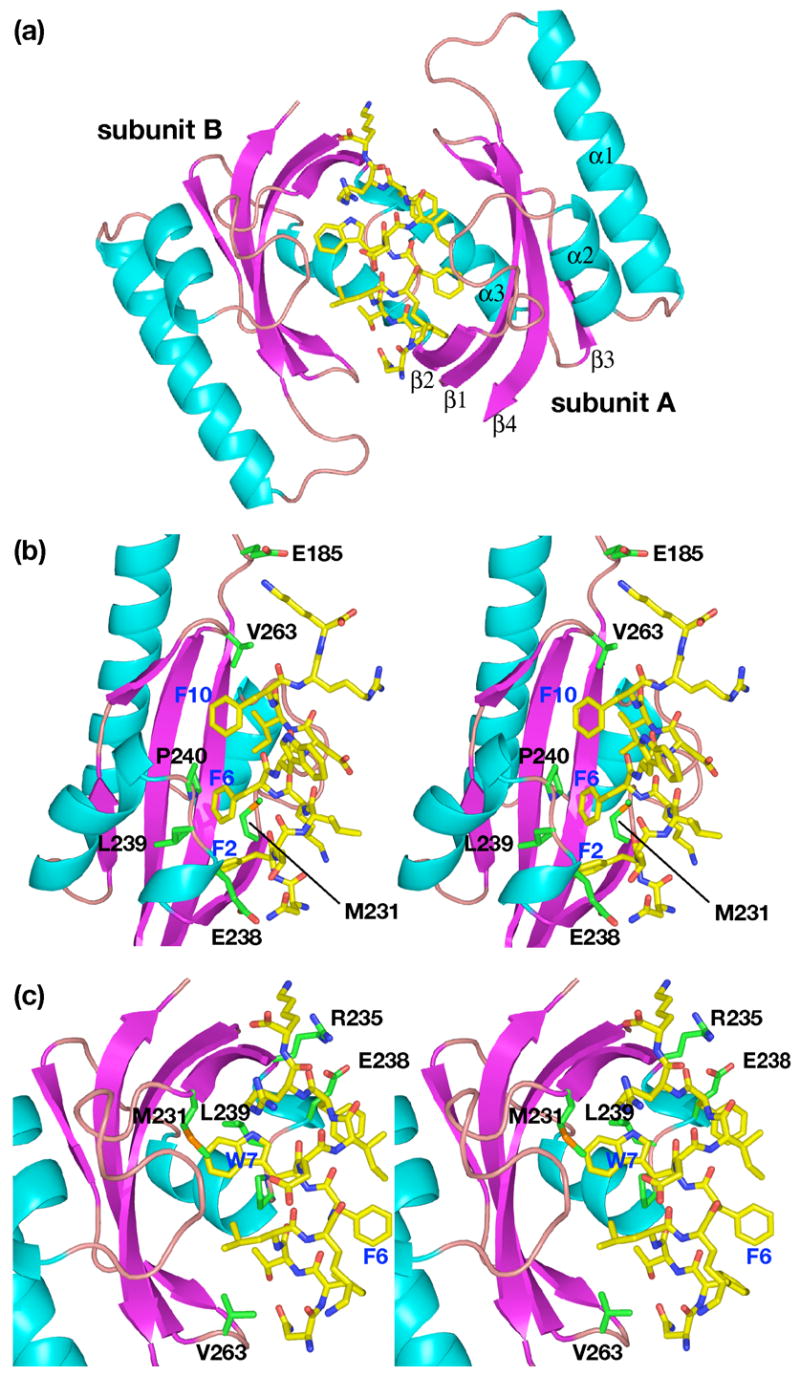
Views of the complex of the C-peptide with SurA-P1. (a) Ribbon drawing of the complex. (b) View showing interactions of peptide with subunit A of SurA-P1 dimer. (c) View showing interactions of peptide with subunit B of SurA-P1 dimer. Colors are same as in Fig. 3.
Figure 5.
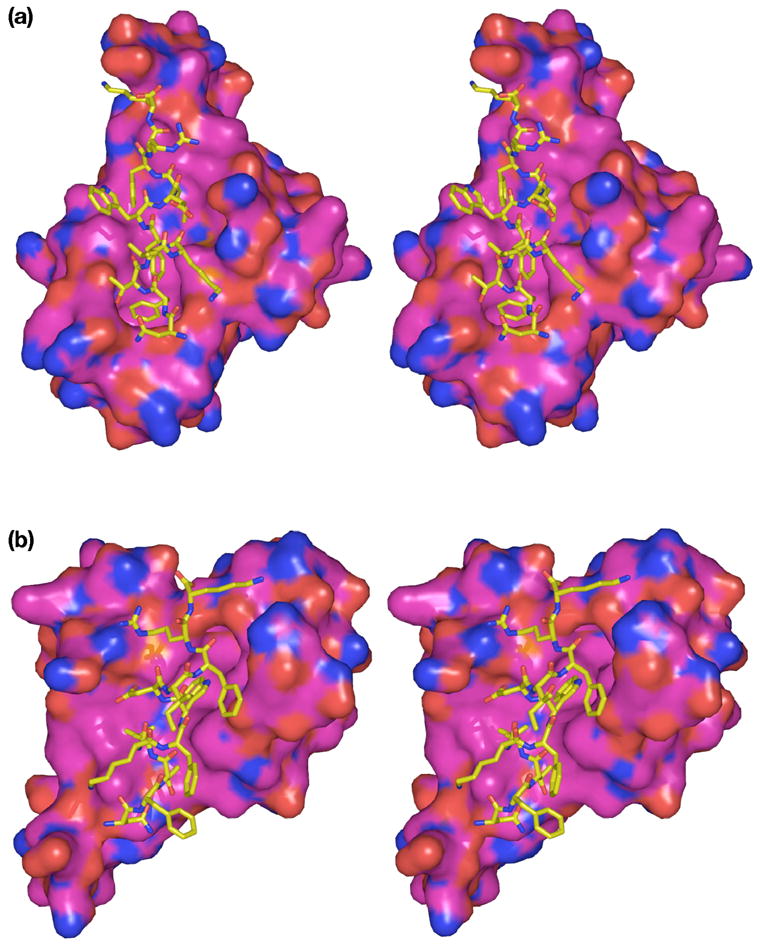
Space-filling views of the complex of the C-peptide with individual subunits of SurA-P1. (a) View of complex between peptide and subunit A of SurA-P1 dimer. (b) View of complex between peptide and subunit B of SurA-P1 dimer. Colors are same as in Fig. 3.
