

Abstract
The inwardly rectifying potassium channel Kir4.1 is widely expressed by astrocytes throughout the brain. Kir4.1 channels are absent in immature, proliferating glial cells. The progressive expression of Kir4.1 correlates with astrocyte differentiation and is characterized by the establishment of a negative membrane potential (> −70 mV) and an exit from the cell cycle. Despite some correlative evidence, a mechanistic interdependence between Kir4.1 expression, membrane hyperpolarization, and control of cell proliferation has not been demonstrated. To address this question, we used astrocyte-derived tumors (glioma) that lack functional Kir4.1 channels, and generated two glioma cell lines that stably express either AcGFP-tagged Kir4.1 channels or AcGFP vectors only. Kir4.1 expression confers the same K+ conductance to glioma membranes and a similar responsiveness to changes in [K+]o that characterizes differentiated astrocytes. Kir4.1 expression was sufficient to move the resting potential of gliomas from −50 to −80 mV. Importantly, Kir4.1 expression impaired cell growth by shifting a significant number of cells from the G2/M phase into the quiescent G0/G1 stage of the cell cycle. Furthermore, these effects could be nullified entirely if Kir4.1 channels were either pharmacologically inhibited by 100 μM BaCl2 or if cells were chronically depolarized by 20 mM KCl to the membrane voltage of growth competent glioma cells. These studies therefore demonstrate directly that Kir4.1 causes a membrane hyperpolarization that is sufficient to account for the growth attenuation, which in turn induces cell maturation characterized by a shift of the cells from G2/M into G0/G1.
Keywords: potassium channel, resting membrane potential, proliferation, glioma cells
INTRODUCTION
The biophysical properties of glial cells have been extensively studied in many systems (Olsen and Sontheimer, 2005; Verkhratsky and Steinhauser, 2000; Walz, 2002) and these studies unexpectedly have demonstrated the presence of a range of voltage-gated ion channels previously suspected to be present only in excitable cells. These studies also confirmed earlier findings showing that astrocytes in particular have an unusually high K+ conductance and consequently very negative resting membrane potentials (Kuffler, 1967; Ransom and Goldring, 1973). Closer examination identified this K+ conductance as being mediated, to a large degree, by inwardly rectifying K+ channels that are sensitive to Ba2+ (Newman, 1993; Ransom and Sontheimer, 1995). This group of channels is comprised of 16 cloned subfamilies (KCNJ1-16) (Nichols and Lopatin, 1997; Olsen et al., 2006) and at least four of these (Kir 2.1, 2.2, 2.3, and 4.1) have been identified in cortical astrocytes (Schroder et al., 2002). Yet, based on studies in transgenic animals, it appears that one member in particular, Kir4.1, is the predominant K+ channel in mature astrocytes and almost solely responsible for establishing the astrocytes negative resting membrane potential (RMP) (Kofuji et al., 2000; Olsen et al., 2006). Astrocytes from Kir4.1 knockout animals demonstrate depolarized RMP to −40 mV in cultured spinal cord astrocytes (Olsen et al., 2006) and to −15 mV in retinal glial cells from these animals in situ (Kofuji et al., 2000). Deletion of Kir4.1 in oligodendrocytes also resulted in depolarized RMP to −35 mV and showed prominent defect in loss of myelination (Neusch et al., 2001).
Preceding the earlier-described molecular studies, a significant effort went into characterizing the functional roles of the various ion channels expressed in astrocytes. These studies (reviewed in Bringmann et al., 2000; Sontheimer, 1992) suggest that the ion channel complement of astrocytes and the lineage of oligodendrocytes change markedly during the late embryonic and early postnatal period. Most notably, immature glial cells lack expression of Kir channels but prominently express delayed rectifying and transient K+ channels and, to a lesser degree, voltage-gated Na+ channels (Bordey and Sontheimer, 1997; MacFarlane and Sontheimer, 2000b; Sontheimer, 1994). These channels are gradually replaced during early postnatal development with Kir channels. This shift in channel expression correlates directly with a negative shift in RMP from about −50 to −80 mV (Bordey and Sontheimer, 1997; Ransom and Sontheimer, 1995). Importantly, it also correlates with a loss of cell proliferation and enhanced differentiation. Indeed, only glial cells with a depolarized resting potential, lacking Kir channels, incorporate BrdU, and thus are actively dividing (Bordey et al., 2001). Moreover, experimental induction of cell differentiation using differentiation agents, such as retinoic acid, caused the precocious induction of Kir channel expression with a negative shift in resting potential as cells exited the cell cycle and became postmitotic (MacFarlane and Sontheimer, 2000a). Similar changes accompany injury-associated cell proliferation where cells, preceding division, lose functional Kir channels resulting in a depolarization of their resting potential (MacFarlane and Sontheimer, 1997). Although the importance of the cell’s RMP as a control switch in cell cycle entry and progression has been debated for many years (Pardo, 2004; Wonderlin and Strobl, 1996), the mechanistic linkage between resting potential and cell proliferation is still missing.
In light of these findings, we used astrocyte-derived tumors, i.e., gliomas as a cellular model system, to further establish a mechanistic link between cell division, cell resting potential, and Kir channel expression. These cells offer several distinct advantages that we exploited. First, glioma cells are highly proliferative and believed to arise from astrocyte or their progenitor cells as astrocytic lineage tumors (Arai et al., 1991; Studer et al., 1985). Second, at the protein level, these gliomas express the same complement of Kir channels found in nonmalignant astrocytes. However, these channels are not functionally expressed at cell surface membrane (Olsen and Sontheimer, 2004). As a result, glioma cells have a resting potential between −20 and −40 mV and are not responsive to changes in K+, thus lacking any significant resting K+ permeability.
Using these cells enabled us to selectively overexpress fluorescently-tagged Kir4.1 channels. Now we asked if Kir expression was sufficient to restore many of the properties of nonmalignant mature astrocytes. Characteristic properties include high K+ permeability, establishment of a negative resting potential along with the ability to take-up K+, and importantly, effectively suppressed proliferation. With this approach, we showed directly that Kir4.1 expression is sufficient to shift the RMP of glioma cells to −80 mV, and that this, in turn, caused growth inhibition by shifting cells from the G2/M into G0/G1 phase of the cell cycle. Inhibition of Kir channels or depolarization of the cell membrane were both sufficient to nullify the growth inhibiting effect of Kir4.1, thus establishing a clear mechanistic link between Kir4.1 (RMP) and cell proliferation.
METHODS
Cell Culture
D54-MG (WHO Grade IV) was a gift from D. D. Bigner, Duke University, Durham, NC). All cell lines including D54-MG-AcGFP or D54-MG-Kir4.1AcGFP cell lines were grown in Dulbecco’s modified Eagle medium (DMEM/F12; Media Tech, University of Alabama at Birmingham, Media Preparation Facility) with 7% fetal bovine serum (Hyclone Logan, UT) and 1% (2 mM) glutamine (Media Tech) in a humidified 37°C and 90% O2/10% CO2 incubator. Primary spinal cord astrocyte cultures were obtained from P0 Sprague Dawley rat pups by an established protocol described previously (Olsen and Sontheimer, 2004).
Construction of the Kir4.1-AcGFP Plasmid and the Generation of D54-MG-AcGFP and D54-MG-Kir4.1AcGFP Cell Lines
The cDNA coding sequence of mouse Kir4.1 (Gene-bank accession number AF 322631) from Kir4.1-pBlue-script (kindly provided by Dr. L. C. Tempi, at the University of California at San Francisco) (Li et al., 2001) was subcloned into EcoR I and Sal I sites of the multiple cloning site of Aequorea coerulescens derived green fluorescence gene (AcGFP) containing pAcGFP-N1 mammalian expression vector (Clontech, Palo Alto, CA). The primer sequences used to subclone Kir4.1cDNA from pBluescript to pAcGFPN-1 were 5′-AAGAATTCTTCG AGCCAAGATGACGT-3′ and 3′-AAAGTCGACCCGACG TTGCTGATGCG-5′. D54-MG glioma cell lines were transiently transfected with an AcGFP as a mock control or Kir4.1AcGFP plasmid. Transfection of suspended cells was achieved by using an Amaxa Nucleofector kit (Amaxa, Gaithersburg, MD) with Amaxa program T-27. At 48-h post-transfection, cells were treated with geneticin (1 mg/mL; Sigma) for 3 weeks to select D54-MG cell colonies that stably carried the AcGFP or Kir4.1AcGFP plasmids containing the kanamycin resistant gene. Colonies were then selected and sorted by fluorescence-activated cell sorter (FACS) and single cells were sorted into each 96-well plates and observed for cloned growth for 2–3 weeks. Single colonies stably expressing D54-MG-AcGFP or D54-MG-Kir4.1AcGFP were then selected to culture for all population studies.
