

1. Introduction
Immune recognition of tumors in an antigen-specific manner was first illustrated by experiments involving transplantation of chemically induced tumors into laboratory mice [1, 2]. Specifically, the growth of a transplanted tumor could be prevented by prior exposure to the same tumor, but not a different tumor. Many investigators have since observed naturally developing tumor-specific T cell responses (reviewed in [3]) which, in patients treated with standard therapies, correlate with improved prognosis [4-10]. Despite this positive correlation, the tumor infiltrating lymphocytes (TIL) do not always control tumor growth. Tumor-specific T cells are ineffective in part due to active regulation and suppression by tumors. For example, tumors produce the tryptophan degrading enzyme indoleamine 2,3-dioxygenase that inhibits T cell proliferation [11]. In addition, tumors produce immune suppressive cytokines such as TGFβ [12, 13] and IL-10 [14]. The mechanism of immune suppression by these cytokines includes the inhibition of proliferation and inflammatory cytokine production by immune cells. For detailed reviews of tumor-induced immune suppression see the other reviews in this issue and [15].
In this review, we focus on another mechanism responsible for the poor reactivity of the tumor-specific T cell repertoire, the low functional avidity of the responding T cells. Functional avidity, or the sensitivity of T cell to antigen, is an important factor influencing the efficacy of a T cell response. Virus-specific cytotoxic T lymphocytes (CTL) with high functional avidity clear viral infections better than T cells with low functional avidity because these CTL are more sensitive to small viral loads [16, 17]. Analysis of the functional avidity of tumor-specific T cells has provided insight into why tumors develop despite the presence of TIL and how these T cells may be harnessed for cancer therapies. In this review we will discuss the factors influencing the functional avidity of CTL and how this affects the T cell response to tumors. Furthermore, we will discuss different approaches aimed at improving the functional avidity of the tumor-specific T cells with the goal of augmenting conventional treatments and T cell therapies against cancer.
2. Affinity, functional avidity, and recognition efficiency of T cells
As mentioned above, T cell functional avidity is defined as the sensitivity of a T cell to activation by an antigenic peptide bound by an MHC molecule. The sensitivity of a T cell to antigen is influenced by multiple factors: the affinity of the TCR-peptide-MHC interaction, the engagement of multiple other receptors on T cells, and the density of these receptors on the T cell surface. The combination of these binding interactions with an APC determines the functional avidity of a T cell. Since avidity is often used to describe the multivalent binding between two molecules rather than the interaction between two cells, the term functional avidity may be misleading and is therefore also referred to as recognition efficiency [18]. We use “functional avidity” since it is used in most of the literature described in this review.
The readout for T cell functional avidity also varies within the field. Functional avidity is frequently determined by the relative capacity of T cells to produce effector cytokines or lyse target cells in an antigen-specific manner. However, as we discuss below, staining intensity of T cells with peptide-loaded MHC tetramers is also a common readout for T cell avidity. Therefore, a comprehensive understanding of what influences T cell function is crucial for understanding and measuring functional avidity of tumor-specific T cells. In the following sections we will dissect the molecular mechanisms that contribute to the activation of T cells and functional avidity.
2.1 TCR-peptide-MHC affinity, kinetics, and T cell functional avidity
Activation of a T cell is initiated by the ligation of the TCR by peptide-MHC complexes on an APC. However, the TCR is not a simple on/off switch, but can be activated to different degrees depending on the binding kinetics. The serial-triggering model of T cell activation explains how a T cell is activated by the low levels of peptide presented on the surface of an APC. This model proposes that one peptide-MHC complex binds multiple TCRs on the surface of the T cell, providing the sustained signal required for activation [19]. One important prediction of this model is that there is an upper limit to the TCR-peptide-MHC binding half-life (t1/2), or dwell time, that results in activation of the T cell. Prolonged binding would prevent the limited numbers of peptide-MHC complexes from binding enough TCRs to transduce a positive signal and, as a result, would inhibit T cell activation [20]. The complementary kinetic-proofreading model proposes that full T cell activation will not occur unless the TCR-peptide-MHC interaction has a long enough half-life for the completion of a series of biochemical intracellular signaling events [21]. If the off-rate is too rapid the T cell will not be fully activated [22, 23].