Table 3.
Interface accessible surface area (ASA) of complex between C-peptide and dimeric SurA-P1. Parameters were computed as described for Table 2. Values in the table are averages of two values for the two complexes in the crystallographic asymmetric unit. For SurA-P1, only values >5% in at least one subunit are shown. Values between 0 and 5% are represented as “--”.
| SurA-P1 residue | % interface ASA in complex,subunits A/C | % interface ASA in complex,subunits B/D | Interface ASA (A2)subunits A/C | Interface ASA (A2)subunits B/D |
|---|---|---|---|---|
| Glu185 | 6.8 | -- | 37.3 | -- |
| Gln224 | 5.9 | 6.7 | 32.5 | 37.2 |
| Met231 | 18.5 | 12.4 | 101.7 | 66.8 |
| Trp233 | 6.0 | 7.6 | 33.0 | 40.6 |
| Arg235 | -- | 5.5 | -- | 29.3 |
| Glu238 | 12.6 | 12.2 | 68.8 | 65.2 |
| Pro240 | 7.3 | 9.5 | 40.1 | 50.9 |
| Gly262 | -- | 5.9 | -- | 31.8 |
| Val263 | 9.7 | 9.9 | 53.3 | 52.9 |
|
| ||||
| C-peptide | % interface ASA in complex,subunits A/C | % interface ASA in complex,subunits B/D | Interface ASA (A2)subunits A/C | Interface ASA (A2)subunits B/D |
|
| ||||
| Asn1 | 7.4 | 10.3 | 48.9 | 65.3 |
| Phe2 | 20.5 | -- | 135.8 | -- |
| Thr3 | 5.8 | 11.1 | 38.3 | 70.5 |
| Leu4 | -- | 13.2 | -- | 83.4 |
| Lys5 | 9.7 | -- | 64.2 | -- |
| Phe6 | 16.0 | -- | 105.9 | -- |
| Trp7 | -- | 24.2 | -- | 153.8 |
| Asp8 | 3.0 | 4.1 | 20.0 | 25.9 |
| Ile9 | 15.4 | -- | 102.3 | -- |
| Phe10 | 15.6 | 10.5 | 103.6 | 66.9 |
| Arg11 | -- | 12.3 | -- | 78.1 |
| Lys12 | 6.4 | 14.2 | 42.7 | 90.1 |
On the other subunit of the dimer, the primary interaction is with Trp7, which accounts for 25% of the peptide interface area with this subunit. The tryptophan side chain binds near the middle of the protein; no side chain is intercalated into the deep cavity in this protomer. Smaller contributions are made by several other residues.
Structure of the Complex of the C-peptide with SurA(ΔP2)
The structure of the complex between SurA(ΔP2) and the C-peptide was initially solved to 4.0 Å resolution using three-wavelength MAD phasing from selenomethionine-labeled protein, as detailed under the experimental procedures. It was then refined to 3.4 Å using data from crystals of native protein. Model building relied on the placement of fragments of the known SurA structure using 12 selenium sites per subunit as tether points. To position the helical C-peptide in the correct register, the refined 1.3 Å structure of the complex between the P1 domain and the C-peptide was placed in the model. Throughout refinement, the model retained relatively high B-factors (Table 5), and segments of polypeptide connecting domains whose relative orientations differed significantly from those of the original SurA structure could not be modeled precisely due to the modest resolution of the data. Consequently, the information in the structure is limited to a description of the overall tertiary fold and quaternary interface of the SurA(ΔP2) dimer.
Table 5.
Data collection and refinement statistics for crystals. Data collection statistics computed in HKL200020, refinement statistics computed in CCP428 and CNS21, as described under “Experimental Procedures”.
| Data Collection | SurA(ΔP2)/C-peptide | P1/C-peptide | P1/N-peptide |
|---|---|---|---|
| Spacegroup | P3221 | P21 | C222 |
| Unit cell parameters (Å) | a=148.33, c=188.68 | a=56.46, b=50.63, c=74.06, β=111.54° | a=51.89, b=98.82,c=41.05 |
| Wavelength (Å) | 1.000 | 1.000 | 1.000 |
| Resolution range (last shell) (Å) | 50.0-3.39 (3.52-3.39) | 50.0-1.30 (1.35-1.30) | 50.0-1.29 (1.34-1.29) |
| Unique reflections | 33486 (3326) | 93257 (8714) | 26586 (2504) |
| Completeness (%) | 99.1 (99.8) | 90.2 (96.2) | 93.8 (98.4) |
| Redundancy | 4.2 (3.5) | 2.2 (2.5) | 4.5 (3.5) |
| Rsyma
|
0.074 (0.422) | 0.045 (0.250) | 0.034 (0.126) |
|
Refinement
|
|||
| Resolution range (last shell) (Å) | 41.8-3.39 (3.48-3.39) | 50.0-1.30 (1.38-1.30) | 50.0-1.30 (1.38-1.30) |
| Rcrystb | 0.286 (0.479) | 0.208 (0.256) | 0.226 (0.250) |
| Rfree | 0.298 (0.456) | 0.227 (0.281) | 0.243 (0.293) |
| Number of reflections (working set) | 31581 (2237) | 81232 (11419) | 23302 (3351) |
| Number of reflections (test set) | 1690 (143) | 9046 (1357) | 2552 (419) |
| Number of protein atoms | 4532 | 3324 | 839 |
| Number of water molecules | 0 | 398 | 132 |
| Average B value, protein (Å2) | 177.0 | AB dimerc: 15.9
CD dimer: 18.0 |
17.3 |
| Average B value, peptide (Å2) | 182.2 | Ec: 16.4
F: 18.8 |
22.3 |
| Rmsd bond length (Å) | 0.010 | 0.006 | 0.009 |
| Rmsd angles (°) | 1.26 | 1.48 | 1.58 |
| Ramachandran analysis | |||
| Residues in most favored regions | 80.5% | 95.0% | 93.3% |
| Residues in additional allowed regions | 17.8% | 5.0% | 6.7% |
| Residues in generously allowed regions | 1.3% | none | none |
| Residues in disallowed regions | 0.4% | none | none |
Rsym = Σ|Ihkl − <Ihkl>|/Σ<Ihkl> where Ihkl = single value of measured intensity of hkl reflection, and <Ihkl> = mean of all measured value intensity of hkl reflection.