Immunocytochemistry
Cells plated on coverslips were washed twice 10 min each with ice-cold phosphate buffer saline (PBS) solution and subsequently fixed with 4% paraformaldehyde for 15 min. Following the fixation, cells were washed four times with ice-cold PBS for 10 min each. Cells were then blocked with blocking buffer (BB, 10% normal horse serum + 0.1% Triton X-100 in PBS) for 1 h. Rabbit anti-Kir4.1 primary antibody (1:500; Alamone Lab, Jerusalem, Israel) was added into diluted BB (1:3 dilution in PBS) and incubated overnight at 4°C. The following day, cells were washed with diluted BB for three times at 10 min each, and the secondary goat anti-rabbit antibody conjugated to Alexa fluoro 596 (1:500 dilution) was incubated for 2 h at room temperature. Cells were then washed twice with diluted BB at 10 min each, and incubated with DAPI for 5 min (0.5 μg/mL; Sigma), followed by two washes and mounted on slides with Gel-Mount (Biomedia, Foster City, CA). Images were acquired by an inverted Zeiss Axiovert 200M microscope (Carl Zeiss, Gottingen, Germany) by an oil immersion 63× magnification objective using Axio-Vision 4.6 software (Carl Zeiss). To further resolve the difference in Kir4.1 channel expression in intracelluar localization between D54-MG-AcGFP and D54-MG-Kir4.1AcGFP cells, we obtained series of z-stacked images utilizing an Olympus IX81 spinning disk microscope (Melville, NY), with images analyzed using Slide Book Image software (Intelligence Innovation Imaging, Denver, CO).
Western Blotting
Cells were homogenized with lysis buffer containing 100 mM Tris, 1% SDS at pH 7.5, supplemented with protease and phosphatase inhibitors. Protein concentration of the homogenate was measured by Lowry protein assay (Sigma, St. Louis, MO) and 25 μg of total protein was loaded onto a 4–20% pre-cast gel (Bio-Red). Gel separation times were 2 h. The gel was transferred to Immobilon PTM PVDF membrane (Millipore, Bedford, MA). The membrane was blocked with 5% nonfat dry milk, 2% goat serum, 2% BSA in TBST solution (Tris-base 20 mM, NaCl 137 mM, 0.1% with Tween 20, pH 7.6) for 2 h at room temperature and then incubated with affinity-purified rabbit polyclonal antibodies for Kir4.1 (1/400 dilution; Alamone lab, Jerusalem, Israel) for 2 h. After washes in TBST buffer, the blots were incubated with 1:2,000 horseradish peroxidase-labeled goat anti-rabbit IgG (Santa Cruz Biochemistry, Santa Cruz, CA) for 1 h at room temperature. The bands were detected with an enhanced chemiluminescent detection kit (ECL+) (Amersham Pharmacia Biotech, Piscataway, NJ) and the signal was detected using a Kodak image-Quant software (Eastman Kodac, Rochester, NY).
Electrophysiology
Whole-cell voltage-clamp recordings of D54-AcGFP or D54-Kir4.1AcGFP cell lines were obtained using standard techniques (Hamill et al., 1981). The patch pipettes for both recording conditions were made from thin-walled borosilicate glass (1.55 mm OD, 1.2 mm ID WPI, TW150F-40) on a PP-83 puller (Narishige, Japan). Patch pipette resistances were 4–6 MΩ when filled with pipette solution (in mM) (145 KCl, 0.2 CaCl2, 1.0 MgCl2, 10 EGTA, and 10 HEPES) at pH 7.2 and 300 mOsm. Bath solutions were made without Mg2SO4, NaH2PO4, or Na2HSO4 (in mM) (5.0 KCl, 125 NaCl, 1.0 CaCl2, 10.5 glucose, and 32.5 HEPES acid) at pH 7.4 and 300 mOsm to prevent precipitation with the Kir inhibitor 100 μM BaCl2. For 20 mM KCl bath solution, 15 mM NaCl was omitted to compensate the osmolarity for a higher KCl. Recordings were performed using a Multiclamp-700A amplifier (Axon Instruments, Foster City, CA). Current signal was filtered at 1 kHz utilizing a Digidata 1322 digitizer. Data acquisition and storage were performed by pClamp 9.0 (Axon Instruments). All cells included for analysis had series resistances of less than 10 MΩ. A 90% series resistance compensation were used to minimize the voltage errors.
Perforated patch recordings were used only to measure the cell’s RMP to eliminate the washout affect seen in whole-cell patch clamp recordings. Pipettes contained amphotericin-B (60 mg/mL) and Alexa fluoro dye 546 (Molecular Probes) to assure membrane integrity (Rae et al., 1991). After establishment of a giga-seal, a slow ramp protocol (− 180 to + 180 mV) was run every 30 s until maximum current amplitude and appropriate input resistance were established (100 MΩ in D54-MG-AcGFP cells and 10–20 MΩ D54-MG-Kir4.1AcGFP cells). Then, the whole cell capacitance and series resistance were adjusted and a reading of the RMP was obtained in current clamp mode at I = 0. D54-MG-AcGFP or D54-MG-Kir4.1AcGFP cells were plated on glass cover-slips with 20,000 cells per wells in a 24-well plate, and recordings were made 2–5 days following the plating. Spinal cord astrocytes were recorded at <5 days range in vitro for immature astrocytes and >8 days range in vitro (DIV) for mature astrocytes.
Cell Proliferation Assay with FACS and BrdU Staining
D54-MG-AcGFP or D54-MG-Kir4.1AcGFP cells were plated onto six-well plates (Becton Dickenson, Bedford, MA) at a density of 5 × 104 cells/mL. For each sample, media were removed on Day 0, washed twice with sterile PBS solution, and treated with media containing 7% serum. BaCl2 or high KCl solution was added directly to each well containing 2 mL of media to reach final concentration of 100 μM or 20 mM, respectively. Media were changed everyday during the duration of 5-day experiment. Cell growth was examined by trypsinizing and resuspending cell pellets into 10 mL PO4−, SO4− free bath solution and then counting cell aliquots using a Coulter Counter (Beckman, Miami, FL) in triplicate, for each time point. The rate of Kir4.1 expressing D54-MG cell proliferation was compared with D54-MG-AcGFP cell proliferation by normalizing to the cell number at Day 0.
BrdU incorporation experiments were performed using 48-h postplated D54AcGFP, D54Kir4.1AcGFP cells with or without 100 μM BaCl2 or 20 mM KCl on coverslips (2 × 104 cells/24-well plates) incubated with BrdU (10 μmol; EMD Chemicals, San Diego, CA) for 6 h. Anti-BrdU staining was performed by a modified protocol of Zupanc (1998). Briefly, cells plated on coverslips were washed twice 10 min each with PBS solution and subsequently fixed with 4% paraformaldehyde for 15 min. Following the fixation, cells were washed four times with PBS for 10 min each. DNA was denatured by 2 M HCl (in PBS with 0.3% Triton) for 30 min at room temperature, rinsed three times with diluted BB (1:3 dilution in PBS), and then blocked with blocking buffer (BB, 10% normal horse serum + 0.1% Triton X-100 in PBS) for 1 h. Mouse anti-BrdU primary antibody (1:100; DAKO, Carpinteria, CA) was added into and incubated overnight at 4°C. The following day, coverslips were washed 3× with diluted BB, and incubated with secondary donkey anti-mouse antibody conjugated to Alexa 594 fluoro for 1 h at room temperature. Cells were then washed twice with diluted BB at 10 min each, and incubated with DAPI for 5 min (0.5 μg/mL; Sigma), followed by two washes and mounted on slides with GelMount (Bio-media). Images of five random fields from each coverslip were acquired by an inverted Zeiss Axiovert 200M microscope (Carl Zeiss) by 20× magnification objective using Axio-Vision 4.6 software (Carl Zeiss). All experiments were performed in triplicate coverslips from each condition on three separate experiments. The % BrdU+ cells was calculated as (number of BrdU+ cells/total number of cells) × 100.