The consequences of both the kinetic-proofreading and serial-triggering models are that TCR-peptide-MHC interactions with too high or low affinity will not activate the T cell; only those with mid-range affinity will result in activation and differentiation of the cell (Figure 1). This conclusion has been verified by experiments showing that the strength of the initial signal received through the TCR, due to antigen concentration [24, 25] or the affinity of the stimulating antigen [20, 26-31], affects the activation of T cell clones. Interactions with exceptionally long half-lives result in impaired T cell activation [20, 25, 32-34]. The KD of a productive TCR-peptide-MHC interaction is on average between 1-10 μM and the t1/2 is 5-20 seconds (reviewed in [35]). The low affinity of TAA-specific TCRs for peptide-MHC complexes has been proposed as a mechanism for preventing efficient recognition of tumors. Interestingly, we have determined that the KD of a TAA-MHC complex for a specific TCR is between 5-7 μM and the t1/2 is 1.5-2 seconds [30, 31] confirming that at least some tumor antigens are weak TCR agonists.
Figure 1. Goldilocks model for the affinity of TCR-peptide-MHC interactions and T cell activation.
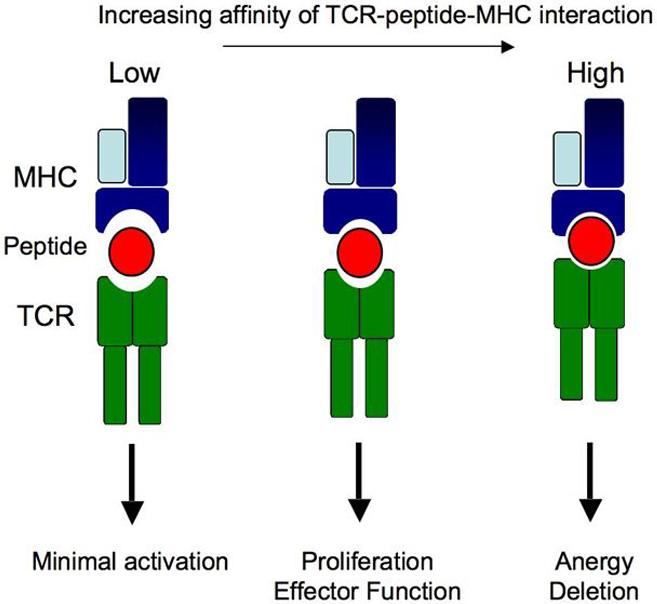
Peptide-MHC complexes that bind TCR with low affinity fail to induce complete intracellular signaling resulting in a lack of T cell activation (left). TCR-peptide-MHC binding interactions with long half-lives prevent serial triggering of the TCR and lead to impaired T cell activation (e.g. anergy or deletion, right). Optimal T cell activation requires an affinity in-between for complete induction of proliferation and acquisition of effector function (middle).
The functional avidity of a T cell is often estimated by the relative staining intensity of MHC tetramers loaded with peptide since TCR-peptide-MHC dwell time correlates with T cell activation. While our group and others have shown that multimer binding correlates with T cell sensitivity to antigen [31, 36-38], this correlation is not always strict [20, 39-41]. In some experiments higher expression levels of TCR on the cell surface increased the multimer-binding intensity [36, 39]. Therefore, MHC tetramer off-rates may better correlate with functional avidity since this controls for TCR expression levels [42]. In addition, as discussed above, peptide-MHC complexes that bind TCRs with a long enough half-life inhibit T cell activation and MHC tetramers of these complexes bind T cells with relatively high intensity [20]. Finally, as we will discuss in the next section the intrinsic affinity of the TCR-peptide-MHC complex is not the only binding interaction that influences the functional avidity of a T cell.
2.2 Other factors that influence the functional avidity of a T cell
The functional avidity of a T cell is also influenced by the overall binding strength between a T cell and APC that results from the additive effects of multiple receptor/ligand interactions (Figure 2). The CD8 co-receptor on CTL binds the alpha 3 domain of the MHC class I molecule, enhancing the binding of the TCR-peptide-MHC complex [43-47]. Since the binding of CD8 by MHC enhances the activation of T cells expressing low affinity TCRs, staining with mutated MHC class I-multimers that inhibit CD8 binding identifies T cells with high-affinity TCRs [48]. The co-stimulatory receptors CD80 (B7-1) and CD86 (B7-2) on APC bind to CD28 on the surface of T cells and enhance the magnitude of TCR signaling, decreasing the level of TCR ligation required for activation. Improved binding between the T cell and APC is also facilitated by the adhesion molecule pairs ICAM-1/LFA-1 [49] and LFA-3/CD2 [50]. Interestingly, TIL have decreased cell-surface expression of LFA-1, CD2, and CD8 suggesting that defects in these T cell binding interactions contribute to the poor functional avidity of TAA-specific T cells [51].