Rcryst = Σ|Fobs−Fcalc|/ΣFobs where Fobs = observed structure factor amplitude and Fcalc = structure factor calculated from model. Rfree is computed in the same manner as Rcryst, using the test set of reflections. Numbers in parentheses are for highest resolution shell.
Letters refer to chain IDs in PDB file 2PV2.
The C-peptide binds to the prolyl isomerase domain of the SurA(ΔP2) fragment the same way it binds to the isolated prolyl isomerase domain, as described above (Figure 6). The peptide binds in a helical conformation, which requires dimerization of SurA(ΔP2); this, in turn, requires a large conformational change within the SurA(ΔP2) protomer. The conformational change in the protomer can be described as the P1 domains dissociating from the N+C fragment of SurA(ΔP2) and binding to the peptide and to each other, while the N+C fragments independently bind to each other. The dimeric interface between the N+C fragments has an interface accessible surface area of 2480 Å2 independently, the P1 domains, with peptide bound, have an interface area of 330 Å2. There is very little interaction between a P1 domain and a N+C fragment. Interface accessible surface areas between the peptide and the A and B subunits, computed from the 3.4 Å structure, are 530 Å2 and 600 Å2 respectively. These values compare well, but are expected to be substantially less precise, than the values computed from the 1.3 Å structure of the complex between the C-peptide and the P1 domain alone, 660 Å2 and 630 Å2 respectively.
Figure 6.
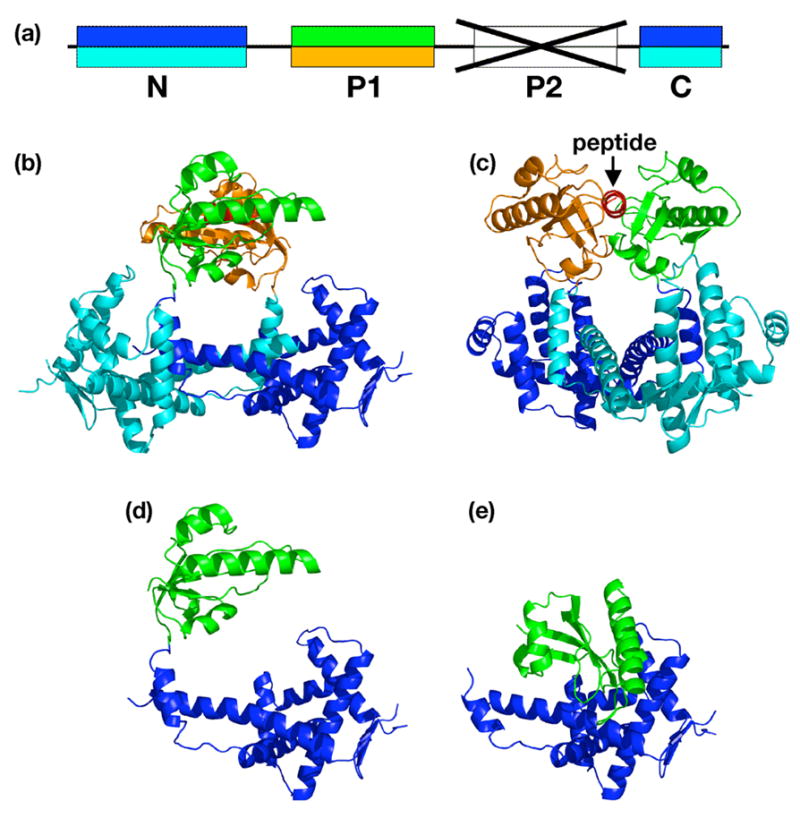
Structure of the complex of the C-peptide with SurA(ΔP2). (a) Color coding of domains in the SurA(ΔP2) fragment. Upper colors, A subunit; lower colors, B subunit. (b) Ribbon drawing of the complex. Peptide is red. (c) Same as B, but rotated 110° around a vertical axis. (d) Conformation of the A subunit alone, in the same orientation as in B. (e) Conformation of the SurA(ΔP2) fragment from the SurA structure, with the N+C domains in the same orientation as D.
Mutagenesis of Residues in the Peptide Interaction Surface of the C-peptide-SurA(ΔP2) Complex
Based on the structure of the complex between the C-peptide and SurA(ΔP2), two residues in the binding site were chosen for mutagenesis : Met231 and Leu129. Each residue was mutated to arginine, thereby introducing a large residue that would potentially clash sterically with the peptide. Mutations M231R and L239R were made for SurA and SurA(ΔP2), and the mutated proteins were expressed and purified. ITC experiments with both the N-peptide and C-peptide were carried out with each mutant for both proteins. In each case, the peptide binding affinity was reduced at least an order of magnitude below that of the wild-type protein, showing that mutation of either of these residues impairs binding of both peptides. We did not attempt to make precise measurements of the binding constants for the mutant proteins.
A Consensus Outer Membrane Protein Carboxy-terminal Peptide does not bind SurA
Many outer membrane proteins have the sequence Y-X-F at the carboxy terminus, where X is typically a hydrophilic amino acid, often glutamine. In selection experiments with phage presenting a carboxy-terminal randomized peptide, no phage emerged that had this terminal motif in their sequence. To directly test whether SurA would bind a peptide having this motif, we synthesized a heptapeptide of sequence VGVNYQF, which is a hybrid of the carboxy-terminal sequences of E. coli OmpF (VGIVYQF) and OmpG (VGVNYSF). ITC experiments with SurA(ΔP2) and SurA-P1 showed that the peptide did not have significant affinity for the protein (data not shown).