Live/Dead Cell Assay
Flow cytometry viability assay protocol from Live/Dead Cytotoxicity Assay Kit (Invitrogen, Carlsbad, CA) was used. Briefly, cells were plated at same density as the cell proliferation assay. At Days 1–5, cells were trypsinized, resuspended with PBS. Eight micromolar of the ethidium bromide homodimer and 0.1 μM calcein AM mixed solution were added to the resuspended pellet and incubated for 20 min at room temperature in the dark. Stained cells were analyzed by flow cytometry sorting. Live cells were indicated by calcein emitting at 530 nm and dead cells were indicated by ethidium bromide intercalated into the nucleus emitting at 585 nm.
Cell Cycle Analysis
Propidium iodide staining followed by FACS cell cycle distribution profiles allowed us to compare the distribution of cells in the G1, S, or G2/M stage of the cell cycle. We compared the glioma cell line that expressed Kir4.1-AcGFP protein with those that expressed AcGFP protein alone. Both D54-Kir4.1AcGFP cells and the D54-AcGFP cells were incubated with or without 100 μM of BaCl2, a specific Kir channel inhibitor that abrogates the effects of the Kir4.1 channel function for 48 h. Both cell lines were also incubated with 20 mM KCl for 48 h, which depolarizes the Kir4.1 expressing glioma cell RMP around −40 mV to test whether proliferation rate could be restored by similar depolarized RMP that is possessed by glioma cells. Cells were plated on 100-mm dishes at 20 × 105 cells per 8 mL media. On the second day (Day 0), media were changed and BaCl2 or KCl solution was added directly to each dishes. Following 5 days (Days 1–5), cells were trypsinized and the pellet was resuspended in PBS solution. Two-milliliter ice cold 70% ethanol was added dropwise while vortexing the resuspended cells and incubated at −20°C for 15 min. Cells were rehydrated with PBS for 30 min, cells were washed with PBS thrice, then the cell pellet was treated with 50 μL (100 μg/mL) ribonuclease, and incubated for 15 min at room temperature. Following the incubation, 200 μL of propidium iodide (50 μg/mL) was added on top of ribonuclease solution, incubated for 30 min, and analyzed by FACS.
Data Analysis
All electrophysiological data acquisition, storage, and statistical analysis were performed by Clampfit and pClamp version 9 (Axon Instruments). One way ANOVA was performed for all the data analysis which had normal distribution. All values are reported as mean ± SEM and significance was denoted with asterisks, when the P < 0.05 or when the P < 0.01.
RESULTS
Kir4.1 Channels Overexpressed in D54-MG Glioma Cells Localize to Discrete Regions on the Cell Surface
To examine growth regulatory effects of Kir4.1 and to functionally link channel expression and cell proliferation, we used astrocyte-derived tumor cells (gliomas) as a model system. Gliomas represent an ideal system to address this question since they are of glial origin, lack functional Kir channels, and are highly proliferative. As a logical first step, we demonstrated an absence of functional Kir4.1 channels in wild-type gliomas and showed that both the protein and functional channels can be established by overexpression of recombinant Kir4.1. We utilized the patient-derived glioma cell line, D54-MG, which we have previously shown to lack functional Kir4.1 channels (Olsen and Sontheimer, 2004). Of note, this cell line expressed the same 52 kDa Kir4.1 protein band that is also found by western blot analysis in cortical and spinal cord astrocytes (Fig. 1D). However, previous immunocytochemistry results (Olsen and Sontheimer, 2004) repeated here (Fig. 1A) demonstrated that endogenous Kir4.1 channels were predominantly found in the nuclear membranes. By contrast, overexpression of Kir4.1AcGFP in these D54-MG cells caused more diffuse Kir4.1 expression through the cytoplasm, and, importantly, caused discrete puncta of Kir4.1 on the cell surface (Figs. 1B,C). To show this difference in subcellular localization more clearly, we present enlarged confocal images in Fig. 1C in which a control, mock transfected glioma cell are shown side-by-side with a Kir4.1 expressing glioma cell.
Fig. 1.
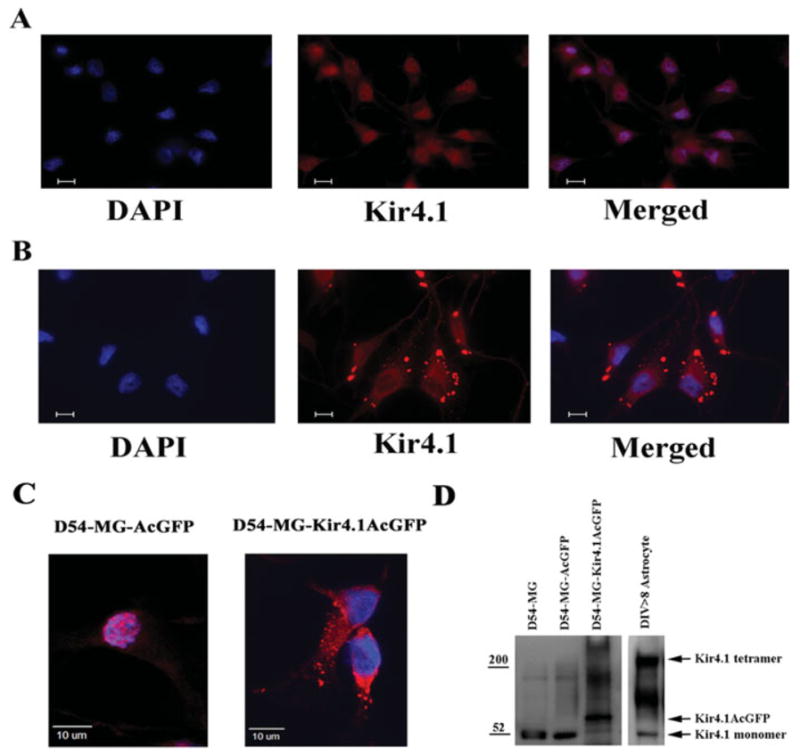
Kir4.1 localizes to discrete regions on the cell membrane. (A) In D54-MG-AcGFP cells, Kir4.1 protein (red Kir4.1 immunoreactivity) localizes to cell nuclei (blue DAPI) and surrounding perinuclear regions. (B) In contrast, D54-MG-Kir4.1AcGFP cells show punctuate membrane associated expression of Kir4.1 protein. Bar represents 10 μm. (C) Magnified confocal image of D54-MG-AcGFP (left: nuclear expression of Kir4.1 channel) and Kir4.1MG-AcGFP (right: diffuse punctuate expression of Kir4.1 channel). (D) Western blot comparison of Kir4.1 expression between lysates from wild-type D54MG, mock transfected D-54-MG-AcGFP, Kir4.1 overexpressing D-54-MG-Kir4.1-AcGFP, and astrocytes.
To show Kir4.1 expression biochemically, we compared control and Kir4.1 transfected glioma cells and astrocytes by western blot in Fig. 1D. As previously reported (Olsen and Sontheimer, 2004), the ~52 kDa Kir4.1 monomer was present in wild-type gliomas and astrocytes. However, the multimeric Kir protein that appeared as a high molecular weight band on the western blot (approximately tetrameric around 200 kDa MW) was lacking in untransfected glioma cells but was prominent in astrocytes and in Kir4.1 overexpressing D54-MG-Kir4.1-AcGFP. Note that in the latter, the monomeric Kir4.1 band was shifted upwards because the AcGFP protein (29 kDa MW) was fused to the Kir4.1 protein. Interestingly, the endogenous Kir4.1 band at 52 kDa was also reduced in Kir4.1 overexpressing D54-MG cells.