Figure 2. Molecular interactions influencing T cell functional avidity.
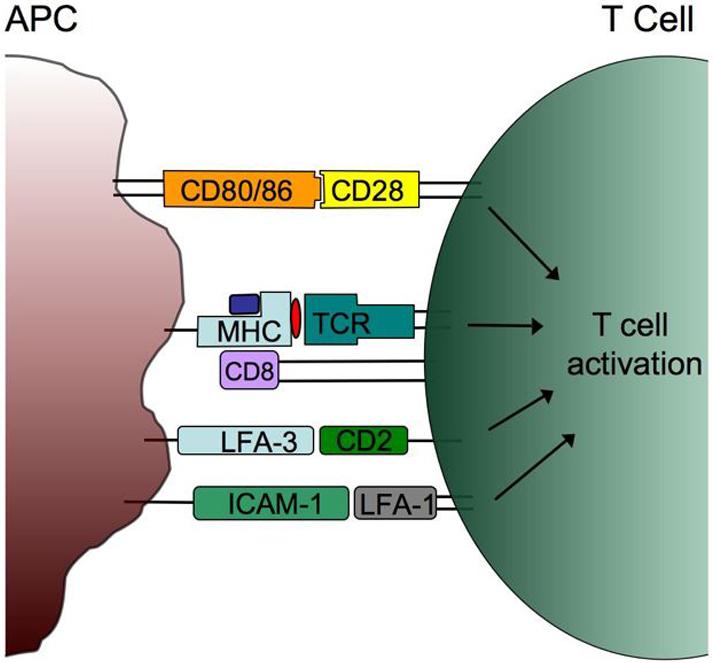
Multiple receptor/ligand interactions between the T cell and APC enhance functional avidity. The affinity and kinetics of the TCR-peptide-MHC interaction, and binding of the CD8 co-receptor, contribute to TCR signaling. Sensitivity to antigen is also affected by ligation of the CD28 co-stimulatory molecule by CD80 (B7-1) and CD86 (B7-2). The adhesion molecule pairs (CD2/LFA-3 and ICAM-1/ LFA-1) enhance functional avidity by increasing the binding strength and intracellular signaling events.
The proximity of TCRs to each other and other membrane-associated molecules on the surface of T cells also affects the functional avidity. Fahmy et al. demonstrated that the increased sensitivity of activated T cells, relative to naive T cells, correlates with increased avidity resulting from TCR reorganization within the cell surface membrane [52]. Increased association of CD8 with TCR on the surface of T cells also enhances T cell activation, likely due to increased co-localization of the CD8-associated Lck kinase [53]. Similarly, increased expression of intracellular Lck following T cell activation increases the sensitivity of T cells to antigen stimulation [54]. Defects in proximal TCR signaling, including Lck activation, are observed in TIL [55]. This signaling blockade may explain the decreased expression of adhesion molecules on TIL since Lck activation is necessary for the expression and activation of these receptors on the cell surface [51]. In summary, numerous molecular interactions between T cells and APC contribute to functional avidity and can be impaired in TIL.
3. Tolerance and the tumor-specific T cell repertoire
The immune system maintains a diverse repertoire of T cells with high avidity for foreign antigen while limiting the activity of T cells that recognize self antigen. Since most tumor antigens are self antigens, tolerance mechanisms greatly influence the quality of the antitumor T cell response. The degree to which tolerance affects tumor-specific T cells differs depending on the TAA, but in many cases both central and peripheral tolerance mechanisms directly influence the functional avidity of T cells for TAA. We discuss both features of TAA and mechanisms of tolerance governing the T cells that recognize these TAA below.
3.1 Tumor antigens recognized by T cells
Tumor antigens can be divided into two basic categories, tumor-specific antigens (TSA) and TAA (see Table I). TSA are often immunogenic since they are derived from viral antigens or neoantigens created by mutations during the transformation process. Many of these mutations contribute to the malignant phenotype of the tumor cells (e.g. RAS, CDK4). While some mutations are found in multiple tumors, often they are unique to the tumor in which they were identified, limiting their clinical value as targets of general tumor immunotherapies.