DISCUSSION
We have shown that the SurA protein can assert its selectivity for aromatic residues in a diversity of peptides by using alternative quaternary structures to bind peptides in different conformations. We are unaware of other examples of molecular chaperones or other proteins that that use alternative structures to bind different substrates.
Initial phage display experiments with a library presenting an amino-terminal heptapeptide of randomized sequence defined a consensus sequence which binds SurA and SurA(ΔP2) with micromolar affinity, Ar-X-Ar-X-P9. Tripeptides fitting the first three residues of the consensus are common in OMPs; pentapeptides fitting the complete consensus, including the proline in the fifth position, are rare. This incongruity presumably arises because the phage display experiment selects the highest-affinity peptides, with the risk that the consensus sequence that emerges may only be an approximation of the sequences with which the protein interacts in natural substrates. However, competition experiments between phage-presented peptides manifesting the consensus and unfolded OMPs revealed the peptides to be reasonable mimics of the presumed biological substrates of SurA10.
Subsequent experiments with peptide arrays derived from representative OMPs revealed a more general binding preference for segments of peptide with a high content of aromatic residues in particular configurations12. Specifically, the preferred patterns included Ar-X-Ar and Ar-Ar, plus elaborations on these short motifs (Ar-X-Ar-X-Ar and Ar-Ar-Ar-Ar) and patterns with a pair of aromatic residues plus a third separated by fine intervening residues, Ar-Ar-X5-Ar and Ar-X5-Ar-X-Ar.
In this work we sought to extend the delineation of the SurA peptide binding specificity using phage display experiments with a library presenting a randomized carboxy-terminal dodecapeptide. One of the higher affinity peptides that emerged, the C-peptide, has an internal Phe-Trp pair of tandem aromatic residues, but otherwise fails to fit any of the proposed binding sequence patterns. ITC measurements showed the C-peptide binds SurA and SurA(ΔP2) with only two- to three-fold lower affinity than the N-peptide characterized previously9. Somewhat surprisingly, binding of the C-peptide to SurA or SurA(ΔP2) induces dimerization of the protein, while binding of the N-peptide does not.
The crystallographic structure of the complex between the C-peptide and SurA(ΔP2) reveals a dimeric protein complex with a single C-peptide bound in an α-helical conformation between the two P1 PPIase domains. Formation of this complex is predicated on a dramatic conformational change in which the P1 domains essentially dissociate from the N and C domains and bind to each other. The N and C domains of the two protomers separately also bind each other to complete the dimer interface.
This structure, albeit at modest resolution, suggested the selectivity for peptide segments with a high content of aromatic residues is encoded in the P1 domain, contrary to previous reports17. To test this suggestion, we isolated the P1 domain alone and characterized its binding to both the C-peptide and the N-peptide. ITC measurements show that the C-peptide and N-peptide bind to the P1 domain with respectively five-fold and fifty-fold higher affinity than to SurA. (It has not been possible to accurately measure peptide binding to a fragment consisting of only the N and C domains, SurA(ΔP1/ΔP2), due to the instability and aggregation propensity of this protein fragment). Further, the C-peptide induces dimerization of the P1 domains, just as it does with SurA and SurA(ΔP2).
High resolution crystal structures of the C-peptide and N-peptide complexed to the P1 domain reveal the peptide-protein interactions unambiguously. Again, a single C-peptide in an α-helical conformation binds a dimer of P1 domains, with a large fraction of the binding interactions with each P1 protomer due to the aromatic side chains that align on one side of the helix. The N-peptide binds in an extended conformation in a 1:1 complex with P1. In this case also, many of the major binding interactions are with the aromatic residues of the peptide. A notable surface feature of the binding region is the deep cavity that appears to be designed to accommodate an aromatic side chain (Trp1 of the N-peptide; Phe2 of the C-peptide). The overall binding surface, including the cavity, shows some plasticity which allows it to adapt to alternative peptide sequences. In particular, the side chain of Met231, which contributes the largest percentage of interface surface area from the protein in each of the complexes, has substantially different conformations in the monomer and dimer structures, resulting in hydrophobic binding surfaces that are molded to accommodate the different peptides. In the monomer structure, the Met231 side chain extends across the binding cleft and across the top face of the tryptophan side chain, while in the subunits of the dimer structure, it is displaced to the side of the binding cleft and interacts with the aromatic rings of Phe6 and Trp7 respectively.
This demonstrates that the SurA P1 domain is adapted to binding side chains of aromatic residues, and that the SurA protein asserts a selectivity for aromatic residues in a variety of sequence configurations within polypeptides by adopting alternative structures. A monomer of SurA binds peptides in an extended conformation, as we observe for a peptide with an Ar-X-Ar sequence motif. For peptides with a configuration of aromatics that would lie on one face of a helix, SurA undergoes a dramatic conformational change and dimerizes.
What is the implication of this result for the recognition of natural substrates in the periplasm in vivo? It may be argued that the dimeric structure in complex with the C-peptide is an artifact. We disfavor this interpretation in view of (a) the similar affinities of the C-peptide and N-peptide for SurA, (b) the specific nature of the dimeric interface between SurA(ΔP2) protomers, which is unlikely to occur in vitro unless it has been mandated by evolutionary constraint in vivo, and (c) the consistency of the structure of the peptide-protein structure with the documented preference of SurA for peptide segments with high aromatic residue content9,12. We would argue that it is likely the in vitro structural and thermodynamic results accurately reflect in vivo activities, and that SurA is specifically adapted to binding peptides in alternative conformations. This hypothesis can be tested in vivo using SurA mutations that disrupt the dimeric interface but that do not interfere with peptide binding by the monomer.