Overexpression of Kir4.1 in D54-MG Glioma Cells Produces Functional Kir Currents with Properties Indistinguishable from Differentiated Astrocytes
To demonstrate currents mediated by Kir4.1 channels, we used whole cell patch clamp recordings directly comparing D54-MG-Kir4.1AcGFP cells with mock transfected control D54-MG-AcGFP cells. Previous studies of glioma cells (Bordey and Sontheimer, 1998; Ransom and Sontheimer, 2001), including ones from D54-MG cells (Olsen and Sontheimer, 2004), wild-type (not shown), and mock transfected D54-MG cells should lack of any evidence of Kir4.1 currents. Representative current traces are shown in Fig. 2A in which currents were recorded in response to step changes in membrane voltage ranging from −180 mV to +100 mV before and after application of the Kir inhibitor Ba2+. Using this voltage step protocol, point-by-point subtraction isolated the currents attributable to Kir channels (Ransom and Sontheimer, 1995). Kir4.1 channels display weak rectification and, as most other Kir channels (Butt and Kalsi, 2006), are highly sensitive to 100 μM Ba2+. Recordings in Fig. 2A illustrate that D54-MG cells lack functional Kir channels. The outward currents are mediated by large conductance KCa channels which are highly expressed in these cells (Liu et al., 2002; Ransom and Sontheimer, 2001).
Fig. 2.
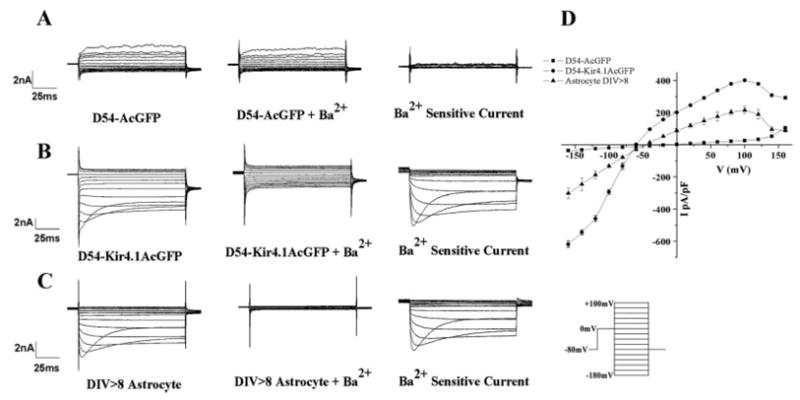
D54-MG cells overexpressing Kir4.1 channel express functional Kir currents. Representative recordings using the same voltage step protocol (inset) shown for (A) D54-MG-AcGFP cells, (B) D54-MG-Kir4.1AcGFP cells, and (C) DIV > 8 spinal cord astrocytes. Large inward current were apparent following Kir4.1 overexpression in D54-MG cells. Although D54-MG-Kir4.1AcGFP cell have larger Kir current density compared with DIV > 8 in vitro spinal cord astrocytes, the step current profile shows similar weak inward rectification and inactivation. (D) IV curves computed from 12 cells under each condition with currents normalized to cell size are presented here.
Figure 2B shows representative recordings from D54-MG cells that express the Kir4.1 gene. Voltage steps activate a large inward current that shows inactivation at very negative potentials, which is a characteristic of Kir4.1 (Newman, 1993; Ransom and Sontheimer, 1995). Importantly, these inward currents were almost completely inhibited in the presence of 100 μM Ba2+ and subtraction isolated currents attributable to the Ba2+ sensitive Kir4.1 channels (Fig. 2B, right). For comparison, we show a typical recording from a cultured spinal cord astrocyte maintained in culture for 8 days (Fig. 2C). As previously shown by others (Neusch et al., 2006; Newman, 1993) and us (Ransom and Sontheimer, 1995), astrocytes show large Ba2+ sensitive currents, and indeed, the vast majority of the overall K+ conductance in spinal cord astrocytes is attributable to Kir4.1 channels (Olsen et al., 2006).
To directly compare currents recorded in astrocytes to glioma cells and glioma cells expressing recombinant Kir4.1, we summarized data from 12 cells each and plotted the recorded currents as a function of voltage in Fig. 2D. Currents were normalized to membrane capacitance, i.e., normalized to cell size. These graphs demonstrate the similarity of currents observed when comparing D54-MG-Kir4.1AcGFP and astrocytes. They illustrate the weak inward rectification, a hallmark feature of Kir4.1 channels. They also illustrate the fact that Kir4.1 D54-MG cells have, on average, two times larger Kir current compared with astrocytes.
An important next step was to demonstrate the functional Kir4.1 channels’ response to changes of extracellular [K+]. This was done by directly comparing D54-MG-Kir4.1AcGFP with spinal cord astrocytes and to mock transfected D54-MG-AcGFP cells that lack functional Kir4.1 channels. Representative examples are illustrated in Fig. 3. Here, cells were recorded using whole-cell voltage clamp recordings while applying a ramped change in voltage, sweeping the voltage from −150 to 150 mV over a 400-ms period. This was repeated after raising extracellular K+ from 5 to 20 mM and after blocking Kir4.1 using 100 μM Ba2+. Identical behavior was observed in D54-MG-Kir4.1-AcGFP (Fig. 3A) and astrocytes (Fig. 3B). Raising K+ from 5 to 20 mM substantially increased the inward currents and caused a rightward shift in the reversal potential by 29 ± 4.1 mV (expected shift based on Nernst potential = 36.1 mV). This behavior has been well documented for Kir currents where K+ conductance is proportional to the square root of K+ concentration (Hagiwara and Takahashi, 1974; Newman, 1993). In both cell types, the K+ current was completely inhibited by 100 μM Ba2+. Neither the mock transfected D54-MG-AcGFP glioma cells (Fig. 3C) nor immature spinal cord astrocytes (DIV < 5; Fig. 3D) show evidence for a similar Ba2+ sensitive inward current that is enhanced with increased K+.
Fig. 3.
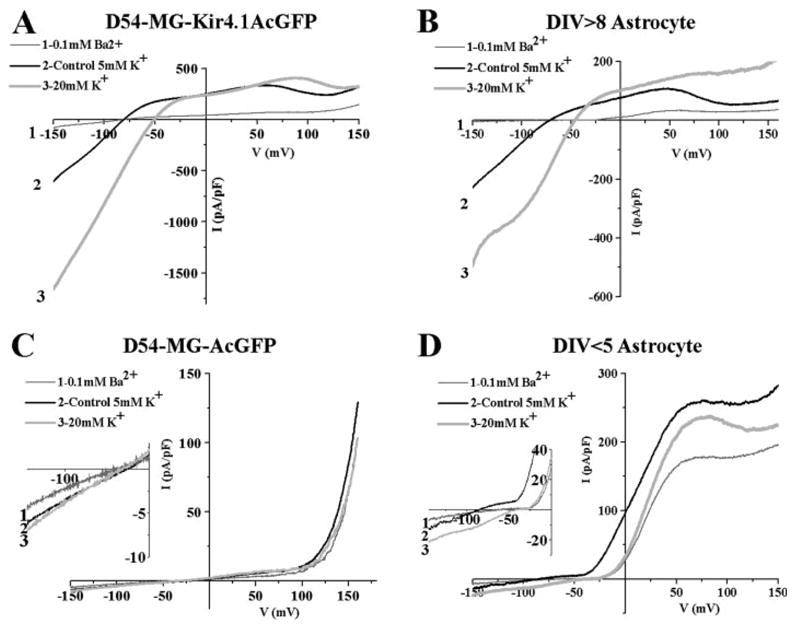
Confirming K+ conductance response in Kir4.1 expressing glioma cells and mature astrocytes. Representative ramp currents (−160 mV to +160 mV) are shown with an application of BaCl2 or 20 mM KCl solution. (A) D54-MG-Kir4.1AcGFP cells with BaCl2 blocked the Kir current similar to (B) DIV > 8 spinal cord astrocytes. Similar right shift of the reversal potential as well as an increased K+ conductance was observed after applying 20 mM KCl. The equivalent response was seen at DIV > 8 in vitro spinal cord astrocytes expressing Kir4.1. (C) Mock control D54-MG-AcGFP cells or (D) DIV < 5 immature astrocytes which do not express Kir4.1 had very small response to 100 μM BaCl2 and modest response to 20 mM KCl applications.
To quantitatively examine and compare Kir4.1 expression between normal and malignant astrocytes before and after overexpression of Kir4.1, we computed the specific conductance attributable to Kir4.1, i.e., sensitive to Ba2+, and normalized that value to cell size and plotted mean data from 12 cells for each population in Fig. 4A. This data show effective expression of Kir channels in transfected gliomas with 10-fold relative conductance difference in mock transfected gliomas (Fig. 4A, P < 0.01) and about twice the relative conductance of spinal cord astrocytes. Not surprisingly, the expression of Kir4.1 also reduced the input resistance of these glioma cells dramatically from 140 ± 11.5 to 19 ± 2.1 MΩ (Fig. 4B, P < 0.01), a value similar to mature astrocytes (25 ± 2.1 MΩ) (Fig. 4B). Taken together, these experiments strongly suggest that overexpression of recombinant Kir4.1 leads to effective insertion of these channels into the cell membrane where they give rise to currents that are indistinguishable, albeit somewhat larger than in nonmalignant astrocytes.