Table I.
Common tumor antigens expressed by murine and human tumors.
| Type of Antigen | Gene | Expression pattern | Ref | |
|---|---|---|---|---|
| Tumor-specific antigens (TSA) | Mutated antigens | b-catentin | Melanoma | [127] |
| CDK4 | Melanoma | [128] | ||
| Myosin | Melanoma | [129] | ||
| RAS | Melanoma | [130] | ||
| Chimeric proteins | Abl-bcr | CML | [131] | |
| ETV6/AML | ALL | [132] | ||
| NPM/ALK | Large cell lymphomas | [133] | ||
| Viral antigens | E6/E7 | Cervical neoplasia | [134] | |
| EBNA-3 | Immunoblastic lymphoma | [135] | ||
| Tumor-associated antigens (TAA) | Cancer-testis antigens | MAGE | Spermatocytes and placenta | [136] |
| BAGE | Spermatocytes | [137] | ||
| GAGE | Spermatocytes and placenta | [138] | ||
| NY-ESO-1 | Spermatocytes and ovary cells | [139] | ||
| Differentiation Antigens | CEA | Embryonic tissue, epithelial cells | [140] | |
| gp100 | Melanocytes | [141] | ||
| Melan-A/MART-1 | Melanocytes | [142] | ||
| NY-BR-1 | Mammary tissue | [143] | ||
| PSA | Prostate gland | [144] | ||
| TRP-1/2 | Melanocytes | [145] | ||
| Tyrosinase | Melanocytes | [146] | ||
| Ubiquitously expressed antigens | AFP | Expressed in multiple tissues | [147] | |
| HER-2/neu | [148] | |||
| hTERT | [149] | |||
| MUC1 | [150] | |||
| p53 | [151] | |||
| p15 | [152] | |||
| SART-1/2/3 | [153-155] | |||
| WT1 | [156] | |||
CML = Chronic myelogenous leukemia, ALL = Acute lymphoblastic leukemia
As stated above, viral antigens expressed by oncogenic viruses are another source of TSA. Viruses that are associated with human cancers include human papilloma virus (HPV) [56], Epstein-Barr virus [57], Kaposi’s sarcoma-associated herpes virus [58] and hepatitis B and C (HBV and HCV) [59, 60]. The increased occurrence of virally associated cancers in immunocompromised patients relative to healthy individuals suggests that the expression of viral antigens by the transformed cells promote antitumor immunity. Vaccines against cancers that result from infections with oncogenic viruses have shown promise. A recently approved vaccine against HPV prevents both infection and the associated cervical neoplasia [61]. Similarly, a decrease in the incidence of hepatocellular carcinoma is observed following vaccination against HBV [62].
Although TSA are attractive targets for immunotherapy against cancers with viral etiology, the majority of tumor antigens from other cancers are TAA. TAA are non-mutated self antigens from proteins expressed in tumors as well as normal tissues (Table I). TAA can be characterized by their expression pattern as tissue-specific or ubiquitously expressed antigens (reviewed in [63]). For example, cancer-testis (CT) antigens are expressed in the testes and sometimes in the placenta, and are reactivated in tumor cells. The low levels of MHC expression in healthy testes and placenta prevent recognition of CT antigens by the immune system making them targets for T cell therapies. Differentiation antigens are present on both the tumor and the tissue from which the tumor arose and not in other tissues. The most studied of these antigens are the melanoma-differentiation antigens (reviewed in [64]). These include the gp100, Mart-1/Melan-A, pMel-17 and tyrosinase antigens that are products in the melanin-production pathway. These TAA are expressed both in melanomas and in normal melanocytes.
While tissue-specific antigens are expressed in a limited number of tissues, ubiquitously expressed antigens are found on most normal tissues but are often overexpressed in transformed cells. For example, overexpression of telomerase occurs in many cancers, see Table I for more examples. The increased expression of these antigens increases the amount of peptide presented by MHC molecules on the cell surface and augmenting T cell recognition.
TAA that are shared between tumors are practical targets for immunotherapies. However, because they are expressed in normal tissues, T cell responses to TAA may promote autoimmunity. Autoimmune destruction of normal melanocytes, or vitiligo, has been observed in both mice and humans following induction of an immune response against melanoma antigens [65-67]. Furthermore, because TAA are self antigens, the immune system is at least partially tolerant of these proteins, affecting the quality of the tumor-specific T cell repertoire.