These results exacerbate the questions of how SurA asserts its chaperone activity, and which domains participate in the activity. It has been shown that genetic constructs expressing a protein in which both PPIase domains are deleted can complement a surA− mutant phenotype almost as well as constructs expressing the full-length SurA protein in an assay that monitors cytoplasmic σE-dependent expression of β-galactosidase. This was taken to imply that the PPIase domains are not essential for in vivo suppression of periplasmic protein misfolding16. Hypothetically, this result could be reconciled with our demonstration that the sequence selectivity of SurA lies in its P1 domain if suppression of polypeptide aggregation and substrate selection are distinct activities. A reasonable model would then be that the extended deep channel of the SurA N+C domains acts as a nonspecific conduit for nascent, unfolded polypeptides once they are translocated into the periplasm, allowing steric shielding from other segments of polypeptide. Preference for unfolded OMPs would be asserted by the amino acid selectivity of the P1 domain. Circumstantial support of this model is suggested by the structural similarity of the N+C domains of SurA to domain III of trigger factor18, a cytoplasmic chaperone which “meets” nascent polypeptides as they emerge from the ribosome. The similar domains of both proteins may suppress protein aggregation through nonspecific sequestering of unfolded segments of polypeptide. It is provocative in this context that in the peptide array data characterizing specific SurA binding peptides, the separation between binding sites (including both high affinity and weak affinity) is generally in the range 10–20 residues, which could readily be sequestered in the ~50 Å channel formed by the N+C domains12. Our structural results provide a framework for clarifying the issues raised here with in vitro and in vivo structure-function studies.
(Parenthetically, it is worth noting that the focus of studies of SurA activity has been on the facilitation of folding and assembly of OMPs1–5. The question of whether SurA also chaperones periplasmic proteins, or periplasmic domains of OMPs, during the folding process has not been addressed thoroughly, although one report cites specific periplasmic proteins which do not require SurA for correct folding1).
Finally, our results resolve the question of whether SurA acts directly as a competitive inhibitor of the unfolded protein response that is signaled via the DegS protease through binding of the Ar-X-Ar motif at the carboxy terminus of OMPs11. A heptapeptide with a hybrid carboxy-terminal OMP sequence ending with YQF failed to bind SurA or SurA(ΔP2) with significant (micromolar) affinity. Thus, SurA does not suppress the unfolded protein response directly by competing with the PDZ domain of DegS for the carboxy-terminal Ar-X-Ar motif of OMPs.
EXPERIMENTAL PROCEDURES
Expression and Purification of Proteins
Mature E.coli SurA (amino acids 21-428) and a core fragment of SurA lacking the second prolyl isomerase domain (SurA(ΔP2), amino acids 21-281 plus 390-428 predicated on an initiator methionine) were expressed and purified as described8,9. An expression vector for the first prolyl isomerase domain (P1, amino acids 172-274 predicated on an initiator methionine) was constructed by amplifying the coding sequence from the SurA expression plasmid and cloning it into the pTYB1 vector using NdeI and KpnI restriction sites. The SurA P1 domain was expressed and purified with essentially the same protocol as the other constructs. Briefly, the expression plasmid was transformed into E. coli BL21(DE3). Cells were grown at 37 °C in Luria-Bertrani (LB) media supplemented with 100 μg/ml ampicillin to a cell density corresponding to A600≈0.6, followed by induction at 25 °C with 0.4 mM isopropyl-B-D-thio-galactoside (IPTG) for 3 hours. After induction, cells were harvested and lysed by sonication. The supernatant was separated by centrifugation and applied to a chitin column (New England Biolabs) at 4 °C. The column was washed extensively with wash buffer (20mM Tris-HCl, 500 mM NaCl, and 2 mM EDTA, pH 7.9), then quickly flushed with 3 column volumes of cleavage buffer (wash buffer plus 30 mM DTT), after which the flow was shut off and the column incubated in cleavage buffer overnight. SurA-P1 protein was the eluted using 3 column volumes of wash buffer.
Seleno-L-methionine (SeMet)-labeled SurA(ΔP2) was expressed in the methionine auxotroph E. coli strain B834(DE3) with 2×M9 minimal salts enriched with 0.4% glucose, 2 mM MgSO4, 25 μg/ml FeSO4·7H2O, 1 μg/ml riboflavin, and 1μg/ml thiamine. Cells were grown at 37°C to a cell density of A600≈0.6, at which point recombinant protein expression was induced by adding 0.4 mM IPTG. Following induction, cells were grown for 4 hours at 25 °C, and then harvested by centrifugation. The SeMet-labeled SurA(ΔP2) was purified by the protocol user for native SurA(ΔP2) except that 1.0 mM dithiothreitol (DTT) was included in all buffers.
Mutations were introduced into the expression plasmids for SurA and SurA(ΔP2) with the “Quik-Change” mutagenesis protocol (Stratagene) using the following primers (mutated bases are underlined ): for M231R, 5′-CGGCGGCCAGCGGGGCTGGGGCCG and its complement; for L239R, 5′-GCCGTATTCAGGAGCGGCCCGGGATCTTCG and its complement. Accuracy of the mutagenesis was confirmed by DNA sequencing of the modified plasmids. Mutant proteins were expressed and purified using the same protocol as for wild-type proteins.
Synthesis of Peptides
Peptides of sequences NFTLKFWDIFRK (“C-peptide”) from selection with a phage library displaying C-terminal peptides (this work) and WEYIPNV (“N-peptide”) from selection with a phage library displaying N-terminal peptides9 were synthesized and purified by high pressure liquid chromatography (HPLC) by the Stanford Protein and Nucleic Acid facility (Stanford, CA). A heptapeptide of sequence VGVNYQF, representing a hybrid of carboxy-terminal sequences of the E. coli outer membrane proteins OmpF and OmpG (carboxy-terminal sequences VGIGYQF and VGVNYSF respectively), was synthesized and purified by HPLC by Genemed Synthesis, Inc (South San Francisco, CA).
Selection of Peptide from C-terminal Phage Display Library
An M13 phage display library presenting 12-residue peptides of randomized sequence on the carboxy terminus of the P8 major coat protein was provided by Dr. Sachdev Sidhu of Genentech14; 19. The phage display selection was performed according to standard protocols13. Phage concentrations were determined spectrophotometrically (A268 = 1.0 for a solution containing 5 × 1012 phage/ml). For each round of selection, phages (1 × 1012) were applied to 8 wells of a 96-well Maxisorp plate precoated with target protein (10μg/well). After 2 hours of incubation, the plate was washed 10 times with Tris-buffered saline/Tween (“TBST”: 50 mM Tris-HCl, 150 mM NaCl, 0.05% Tween-20, pH 7.6). Bound phages were eluted with 0.1 M HCl for 5 minutes and the eluant was neutralized with 1.0 M Tris-HCl, pH 8.0. Eluted phages were amplified in E. coli XL1-blue with M13KO7 helper phage for further rounds of selection. After four rounds of selection, individual phage clones were isolated and analyzed by DNA sequencing.