Fig. 4.
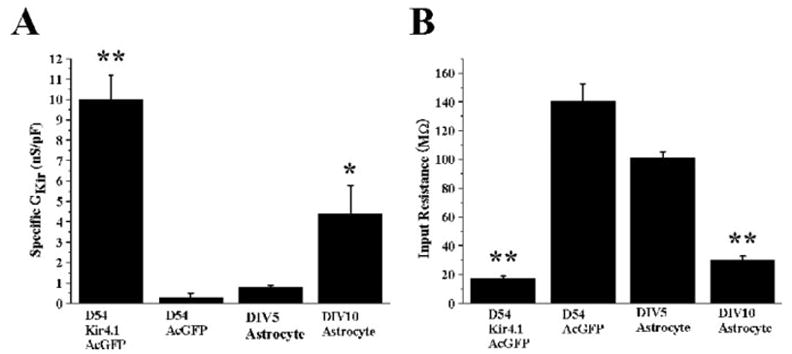
Kir4.1 expression yields high K+ conductance and low input resistance in glioma cells. (A) D54-MG-Kir4.1AcGFP cells have considerably higher Ba2+ sensitive Kir specific conductance (GKir at −140 mV) compared with cells not expressing Kir4.1 (P < 0.01). Similarly DIV > 8 astrocytes have notably higher Ba2+ sensitive Kir specific conductance (P < 0.05). (B) D54-MG-Kir4.1AcGFP cells have significantly low input resistance similar to DIV > 8 astrocytes compared with cells that does not have functional Kir4.1 expression (P < 0.01, D54-MG-Kir4.1AcGFP, P < 0.01, DIV > 8 astrocytes).
We next sought to determine whether Kir4.1 overexpression would predictably alter the resting potential of glioma cells, since the RMP of mature astrocyte is primarily determined by the activity of Kir4.1. We used perforated patch-clamp techniques with amphotericin-B in current clamp recording for this purpose so as to not disturb the ionic milieu of the cell and to gain an accurate measurement of the cell’s true resting potential. Based on recordings taken from 12 cells each from the 4-cell preparations, we found that overexpresion of Kir4.1 in D54-MG caused a negative shift of the RMP from −50 ± 4.1 to −80 ± 4.4 mV (Fig. 5A, P < 0.05). The D54-MG cells expressing functional Kir4.1 was similar to that of mature spinal cord astrocytes, whereas glioma cells lacking Kir4.1 channels were close to that of immature astrocytes, which do not yet express Kir channels to a significant extent. Moreover, application of 100 μM BaCl2 to D54-MG-Kir4.1AcGFP cells caused an acute 15 ± 2.4 mV depolarization (Fig. 5B). This change in RMP was equivalent to the change in RMP observed in DIV > 8 spinal cord astrocytes (16 ± 3.3 mV, Fig. 5B). By comparison, BaCl2 application had an insignificant effect on either D54-MG-AcGFP cells (2 ± 2.1 mV) or DIV < 5 immature proliferating spinal cord astrocytes (4 ± 4.3 mV), not yet expressing large inward Kir currents (Fig. 5B). Taken together, this data show that overexpression of Kir4.1 in glioma cells is sufficient to establish the same resting K+ conductance and negative RMP that also characterizes differentiated astrocytes.
Fig. 5.
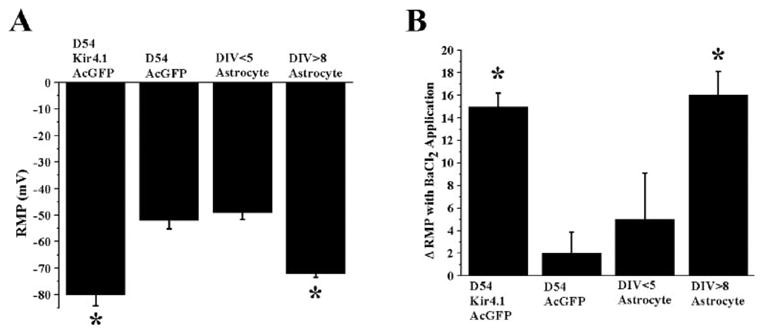
Kir4.1 contributes to the resting membrane potential of astrocytes and Kir4.1 expressing glioma cells. (A) Mean data for 12 cells from four different cells lines were summarized to demonstrate differences in resting membrane potential. Functional expression of Kir4.1 in D54-MG cells RMP was significantly hyperpolarized (P < 0.05, compared with mock transfected D54-MG cells), equivalent to DIV > 8 spinal cord astrocytes expressing Kir4.1 channel. (B) The effects of BaCl2 on the RMP of glioma cells with and without Kir4.1 versus immature and mature astrocytes. D54-MG-AcGFP cells that do not express functional Kir4.1 do not respond to 100 μM BaCl2 application, whereas D54-MG-Kir4.1AcGFP cells respond to BaCl2 treatment with a depolarization.
Persistent Expression of Kir4.1 in D54-MG Cells Attenuates Cell Growth
Since previous studies attribute the expression of Kir channels with an exit from the cell cycle and a differentiation of astrocytes (MacFarlane and Sontheimer, 2000b), we were logically interested in whether overexpression of Kir4.1 in glial tumor cells would similarly attenuate their growth. Using D54-MG-Kir4.1AcGFP cells that stably express the Kir4.1 gene, we obtained growth curves over a 5-day period and directly compared them with cells which stably expressed just the AcGFP vector alone. Mean data from four experiments are shown in Fig. 6A with representative photomicrographs of the mock-control cells in Fig. 6C and the Kir4.1 expressing D54-MG-Kir4.1AcGFP in Fig. 6D (both at 5 days). As is clearly evident from the growth curves, D54-MG-AcGFP glioma cells that lacked Kir expression showed robust growth and expanded 32-fold by 5 days. In contrast, the D54-MG-Kir4.1AcGFP cells grew more slowly and over a 5-day period only expanded eightfold, a ~400% reduction in growth rate compared with control. Importantly, however, when Kir function was inhibited in these cells by the chronic application of 100 μM Ba2+, cell growth was restored significantly albeit not completely. Similarly, depolarization of the Kir4.1 expressing cells using 20 mM K+, which depolarizes the resting potential to the similar value as the D54-MG-AcGFP cells (Fig. 6B), restored growth rate within 80% of the control cells. Of note, D54-MG-AcGFP and D54-MG cell lines have a similar growth curve, suggesting persistent expression of AcGFP protein alone did not affect D54-MG cell growth (data not shown). Moreover, D54-MG-AcGFP cells treated with 100 μM BaCl2 (Fig. 6A) or 20 mM KCl (data not shown) did not affect the rate of growth.
Fig. 6.