3.2 T cell tolerance to tumor-associated antigens
The T cell repertoire must include TCRs that recognize foreign antigens and protect the host from autoimmunity. This balance is achieved through central and peripheral tolerance mechanisms. Central tolerance removes autoreactive T cells from the developing repertoire by negative selection in the thymus [68-70]. If a T cell expresses a TCR with avidity for MHC and self-peptide above a certain threshold, the cell is deleted before reaching the periphery.
Given that TAA are self-proteins, the majority of high-avidity TAA-specific T cells are eliminated in the thymus. Endogenous expression of the p53 tumor antigen results in the deletion of high-avidity p53-reactive T cells that are not deleted in a p53-null mouse [71]. Similarly, mice with a deleted tyrosinase gene have CTL with increased functional avidity for TAA after vaccination with tyrosinase compared to mice sufficient for tyrosinase expression [67]. We have also observed similar results in studies of the gp70 TAA. T cells from mice lacking the gp70 gene bind MHC tetramers loaded with gp70423-431 with increased intensity following vaccination with irradiated tumor compared to those from wild type mice (unpublished data). In each of these models antitumor immunity correlates with the detection of high-avidity TAA-specific T cells. Therefore, elimination of these T cells during negative selection in the thymus likely contributes to the low tumor-reactivity of the peripheral T cell repertoire.
3.3 T cell escape from central tolerance
Although negative selection eliminates a significant portion of the self-reactive repertoire, self-reactive T cells do escape deletion and are found in the periphery. Some T cells escape due to a lack of exposure to antigen in the thymus. In a model of experimental autoimmune encephalomyelitis, T cells specific for the proteolipid protein evade deletion because a shorter splice-variant of the protein is preferentially expressed in the thymus [72, 73]. Self-reactive T cells may also escape negative selection because of poor antigen presentation by MHC [74, 75]. For example, the human melanoma antigen gp100280-288 has a fast dissociation rate from HLA-A*0201 [74]. This poor MHC-binding prevents efficient presentation of antigen in the thymus and non-tolerized T cells enter the periphery. However, the low affinity of gp100280-288 for MHC also results in poor presentation on peripheral tissues and tumors, preventing recognition by peripheral T cells.
T cells also escape deletion in the thymus if they have low avidity for self antigens. For example, influenza nucleoprotein (NP)-specific T cells from transgenic mice that express NP bind less MHC tetramer relative to T cells from wild type mice, suggesting that they escaped deletion as a result of low avidity for antigen [76]. Similar low-avidity T cells are detected in OT-I transgenic mice that express a TCR specific for ovalbumin257-264 and express ovalbumin driven by the insulin promoter. These T cells not only bind MHC tetramers with lower intensity, but also have decreased functional avidity since they require ∼ 10-fold more peptide to produce IFNγ [77]. Theobold et al. showed that, although expression of the p53 results in complete loss of reactivity towards the dominant epitope, CTL specific for a cryptic epitope are detected [71]. The T cells specific for the cryptic epitope are low avidity, i.e. they require more peptide antigen for CTL-mediated lysis than T cells specific for the dominant epitope from mice that are not tolerized to p53. These experiments demonstrate that although T cells with high avidity are deleted, some T cells with low avidity evade deletion in the thymus.
The presence of self-reactive T cells in the periphery does not guarantee destruction of tissues expressing the antigen as illustrated in the following examples. In a transgenic mouse expressing a TCR Vβ chain specific for self antigen, tissue destruction does not occur, despite approximately 4% of the natural repertoire of CD8+ T cells being self-reactive [77]. Furthermore, in healthy individuals up to 2% of the CD8+ T cells are Melan-A specific without any evidence of autoimmune destruction of melanocytes [78]. These observations highlight the role of peripheral tolerance mechanisms in preventing autoimmunity and tumor immunity by the low avidity T cells. However, these mechanisms can be overcome. For example, in cancer patients receiving immunotherapy, tumor regression was observed when 80-90% of the CD8+ T cells were tumor-specific [79].