Affinity Evaluation of Clones by Phage ELISA
Maxisorp 96 well plates were coated overnight at 4 °C in a humidified closed container with 100 μl/well of 100 μg/ml SurA protein in 100 mM NaHCO3, pH 8.6. Protein was omitted from one row of wells as a control to identify plastic-binding phage. The following day, protein solutions were discarded and wells were blocked for 1 hour at room temperature with blocking buffer (100 mM NaHCO3, 5 mg/ml bovine serum albumin, pH 8.6). Plates were washed eight times with TBST. In a separate 96-well plate, serial dilutions of phage into blocking buffer were made, and 100 μl of each dilution was transferred to the plate with protein-coated wells. After 1 hour incubation, plates were washed eight times with TBST, incubated with 100 μl of 1:5000 horseradish peroxidase-conjugated anti-M13 antibody (GE Healthcare) in blocking buffer for 1 hour, and then washed six times with TBST and two times with TBS (=TBST minus Tween-20). To each well, 100 μl of 1-step™ 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) (ABTS) substrate (Pierce) was added and incubated at room temperature for 15 min. Product of the horseradish peroxidase reaction was monitored as optical absorbance at λ=405 nm with a microplate reader.
For peptide competition assays, equal volumes of N-peptide (WEYIPNV) at variable concentration (0, 0.5, 1.0, 1.5, 2.0, 2.5, 5.0, 10.0, 15.0, 20.0, and 25.0 μM) and phage presenting the C-peptide NFTLKFWDIFRK at an invariant concentration were mixed and then aliquoted to the wells of a SurA-coated 96 well plate. ELISA assays were then carried out as described above.
Isothermal Titration Calorimetry
Calorimetry measurements were performed with a VP-ITC microcalorimeter (MicroCal Inc.). For each titration experiments, 1.4 ml protein solution (30–50 μM) in 50 mM sodium acetate, pH 5.0 was added to the sample cell. The peptide was dissolved in the same buffer as the protein at a concentration 0.6–1.0 mM and added to the injection syringe. An initial injection of 1 μl was made, followed by 23 injections of 7 μl at an injection rate of 0.5 μl/s at 25 °C. The equilibration interval was 240s between injections, and the stirring speed was 310 rpm. Binding isotherms were plotted and analyzed using Origin Software (MicroCal Inc.).
Preparation and Crystallization of Peptide-Protein Complexes
Complexes were formed by incubation mixtures of proteins with excess peptides at room temperature for a few minutes. Excess peptides were removed by gel filtration on Superdex-75 (GE Healthcare) and the pooled peaks of protein-peptide complexes were concentrated to 30 mg/ml (for SurA(ΔP2) with C-peptide) or 10 mg/ml (for P1 with either C-peptide or N-peptide). Crystals of the SurA(ΔP2)/C-peptide complex were grown by the hanging drop vapor diffusion method against 0.1 M MES (pH 6.5), 0.1 M KH2PO4, 0.1 M NaH2PO4, and 1.4–1.8 M NaCl during 2–3 days at 18°C. Crystals were transferred to a stabilizing solution containing 0.1 M MES (pH 6.5), 0.1 M KH2PO4, 0.1 M NaH2PO4, and 1.6 M MgSO4 plus 15% ethylene glycol as cryoprotectant and flash-frozen in liquid nitrogen. SeMet-labeled crystals were grown and frozen as above, except that 1 mM DTT and 3% DMSO were included in the crystallization drop.
Crystals of the SurA-P1/C-peptide complex were grown in hanging drops using 24% polyethylene glycol of average molecular weight 8,000 (PEG-8K), 0.1 M imidazole (pH 8.0) as a precipitant over a period of several days at room temperature. Crystals were transferred to a stabilizing solution containing 0.1 M imidazole (pH 8.0) and 24 % PEG 8K plus 20% ethylene glycol as cryoprotectant and flash-frozen in liquid nitrogen. Crystals of the P1/N-peptide complex were grown by sitting drop vapor diffusion method against 0.1 M MES (pH 6.5), 0.2 M (NH4)2SO4, and 25~28% polyethylene glycol monomethylether of average molecular weight 5,000 (PEG-MME-5K) during 3~4 weeks at room temperature. Crystals were transferred to a stabilizing solution containing 0.1 M MES (pH 6.5), 0.2 M (NH4)2SO4, and 30% PEG-MME-5K plus 5% ethylene glycol as cryoprotectant and flash-frozen in liquid nitrogen.
Data Collection and Structure Determination
Diffraction data were collected on beamline 4.2.2 of the Advanced Light Source (ALS) and beamlines 11-1 and 9-2 of the Stanford Synchrotron Radiation Laboratory (SSRL). For crystals of native SurA(ΔP2)/C-peptide complex, data to 3.4 Å resolution were collected on beamline 9-2 and processed with HKL200020. Crystals were trigonal, space group P3221 or P3121, with unit cell parameters a =148.33 Å, c = 188.68 Å. Molecular replacement trials were attempted by using the core fragment of SurA structure (PDB ID 1M5Y) with CNS21 and Molrep22. However, the molecular replacement failed to find a correct solution for the complex. Multiwavelength anomalous dispersion (MAD) data were collected at three wavelengths on ALS beamline 4.2.2 on crystals of SeMet-labeled protein and processed with d*TREK23. Using the direct methods program “Shake-n-Bake”24; 25, 23 Se atoms per asymmetric unit were located and the space group ambiguity (P3221 rather than P3121) was resolved. Se sites were refined and phases were computed to 4.0 Å resolution using solve/resolve26; phasing statistics are summarized in Table 4. (Data were processed to different outer resolutions for the three wavelengths; subsequently, the maximum resolution of useful phasing was determined to be 4.0Å, so that the data were truncated to that resolution. It was not possible to re-process the data remotely with a 4.0 Å cutoff for consistency with the phasing; consequently, the high values of Rsym in Table 4 overestimate the imprecision of the data actually used for phasing). Clear secondary structure was apparent in the resulting experimental map; using selenium sites as tether points, molecular fragments from the SurA crystal structure8 were manually placed in the map. The model was rebuilt using the program Coot27. Further model refinement was performed with CCP428. Eventually, models of the refined domains and peptide from the SurA-P1/C-peptide structure (see below) were placed in the SurA(ΔP2) structure. The final model was refined to 3.4 Å resolution using native data. A representative difference map is shown in Figure 7; data collection and refinement statistics are summarized in Table 5.