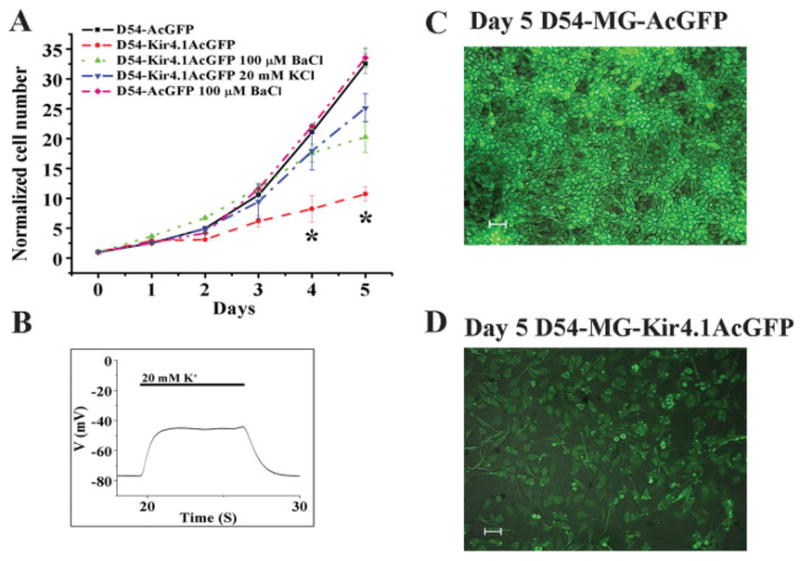
Functional Kir4.1 expression in human glioma cells attenuates cell growth. (A) Five-day proliferation assays demonstrate Kir4.1 expressing D54-MG cell line significantly attenuated its proliferation rate at Days 4 and 5 (P < 0.05). Treatment with 100 μM BaCl2 or 20 mM KCl restored the proliferation rate similar to wild-type D54-MG or D54-MG-AcGFP cells. (B) Current clamp recording demonstrate a depolarizing D54-MG-Kir4.1AcGFP cell RMP by applying 20 mM extracellular K+ to restore RMP, similar to D54-MG-AcGFP cells during high K+ treatment. (C) Top panel: D54-MG-AcGFP cells at Day 5 proliferation. (D) Bottom panel: D54-MG-Kir4.1AcGFP cells at Day 5 proliferation. Both cell lines were plated at the density of 50,000 cells per well. The rate of proliferation of D54-MG-Kir4.1AcGFP cells and D54-MG-AcGFP cells were determined by normalizing the cell number at Day 0. White bar represents 50 μm.
The attenuation of growth observed in stably expressing Kir4.1 in glioma cells could also be attributed to enhanced cell death because population growth is a balance of cell division and cell death. To evaluate this possibility, we used the live and dead assay kit from Molecular Probes to quantitatively assess the relative amount of cell death in D54-MG-AcGFP and D54-MG-Kir4.1-AcGFP cells. Both cell lines did not differ significantly, each showing a similar percentage of cell viabilities (85.1% ± 1.4% D54-MG-Kir4.1 cells compared with 87.4% ± 1.7% mock control, n = 4) and cell death (4.1% ± 1.1% D54-MG-Kir4.1 cells compared with 4.4% ± 0.95% mock control, n = 4) through the 5-day assay period. These results suggest that glioma cell proliferation was significantly reduced by functional Kir4.1 expression, and that this effect was most likely mediated by the establishment of a hyperpolarized RMP.
Overexpression of Kir4.1 in D54-MG Increases the Percentage of Cells in G0/G1 and Reduces the Percentage of Cells in G2/M Phase
To investigate the effect of Kir4.1 expression on cellular growth in greater detail, we analyzed cell cycle distribution of Kir4.1 expressing vs. mock transfected glioma cells by staining fixed cells with propidium iodide and then sorting cells based on this staining with FACS. This approach allowed us to determine the percentage of cells in each sample that were attributed to the G0/G1, G2/M, and S-phase of the cell cycle, respectively, based on DNA content. Propidium iodide intercalates into dividing cells chromosome (duploid cells, G2/M phase) at twice the intensity compared with nondividing cells (haploid cells G0/G1 phase). D54-MG-Kir4.1AcGFP cells showed a significantly greater percentage (Fig. 7A; Day 1, 71.9% ± 0.39%, to Day 5, 75.9% ± 0.96%) of cells in the G0/G1-phase compared with D54-MG-AcGFP cells (Day 1, 67.8% ± 0.77%, to Day 5, 66.5% ± 0.49%) (Fig. 7A; Day 1 to Day 5, P < 0.05). This corresponded to an average decrease of 37.2% in G2/M phase cells that persisted throughout the 5 days that we examined (Fig. 7B). Notably this shift of cells from the growth phase into the quiescent phase was completely nullified when Kir channels were chronically inhibited by 100 μM Ba2+ or when cells were depolarized by 20 mM KCl. Neither drug nor high KCl application altered the cell cycle profile of the mock transfected controls (data not shown).
Fig. 7.
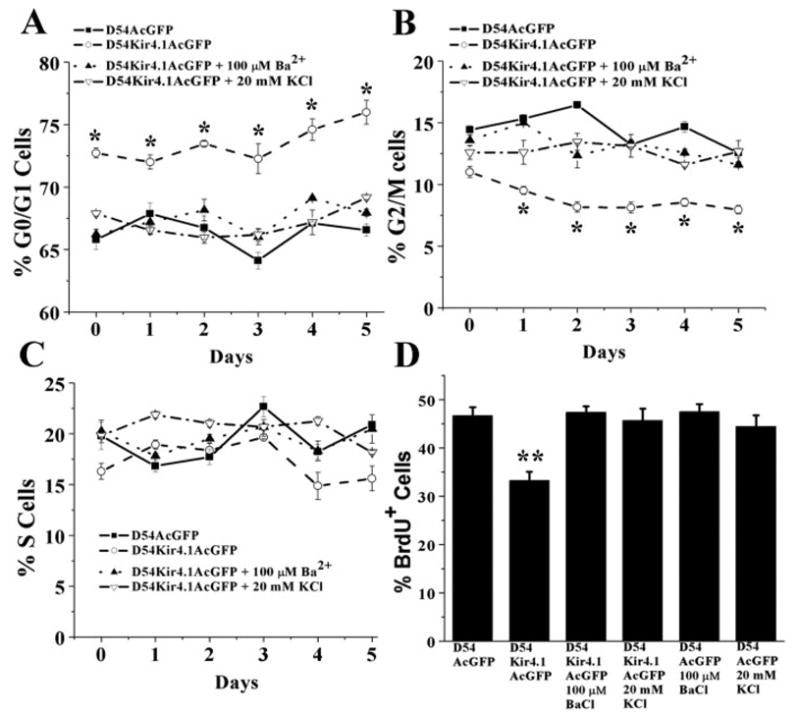
Kir4.1 expression increases the percentage of cells in the G0/G1 phase of the cell cycle. FACS analysis was used to determine the relative percentage of cells found in each of the three discernable cell cycle stages. G0/G1 cells in (A) show an increased percentage of D54-MG-Kir4.1AcGFP cells in the G0/G1 phase. (B) Kir4.1 expressing cells showed a decrease in the G2/M phase without notable changes in S phase. (C) Application of 100 μM BA2+ or 20 mM high KCl solution revert this cell cycle change back to D54-MG-AcGFP control cells. (D) Significant reduction in % BrdU+ cell number is revealed in D54Kir4.1AcGFP cell line compared with D54-MG-AcGFP cells (P < 0.01). Treatment with 100 μM BA2+ or 20 mM KCl to D54-MG-Kir4.1-AcGFP cells restore % BrdU+ cell number similar to D54-MG-AcGFP mock transfected cells.
Further analysis using cellular BrdU incorporation also confirmed that D54-MG-Kir4.1AcGFP cells showed a significantly lesser percentage of cells incorporating BrdU following 6-h incubation (Fig. 7D; 33.2% ± 1.76% D54-MG-Kir4.1AcGFP cells compared with 46.7% ± 1.74% D54-MG-AcGFP mock control cells, P < 0.01). Application of 100 μM Ba2+ or 20 mM KCl to D54-MG-Kir4.1AcGFP cells reverted % BrdU+ cells similar to mock transfected control cell number. Application of drug or high KCl did not alter D54-MG-AcGFP cells BrdU % incorporation. These data again support the conclusion that the negative RMP established by Kir4.1 is directly involved in regulating the growth of these cells.
DISCUSSION
This study significantly extends previous investigations in defining the role of K+ channels in glial growth control. While previous studies have suggested that Kir channels are developmentally regulated in astrocytes (Bringmann et al., 1999; Kressin et al., 1995) and particularly upregulated in astrocytes that have differentiated and no longer divide (MacFarlane and Sontheimer, 2000a), we now show that Kir4.1, the major astrocytic Kir channel (Li et al., 2001), is sufficient to inhibit glial growth. Glial-derived tumor cells provided an ideal model system to study Kir channels in glial cell proliferation. These cells do not express functional Kir channels and we (Bordey and Sontheimer, 1998; Olsen and Sontheimer, 2004) have previously suggested that the absence of Kir channels may facilitate their unrestrained growth. Many studies have reported on a depolarized RMP as being common among proliferating cells including many cancer cells (Cone, 1970). Moreover, several studies have shown an interdependence of cell proliferation and depolarized RMP (for review see: Kunzelmann, 2005; Pardo et al., 2005). Also, recently, overexpressing delayed rectifying potassium channels Kv1.3 and 1.4, highly expressed in oligodendrocyte progenitors cells, were showed to promote cell proliferation (Vautier et al., 2004). Proliferative oligodendrocyte progenitor cells express large delayed rectifying potassium channels, however; similar to astrocytic development, these current become smaller in mature oligodendrocyte with concomitant acquisition of Kir currents and RMP hyperpolarization (Knutson et al., 1997; Sontheimer et al., 1989).