3.4 Peripheral tolerance and the tumor-specific T cell repertoire
Peripheral tolerance is achieved through a variety of mechanisms. In some cases the avidity of the T cells is too low to respond to the endogenous antigen found in peripheral tissues. This passive tolerance is demonstrated in tumor models in which growth of a spontaneous tumor is not sufficient to activate the small numbers of TAA-specific T cells [80, 81]. Similarly, analysis of melanoma-infiltrating lymphocytes shows that the tumor-specific T cells are activated by target cells loaded with high concentrations of peptide but are too low avidity to be fully activated by melanoma cells that express much lower concentrations of antigen [82].
Although passive tolerance explains the lack of autoimmunity in some cases, in many studies autoreactive T cells have proliferated and display activation markers suggesting that they respond to antigen in the periphery [83-85]. However, rather than becoming fully activated, these T cells become anergic upon encountering self antigen. In one study, TAA-specific T cells isolated from melanoma patients were non-cytolytic and did not produce cytokines in response to antigen [83]. The induction of anergy is in part due to activation of T cells in a non-inflammatory environment where they encounter immature dendritic cells ([86], reviewed in [87]). Therefore, TAA-specific T cells may also be rendered anergic because the tumor may not provide appropriate inflammatory stimuli. Induction of anergy in TAA-specific T cells in a number of animal models suggests that chronic inflammation of the tumor environment does not activate TIL [88, 89]. Other studies show that tumor growth can induce the activation of tumor-specific T cells, although the T cells do not prevent tumor growth [90]. Why T cell activation occurs in some tumor systems is not clear, but it may be influenced by the frequency of tumor-reactive T cells at the time of tumor growth. Transfer of large numbers of TAA-specific T cells leads to improved T cell activation by the tumor due to a reduced requirement for CD4-T cell help [80].
Autoreactive T cells, specifically those of higher avidity, may be deleted following antigen stimulation in the periphery [91-93]. Molldrem et al. demonstrated that T cells with both high and low avidity for the PR1 leukemia antigen can be cultured from healthy individuals, but high-avidity PR1-specific T cells are deleted in leukemia patients due to the high expression levels of PR1 by tumors [94]. Interestingly, patients in remission retained a population of T cells with high avidity for the tumor antigen. Since deletion of PR1 cells was only observed at high antigen doses in vitro it would be interesting to determine if the lack of T cell deletion in the patients in remission correlates with low expression of PR1 by their tumors.
Finally, the tumor-reactive T cell repertoire can also be actively inhibited by regulatory T cells (Tregs) (reviewed in [12]). This inhibition occurs as a result of both direct contact and the production of soluble factors such as TGFβ. Interestingly, Tregs may specifically inhibit T cells with high avidity for tumor antigen. In the HER-2/neu transgenic (neu-N) mouse model of breast cancer, vaccination against the HER-2/neu tumor antigen following removal of Tregs elicited T cells that bind MHC tetramers with higher intensity [95].
In summary, both central and peripheral tolerance mechanisms lead to deletion and inactivation of T cells with the highest avidity for TAA. Low-avidity populations persist in the periphery although it is unlikely that they recognize the low levels of TAA expressed by tumors in vivo. These explanations are consistent with why, despite large numbers of tumor-specific T cells in some studies, tumor growth is uninhibited [3]. These results provide a rationale for the development of immunotherapies aimed at improving the avidity and activation of the tumor-specific T cell repertoire.
4. Enhancing the T cell response to tumors
Can the low-avidity tumor-specific T cell repertoire be manipulated to enhance the immune response to tumors? Typically, the low avidity of TAA-specific T cells for antigen prevents activation of these T cells in response to endogenous levels of tumor antigens. Therapies that enhance antigenic priming of tumor-specific T cells will likely elicit a more productive antitumor response. A number of strategies are being developed to improve the function of these T cells so that they may be used prophylactically or therapeutically against cancer. These strategies exploit the binding properties of T cells for tumors both antigen-specifically and non-specifically.
4.1 Vaccination with peptide mimotopes
Vaccination with TAA to elicit functionally avid tumor-specific T cells is one strategy for augmenting antitumor responses. In fact, in one study vaccination of a melanoma patient with the Melan-A peptide elicited a population of T cells with increased avidity for antigen compared to the preimmmune T cells, but these cells did not prevent disease progression [96]. Unfortunately, in the majority of studies vaccination with the TAA increases the frequency of T cells that recognize tumor, but does not improve functional avidity and is insufficient to control tumor growth [97-99]. One possibility is that the affinity of the TCR for TAA-MHC is too low to sufficiently activate the tumor-specific T cells in these cases. Alternatively, the chemical structure of synthetically produced peptides may not adequately mimic naturally presented TAA, as the T cells elicited by peptide vaccines do not always recognize the tumor [100-102].