Table 4.
Data collection and phasing statistics for SeMet-labeled SurA(ΔP2)/C-peptide.
| Data collection statistics | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Wavelength (Å) | Resolution (highest shell) (Å) | Completeness (%) | Rsyma | f′ | f″ | ||||
| 0.9790 | 48.55–3.30 (3.42–3.30) | 99.9 (100) | 0.189 (0.767) | −8.7 | 5.9 | ||||
| 0.9794 | 48.31–3.80 (3.94–3.80) | 99.8 (100) | 0.123 (0.566) | −10.6 | 3.6 | ||||
| 0.9950 | 48.61–3.60 (3.73–3.60) | 99.8 (100) | 0.115 (0.533) | −3.6 | 0c | ||||
|
| |||||||||
| Diffraction Ratios and Phasing Statistics | |||||||||
|
| |||||||||
| Anomalous Diffraction Ratios | Anomalous Phasing Power | ||||||||
|
|
|||||||||
| Wavelength (Å) | λ1 | λ2 | λ3 | λ1 | λ2 | λ3 | |||
|
| |||||||||
| λ1=0.9790 | 0.1032 | 0.1186 | 0.1046 | 1.9 | 0.3 | 0.9 | |||
| λ2=0.9794 | 0.1137 | 0.1540 | 0.9 | 0.9 | |||||
| λ3=0.9950 | 0.0790 | 0c | |||||||
|
| |||||||||
| Figure of Merit<m> | |||||||||
|
| |||||||||
| Resolution (Å) | 14.2–9.0 | 9.03–7.0 | 7.08–6.0 | 6.01–5.3 | 5.31–4.8 | 4.81–4.4 | 4.43–4.1 | 4.13-4.0 | overall |
| 3 | 8 | 1 | 1 | 1 | 3 | 3 | |||
|
|
|||||||||
| <m> | 0.83 | 0.82 | 0.78 | 0.73 | 0.70 | 0.66 | 0.58 | 0.52 | 0.67 |
Rsym = Σ|Ihkl − <Ihkl>|/Σ<Ihkl> where Ihkl = single value of measured intensity of hkl reflection, and <Ihkl> = mean of all measured value intensity of hkl reflection. Bijvoet measurements were treated as independent reflections for the MAD phasing data sets.
Values of f′ and f″ were initially estimated from an EXAFS scan and refined in SOLVE26.
Taken as the reference for phasing in SOLVE.
Figure 7.
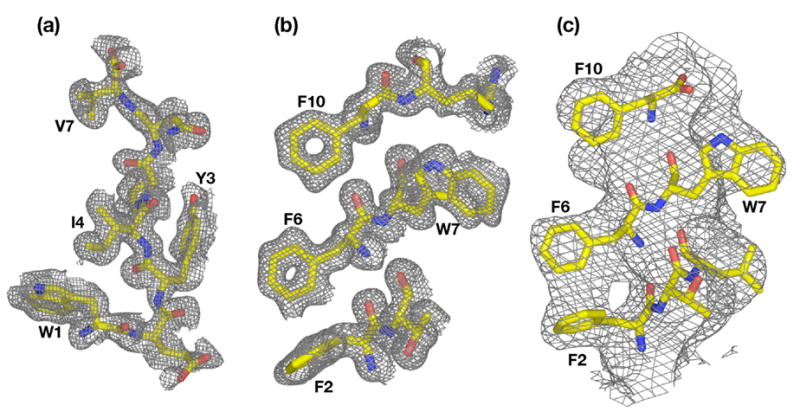
Electron density maps. (a) Fo–Fc simulated annealing omit map of peptide in N-peptide~SurA-P1 structure, computed at 1.3 Å resolution and contoured at 1.0σ. (b) Fo–Fc simulated annealing omit map of a slab of peptide in C-peptide~SurA-P1 structure, computed at 1.3 Å resolution and contoured at 1.0 σ. (c) Fo–Fc simulated annealing omit map of peptide in C-peptide~SurA(ΔP2) structure, computed at 3.4 Å resolution and contoured at 1.0 σ. In each case, the peptide was omitted from the model for simulated annealing.
Data were collected on crystals of SurA-P1/C-peptide and SurA-P1/N-peptide complexes on SSRL beamline 11-1 and 9-2 respectively and processed to 1.3 Å resolution in both cases with HKL200020. Both structures were solved by molecular replacement using PHASER29 with the P1 domain of SurA (PDB ID 1M5Y) at 2.0 Å resolution. Following models were built using Coot27 and refined to 1.3 Å resolution with CNS21. Representative simulated-annealing omit difference maps from the two structures are shown in Figure 7. Data collection and refinement statistics are summarized in Table 5.
Molecular structure figures were prepared with PYMOL (http://pymol.sourceforge.net). Coordinates and structure factors have been deposited in the Protein Data Bank, PDB IDs 2PV1 (SurA-P1/N-peptide), 2PV2 (SurA-P1/C-peptide), and 2PV3 (SurA(ΔP2)/C-peptide).