In this study, we considerably extend these prior works by showing a mechanistic interdependence between Kir4.1 channel function, cell RMP, and cell proliferation. The finding that overexpression of Ba2+ sensitive Kir4.1 channel hyperpolarizes the membrane, leading to a more negative RMP was expected and is consistent with previous studies from Kir4.1 gene knockout studies in astrocytes (Neusch et al., 2006; Olsen et al., 2006) and in retinal glial cells (Kofuji et al., 2000). However, the finding that this overexpression is sufficient to inhibit cell proliferation and does so by promoting cells to move into the quiescent G0/G1 phase of the cell cycle has not been demonstrated before. More importantly we demonstrate that these positive effects of Kir4.1 promoting cell differentiation and inhibiting growth can be completely overcome if either the channel is pharmacologically turned off, or the resting potential is experimentally reset to the same depolarized potential that characterizes proliferating glioma cells. This provides very strong mechanistic evidence that the link between Kir4.1 expression and growth regulation is indeed the cells RMP. This supports the conclusion that persistent hyperpolarized resting potential of −70 to −90 mV is not conducive for cell proliferation. Kir4.1 expression in glial cells, therefore, induces a nonfavorable RMP for cell cycle progression.
The mechanistic link between the cells resting potential and cell cycle regulation is not well understood and requires further research. It is, however, largely accepted that most cell types undergo a transient hyperpolarization at the exit of G1 phase of the cell cycle, and that tumor cells are, on average, depolarized compared with nontumor cells (Ghiani et al., 1999). A depolarized RMP is typically linked to intracellular alkalization (Grichtchenko and Chesler, 1994), favored by protein, RNA, and DNA synthesis (Erecinska et al., 1995). Glioma cells are indeed more alkaline than nonmalignant brain cells (McLean et al., 2000). Membrane depolarization has been shown to cause an accumulation of the cyclin-dependent kinase inhibitors p27 and p21 in glial progenitors cells and, hence, may directly intersect with signals involved in cell cycle checkpoint control (Ghiani et al., 1999). A depolarized membrane potential facilitates the entry of Ca2+, and increases in Ca2+ have been shown to be required for cell replication (Schreiber, 2005). At a more homeostatic level, the membrane potential maintains the driving force for the transport of nutrients, and hence, its regulation is of primordial importance for rapidly proliferating cells (Pardo et al., 2005). Changes in cell volume are fundamental to cell division (Rouzaire-Dubois et al., 2000) and require the movement of ions across the cell membrane. Owing to their charge, ion movement and volume changes are directly responsive to changes in membrane potential. As these examples illustrate, a multitude of intersecting pathways exist, which could mechanistically link changes in membrane potential to cell division, yet all remain speculative at this time.
While the major motivation for this study was to unravel if and how Kir channels are involved in cell cycle regulation, an important additional outcome pertains to the lack of functional Kir channels in glioma cells. Their unrestricted growth and rapid expansion makes these tumors almost impossible to treat, and many patients die within 9 months of diagnosis (Burton and Prados, 2000). Chemotherapeutic drugs that interfere with DNA replication have been shown to be largely ineffective, in part because of efficient DNA repair mechanisms (Jiang et al., 2006). Our findings suggest that tumor growth could be effectively inhibited by membrane expression of Kir4.1. This channel is indeed found in patient biopsies from gliomas yet due to an unknown trafficking defect these channels are not properly inserted into the cell membrane but instead mislocalize to the nuclear membrane (Olsen and Sontheimer, 2004). Hence, if one could identify ways in which to correct for this mislocalization of Kir4.1, one would expect to see a significant slowing in tumor growth. The possibility to exploit this as a therapeutic approach clearly warrants further study. Alternatively, one could explore ways of delivering Kir4.1, using a viral vector system. While highly speculative at this point, similar approaches are being undertaken with other genes in early clinical studies (Sonabend et al., 2006). Given the severity of the disease, both avenues deserve further consideration.
Acknowledgments
The authors thank Dr. L. C. Tempi at UCSF for providing mouse Kir4.1cDNA plasmid. Authors also acknowledge the valuable input provided by Drs. A. W. Kendall, M. L. Olsen, and S. A. Lyons.
Grant sponsor: NIH; Grant number: RO1 NS31234.
References
- Arai K, Horie Y, Kurimoto M, Endoh S, Hiraga K, Takaku A. A cell line of human malignant astrocytoma producing autocrine growth factor. In Vitro Cell Dev Biol A. 1991;27A:606–614. doi: 10.1007/BF02631103. [DOI] [PubMed] [Google Scholar]
- Bordey A, Lyons SA, Hablitz JJ, Sontheimer H. Electrophysiological characteristics of reactive astrocytes in experimental cortical dysplasia. J Neurophysiol. 2001;85:1719–1731. doi: 10.1152/jn.2001.85.4.1719. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H. Postnatal development of ionic currents in rat hippocampal astrocytes in situ. J Neurophysiol. 1997;78:461–477. doi: 10.1152/jn.1997.78.1.461. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H. Electrophysiological properties of human astrocytic tumor cells in situ: Enigma of spiking glial cells. J Neurophysiol. 1998;79:2782–2793. doi: 10.1152/jn.1998.79.5.2782. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Biedermann B, Reichenbach A. Expression of potassium channels during postnatal differentiation of rabbit Muller glial cells. Eur J Neurosci. 1999;11:2883–2896. doi: 10.1046/j.1460-9568.1999.00706.x. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Francke M, Pannicke T, Biedermann B, Kodal H, Faude F, Reichelt W, Reichenbach A. Role of glial K(+) channels in ontogeny and gliosis: A hypothesis based upon studies on Muller cells. Glia. 2000;29:35–44. doi: 10.1002/(sici)1098-1136(20000101)29:1<35::aid-glia4>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Burton EC, Prados MD. Malignant gliomas. Curr Treat Options Oncol. 2000;1:459–468. doi: 10.1007/s11864-000-0073-2. [DOI] [PubMed] [Google Scholar]
- Butt AM, Kalsi A. Inwardly rectifying potassium channels (Kir) in central nervous system glia: A special role for Kir4.1 in glial functions. J Cell Mol Med. 2006;10:33–44. doi: 10.1111/j.1582-4934.2006.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone CD., Jr Variation of the transmembrane potential level as a basic mechanism of mitosis control. Oncology. 1970;24:438–470. doi: 10.1159/000224545. [DOI] [PubMed] [Google Scholar]
- Erecinska M, Deas J, Silver IA. The effect of pH on glycolysis and phosphofructokinase activity in cultured cells and synaptosomes. J Neurochem. 1995;65:2765–2772. doi: 10.1046/j.1471-4159.1995.65062765.x. [DOI] [PubMed] [Google Scholar]
- Ghiani CA, Yuan X, Eisen AM, Knutson PL, DePinho RA, McBain CJ, Gallo V. Voltage-activated K+ channels and membrane depolarization regulate accumulation of the cyclin-dependent kinase inhibitors p27(Kip1) and p21(CIP1) in glial progenitor cells. J Neurosci. 1999;19:5380–5392. doi: 10.1523/JNEUROSCI.19-13-05380.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grichtchenko II, Chesler M. Depolarization-induced acid secretion in gliotic hippocampal slices. Neuroscience. 1994;62:1057–1070. doi: 10.1016/0306-4522(94)90343-3. [DOI] [PubMed] [Google Scholar]