These studies with TAA peptides provide a rationale for the design of peptide-mimotope vaccines. Mimotopes are mimics of peptide epitopes also known as peptide analogues, agonists, heteroclitic peptides, or altered peptide ligands. Peptide mimotopes contain amino acid substitutions in the TAA that either enhance binding to the MHC [103-108] and/or improve the affinity of the TCR-peptide-MHC complex [30, 38, 109]. Mimotopes are hypothesized to enhance activation of tumor-specific T cells by providing optimal antigen presentation and stimulation of T cells, allowing for the acquisition of full effector function. In addition, mimotope vaccines target T cells that are not activated by the endogenous antigen, but cross-react with it once activated.
Both animal models (see Table II) and clinical trials (see Table III) of mimotope vaccines show increased numbers of tumor-specific T cells and, in some cases, improved antitumor immunity. In some studies, mimotope vaccines also elicit T cells with increased functional avidity for tumor antigen compared to those elicited by vaccination with the endogenous antigen [67, 106, 110, 111]. However, in other studies T cells with decreased functional avidity are detected following vaccination. Comparison of T cell clones from mimotope-vaccinated or unvaccinated melanoma patients show that the vaccine-elicited T cells do not lyse melanoma targets whereas T cells from the endogenous response do [112]. In these patients, mimotope vaccines preferentially expanded T cells with low functional avidity that could only lyse target cells coated with high concentrations of TAA peptide. It is proposed that some high-affinity mimotopes induce deletion of the highest-avidity T cells leaving only low-avidity T cells in the periphery. This hypothesis is consistent with experiments that show deletion of autoreactive T cells after vaccination with high-affinity peptide mimotopes in autoimmunity models [113].
Table II.
Antitumor activity elicited by mimotope vaccines in mice.
| Tumor Antigen | MHC haplotype/epitope | Mimotope | Improved binding to MHC or TCR | Tumor assay | Response | Ref |
|---|---|---|---|---|---|---|
| gp70423-431 | H-2Ld SPSYVYHQF |
SPSYAYHQF | TCR | Prophylactic | 50% protection | [30] |
| MNKYAYHML | TCR | Prophylactic | 50% protection | [31] | ||
| mTERT988-997 | HLA-A*0201† DLQVNSLQTV |
YLQVNSLQTV | MHC | Prophylactic | 33% protection | [75] |
| Her2/Neu773-782 | HLA-A*0201†† VMAGVGSPYV |
FMANVAIPHL FMHNVPIPYL FYANVPSPHL |
n.d. | Therapeutic | 40-50% delayed tumor growth | [157] |
| Her2/Neu435-443 | HLA-A*0201† ILHDGAYSL |
ILHNGAYSL | TCR | Therapeutic | 30% protection | [158] |
| TRP1222-229 | H-2Kb TWHRYHLL |
TAYRYHLL | MHC | Prophylactic | 90-100% protection | [103] |
| TRP2180-188 | H-2Kb SVYDFFVWL |
SIYDFFVWL | TCR | Therapeutic | No protection | [159] |
| gp10025-33 | H-2Db EGSRNQDWL |
KVPRNQDWL | MHC | Therapeutic (ACT and IL-2) | 80-100% tumor inhibition | [65] |
| Therapeutic (ACT) | 100% delayed tumor growth | [104] |
n.d.= not determined
Antitumor activity evaluated in HLA-A*0201-transgenic HHD mice.
Antitumor activity evaluated in double transgenic mice: HLA-A*0201 and neu-N.
Table III.