Acknowledgments
This work was supported by grant GM39928 from the National Institutes of Health to DBM. We thank Dr. Sachdev Sidhu of Genentech (South San Francisco) for providing the carboxy-terminal phage display library and for helpful discussions. Parts of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Science. The SSRL Structural Molecular Biology Program is supported by the Department of Energy and by the National Institutes of Health. Parts of this research were carried out at the Advanced Light Source of the Lawrence Berkeley Laboratory.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lazar SW, Kolter R. SurA assists the folding of Escherichia coli outer membrane proteins. J Bacteriol. 1996;178:1770–1773. doi: 10.1128/jb.178.6.1770-1773.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Missiakas D, Betton JM, Raina S. New components of protein folding in extracytoplasmic compartments of Escherichia coli SurA, FkpA and Skp/OmpH. Mol Microbiol. 1996;21:871–884. doi: 10.1046/j.1365-2958.1996.561412.x. [DOI] [PubMed] [Google Scholar]
- 3.Rouvière PE, Gross CA. SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev. 1996;10:3170–3182. doi: 10.1101/gad.10.24.3170. [DOI] [PubMed] [Google Scholar]
- 4.Justice SS, Hunstad DA, Harper JR, Duguay AR, Pinkner JS, Bann J, Frieden C, Silhavy TJ, Hultgren SJ. Periplasmic peptidyl prolyl cis-trans isomerases are not essential for viability, but SurA is required for pilus biogenesis in Escherichia coli. J Bacteriol. 2005;187:7680–7686. doi: 10.1128/JB.187.22.7680-7686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ureta AR, Endres RG, Wingreen NS, Silhavy TJ. Kinetic analysis of the assembly of the outer membrane protein LamB in Escherichia coli mutants each lacking a secretion or targeting factor in a different cellular compartment. J Bacteriol. 2007;189:446–454. doi: 10.1128/JB.01103-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tormo A, Almiron M, Kolter R. surA, an Escherichia coli gene essential for survival in stationary phase. J Bacteriol. 1990;172:4339–4347. doi: 10.1128/jb.172.8.4339-4347.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahfeld JU, Rucknagel KP, Schelbert B, Ludwig B, Hacker J, Mann K, Fischer G. Confirmation of the existence of a third family among peptidyl-prolyl cis/trans isomerases. Amino acid sequence and recombinant production of parvulin. FEBS Lett. 1994;352:180–184. doi: 10.1016/0014-5793(94)00932-5. [DOI] [PubMed] [Google Scholar]
- 8.Bitto E, McKay DB. Crystallographic structure of SurA, a molecular chaperone that facilitates folding of outer membrane porins. Structure. 2002;10:1489–1498. doi: 10.1016/s0969-2126(02)00877-8. [DOI] [PubMed] [Google Scholar]
- 9.Bitto E, McKay DB. The periplasmic molecular chaperone protein SurA binds a peptide motif that is characteristic of integral outer membrane proteins. J Biol Chem. 2003;278:49316–49322. doi: 10.1074/jbc.M308853200. [DOI] [PubMed] [Google Scholar]
- 10.Bitto E, McKay DB. Binding of phage-display-selected peptides to the periplasmic chaperone protein SurA mimics binding of unfolded outer membrane proteins. FEBS Lett. 2004;568:94–98. doi: 10.1016/j.febslet.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 11.Walsh N, Alba B, Bose B, Gross C, Sauer R. OMP Peptide Signals Initiate the Envelope-Stress Response by Activating DegS Protease via Relief of Inhibition Mediated by Its PDZ Domain. Cell. 2003;113:61–71. doi: 10.1016/s0092-8674(03)00203-4. [DOI] [PubMed] [Google Scholar]
- 12.Hennecke G, Nolte J, Volkmer-Engert R, Schneider-Mergener J, Behrens S. The periplasmic chaperone SurA exploits two features characteristic of integral outer membrane proteins for selective substrate recognition. J Biol Chem. 2005;280:23540–23548. doi: 10.1074/jbc.M413742200. [DOI] [PubMed] [Google Scholar]
- 13.Sidhu SS, Lowman HB, Cunningham BC, Wells JA. Phage display for selection of novel binding peptides. Meth Enzymol. 2000;328:333–363. doi: 10.1016/s0076-6879(00)28406-1. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Yeh S, Appleton BA, Held HA, Kausalya PJ, Phua DC, Wong WL, Lasky LA, Wiesmann C, Hunziker W, Sidhu SS. Convergent and divergent ligand specificity among PDZ domains of the LAP and zonula occludens (ZO) families. J Biol Chem. 2006;281:22299–22311. doi: 10.1074/jbc.M602902200. [DOI] [PubMed] [Google Scholar]
- 15.Ranganathan R, Lu KP, Hunter T, Noel JP. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell. 1997;89:875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 16.Behrens S, Maier R, de Cock H, Schmid FX, Gross CA. The SurA periplasmic PPIase lacking its parvulin domains functions in vivo and has chaperone activity. EMBO J. 2001;20:285–294. doi: 10.1093/emboj/20.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Webb HM, Ruddock LW, Marchant RJ, Jonas K, Klappa P. Interaction of the periplasmic peptidylprolyl cis-trans isomerase SurA with model peptides. The N-terminal region of SurA id essential and sufficient for peptide binding. J Biol Chem. 2001;276:45622–45627. doi: 10.1074/jbc.M107508200. [DOI] [PubMed] [Google Scholar]
- 18.Ludlam AV, Moore BA, Xu Z. The crystal structure of ribosomal chaperone trigger factor from Vibrio cholerae. Proc Natl Acad Sci U S A. 2004;101:13436–13441. doi: 10.1073/pnas.0405868101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuh G, Pisabarro MT, Li Y, Quan C, Lasky LA, Sidhu SS. Analysis of PDZ domain-ligand interactions using carboxyl-terminal phage display. J Biol Chem. 2000;275:21486–21491. doi: 10.1074/jbc.275.28.21486. [DOI] [PubMed] [Google Scholar]
- 20.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meth Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 21.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 22.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 23.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr. 1999;D55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 24.Miller R, Gallo SM, Khalak HG, Weeks CM. SnB: Crystal-structure determination via Shake-and-Bake. J Appl Cryst. 1994;27:613–621. [Google Scholar]
- 25.Weeks CM, Miller R. The design and implementation of SnB version 2.0. J Appl Cryst. 1999;32:120–124. [Google Scholar]
- 26.Terwilliger TC, Berendzen J. Automated Structure Solution for MIR and MAD. Acta Crystallogr. 1999;D55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Bailey S. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 29.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr. 2005;D61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]