- Hagiwara S, Takahashi K. The anomalous rectification and cation selectivity of the membrane of a starfish egg cell. J Membr Biol. 1974;18:61–80. doi: 10.1007/BF01870103. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Jiang H, Alonso MM, Gomez-Manzano C, Piao Y, Fueyo J. Oncolytic viruses and DNA-repair machinery: Overcoming chemoresistance of gliomas. Expert Rev Anticancer Ther. 2006;6:1585–1592. doi: 10.1586/14737140.6.11.1585. [DOI] [PubMed] [Google Scholar]
- Knutson P, Ghiani CA, Zhou JM, Gallo V, McBain CJ. K+ channel expression and cell proliferation are regulated by intracellular sodium and membrane depolarization in oligodendrocyte progenitor cells. J Neurosci. 1997;17:2669–2682. doi: 10.1523/JNEUROSCI.17-08-02669.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: Phenotypic impact in retina. J Neurosci. 2000;20:5733–5740. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kressin K, Kuprijanova E, Jabs R, Seifert G, Steinhauser C. Developmental regulation of Na+ and K+ conductances in glial cells of mouse hippocampal brain slices. Glia. 1995;15:173–187. doi: 10.1002/glia.440150210. [DOI] [PubMed] [Google Scholar]
- Kuffler SW. Neuroglial cells: Physiological properties and a potassium mediated effect of neuronal activity on the glial membrane potential. Proc R Soc London B Biol Sci. 1967;168:1–21. doi: 10.1098/rspb.1967.0047. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K. Ion channels and cancer. J Membr Biol. 2005;205:159–173. doi: 10.1007/s00232-005-0781-4. [DOI] [PubMed] [Google Scholar]
- Li L, Head V, Timpe LC. Identification of an inward rectifier potassium channel gene expressed in mouse cortical astrocytes. Glia. 2001;33:57–71. doi: 10.1002/1098-1136(20010101)33:1<57::aid-glia1006>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Liu X, Chang Y, Reinhart PH, Sontheimer H. Cloning and characterization of glioma BK, a novel BK channel isoform highly expressed in human glioma cells. J Neurosci. 2002;22:1840–1849. doi: 10.1523/JNEUROSCI.22-05-01840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane SN, Sontheimer H. Electrophysiological changes that accompany reactive gliosis in vitro. J Neurosci. 1997;17:7316–7329. doi: 10.1523/JNEUROSCI.17-19-07316.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane SN, Sontheimer H. Changes in ion channel expression accompany cell cycle progression of spinal cord astrocytes. Glia. 2000a;30:39–48. doi: 10.1002/(sici)1098-1136(200003)30:1<39::aid-glia5>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- MacFarlane SN, Sontheimer H. Modulation of Kv1.5 currents by Src tyrosine phosphorylation: Potential role in the differentiation of astrocytes. J Neurosci. 2000b;20:5245–5253. doi: 10.1523/JNEUROSCI.20-14-05245.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean LA, Roscoe J, Jorgensen NK, Gorin FA, Cala PM. Malignant gliomas display altered pH regulation by NHE1 compared with nontransformed astrocytes. Am J Physiol Cell Physiol. 2000;278:C676–C688. doi: 10.1152/ajpcell.2000.278.4.C676. [DOI] [PubMed] [Google Scholar]
- Neusch C, Papadopoulos N, Muller M, Maletzki I, Winter SM, Hirrlinger J, Handschuh M, Bahr M, Richter DW, Kirchhoff F, Hulsmann S, et al. Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: Impact on extracellular K+ regulation. J Neurophysiol. 2006;95:1843–1852. doi: 10.1152/jn.00996.2005. [DOI] [PubMed] [Google Scholar]
- Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci. 2001;21:5429–5438. doi: 10.1523/JNEUROSCI.21-15-05429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Inward-rectifying potassium channels in retinal glial (Muller) cells. J Neurosci. 1993;13:3333–3345. doi: 10.1523/JNEUROSCI.13-08-03333.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annu Rev Physiol. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- Olsen ML, Higashimori H, Campbell SL, Hablitz JJ, Sontheimer H. Functional expression of Kir4.1 channels in spinal cord astrocytes. Glia. 2006;53:516–528. doi: 10.1002/glia.20312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ML, Sontheimer H. Mislocalization of Kir channels in malignant glia. Glia. 2004;46:63–73. doi: 10.1002/glia.10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ML, Sontheimer H. Voltage activated ion channels in glial cells. In: Ransom BR, Kettenmann H, editors. Neuroglia. New York: Oxford University Press. Inc; 2005. pp. 112–130. [Google Scholar]
- Pardo LA. Voltage-gated potassium channels in cell proliferation. Physiology (Bethesda) 2004;19:285–292. doi: 10.1152/physiol.00011.2004. [DOI] [PubMed] [Google Scholar]
- Pardo LA, Contreras-Jurado C, Zientkowska M, Alves F, Stuhmer W. Role of voltage-gated potassium channels in cancer. J Membr Biol. 2005;205:115–124. doi: 10.1007/s00232-005-0776-1. [DOI] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Ransom BR, Goldring S. Slow hyperpolarization in cells presumed to be glia in cerebral cortex of cat. J Neurophysiol. 1973;36:879–892. doi: 10.1152/jn.1973.36.5.879. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Sontheimer H. Biophysical and pharmacological characterization of inwardly rectifying K+ currents in rat spinal cord astrocytes. J Neurophysiol. 1995;73:333–346. doi: 10.1152/jn.1995.73.1.333. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Sontheimer H. BK channels in human glioma cells. J Neurophysiol. 2001;85:790–803. doi: 10.1152/jn.2001.85.2.790. [DOI] [PubMed] [Google Scholar]
- Rouzaire-Dubois B, Milandri JB, Bostel S, Dubois JM. Control of cell proliferation by cell volume alterations in rat C6 glioma cells. Pflugers Arch. 2000;440:881–888. doi: 10.1007/s004240000371. [DOI] [PubMed] [Google Scholar]
- Schreiber R. Ca2+ signaling, intracellular pH and cell volume in cell proliferation. J Membr Biol. 2005;205:129–137. doi: 10.1007/s00232-005-0778-z. [DOI] [PubMed] [Google Scholar]
- Schroder W, Seifert G, Huttmann K, Hinterkeuser S, Steinhauser C. AMPA receptor-mediated modulation of inward rectifier K+ channels in astrocytes of mouse hippocampus. Mol Cell Neurosci. 2002;19:447–458. doi: 10.1006/mcne.2001.1080. [DOI] [PubMed] [Google Scholar]
- Sonabend AM, Ulasov IV, Han Y, Lesniak MS. Oncolytic adenoviral therapy for glioblastoma multiforme. Neurosurg Focus. 2006;20:E19. [PubMed] [Google Scholar]
- Sontheimer H. Astrocytes, as well as neurons, express a diversity of ion channels. Can J Physiol Pharmacol. 1992;70(Suppl):S223–S238. doi: 10.1139/y92-266. [DOI] [PubMed] [Google Scholar]
- Sontheimer H. Voltage-dependent ion channels in glial cells. Glia. 1994;11:156–172. doi: 10.1002/glia.440110210. [DOI] [PubMed] [Google Scholar]
- Sontheimer H, Trotter J, Schachner M, Kettenmann H. Channel expression correlates with differentiation stage during the development of oligodendrocytes from their precursor cells in culture. Neuron. 1989;2:1135–1145. doi: 10.1016/0896-6273(89)90180-3. [DOI] [PubMed] [Google Scholar]
- Studer A, de Tribolet N, Diserens AC, Gaide AC, Matthieu JM, Carrel S, Stavrou D. Characterization of four human malignant glioma cell lines. Acta Neuropathol (Berl) 1985;66:208–217. doi: 10.1007/BF00688585. [DOI] [PubMed] [Google Scholar]
- Vautier F, Belachew S, Chittajallu R, Gallo V. Shaker-type potassium channel subunits differentially control oligodendrocyte progenitor proliferation. Glia. 2004;48:337–345. doi: 10.1002/glia.20088. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Steinhauser C. Ion channels in glial cells. Brain Res Brain Res Rev. 2000;32:380–412. doi: 10.1016/s0165-0173(99)00093-4. [DOI] [PubMed] [Google Scholar]
- Walz W. Chloride/anion channels in glial cell membranes. Glia. 2002;40:1–10. doi: 10.1002/glia.10125. [DOI] [PubMed] [Google Scholar]
- Wonderlin WF, Strobl JS. Potassium channels, proliferation and G1 progression. J Membr Biol. 1996;154:91–107. doi: 10.1007/s002329900135. [DOI] [PubMed] [Google Scholar]
- Zupanc GK. An in vitro technique for tracing neuronal connections in the teleost brain. Brain Res Brain Res Protoc. 1998;3:37–51. doi: 10.1016/s1385-299x(98)00019-1. [DOI] [PubMed] [Google Scholar]