Clinical trials of therapeutic cancer vaccines using modified HLA-A*0201-restricted tumor antigens
| Cancer | Antigen | Adjuvant | Positive in vitro responses | Clinical response | Ref |
|---|---|---|---|---|---|
| Extensive metastatic melanoma | gp100209-217 gp100209-217(210M) gp100209-217(210M) |
IFA IFA IFA+IL-2 |
2/8 IFNγ* 10/11 IFNγ* 3/19 IFNγ* |
1/9 CR 0/11 CR, 3/11 MR 8/19 CR, 3/19 MR, 3/19 SD |
[97] |
| Stage I-III melanoma | gp100209-217(210M) | IFA +/- IFNα** |
28/29 tetramer 9/9 IFNγ* |
Not Reported | [160] |
| Melanoma | gp100209-217(210M) | IFA IFA+IL-12 IFA+IL-2 |
7/7 tetramer* 4/5 tetramer* 2/11 tetramer* |
Not Reported | [161] |
| Stage III/IV melanoma | gp100209-217(210M) tyrosinase368-376 (370D) |
IFA+/- IL-12 |
34/40 Skin test*‡ 33/38 IFNγ* 37/42 tetramer* |
24/48 relapsed, 10 died | [162] |
| Stage IV melanoma | gp100209-217(210M) tyrosinase368-376 (370D) |
DCs | 2/16 Skin test* 11/16 IFNγ* 0/16 tetramer* |
1/16 CR 2/16 MR, 2/16 SD 10/16 died |
[163] |
| Stage IIA/IIB melanoma | gp100209-217(210M) tyrosinase368-376(370D) |
IFA+/- GM-CSF |
34/39 ELISA 37/42 tetramer |
After 24 months: “favorable:” 7 relapsed, 2 died | [164] |
| Recurrent small cell lung or colon cancer | CEA605-613(610D) | Flt3 | 7/12 CTL 10/12 tetramer* |
2 CR, 7 PD 2 SD, 1 MR |
[165] |
CR=Complete response, PD=progressive disease, SD=Stable disease, MR=Mixed response, IFA=Incomplete Freund’s adjuvant, DCs=dendritic cells
specific for native peptide
response measured to gp100, not tyrosinase
IFNα does not interfere with the antigen-specific response.
Using the CT26 tumor model we showed that vaccination with mimotopes that increase the binding of the TCR-peptide-MHC interaction above a certain threshold results in a loss of tumor protection [31]. Direct ex vivo analyses of TIL showed that these mimotopes elicit T cells that do not produce IFNγ upon stimulation with either the TAA or the mimotope used for priming, at any concentration of peptide. We also identified mimotopes that elicited functional CTL that produced IFNγ and successfully elicited antitumor immunity. The affinity of the TCR-peptide-MHC interaction with these mimotopes is between that of the TAA and the mimotopes that anergized the T cells. Interestingly, TAA-loaded MHC tetramer binds T cells responding to both the high- and intermediate-affinity mimotopes with the same intensity, further indicating that the T cells elicited by these mimotope vaccines are anergic rather than low avidity.
Thus, vaccination with peptide mimotopes shows significant promise for improving antitumor responses, especially when used in combination with other therapies [65]. However, recent research suggests that there is an upper boundary for the affinity of peptide vaccines, above which the expansion of functionally unresponsive T cells occurs. Improved efficacy of these vaccines requires optimization to elicit T cells that recognize tumor with high functional avidity.
4.2 Adoptive cellular therapy
Another approach aimed at improving the T cell sensitivity to tumor is adoptive cellular therapy (ACT). In ACT tumor-specific T cells are removed from the patient, cultured in vitro, and transferred back into the patient. The T cells may be sorted, expanded, and manipulated to enhance their antitumor activity. Selective expansion of high-avidity CTL is achieved by in vitro stimulation with low concentrations of peptide [16, 114]. These CTL have superior antitumor activity as shown by improved protection against challenge with the B16 murine melanoma [115]. Furthermore, antigen-specific ex vivo expansion of T cells in combination with IL-2 can reverse the non-functional state of TIL [116]. In a clinical trial, adoptive transfer of melanoma-specific T cells in combination with IL-2 therapy and lymphodepletion resulted in clinical responses in 50% of the patients treated, demonstrating the potential of this therapy [79].
With the goal of increasing the functional avidity of T cells or providing TAA-specific T cells for patients without endogenous T cell responses, T cells are being genetically engineered to express high affinity TAA-specific TCRs. Human PBMC transduced with TAA-specific TCRs recognize and kill tumors expressing the antigen in vitro [117-119] and were recently tested in vivo for the treatment of metastatic melanoma [120]. PBMC were retrovirally transduced with a high-affinity MART-1-specific TCR and transferred into patients. Although the frequency of circulating MART-1-specific T cells sometimes remained high for up to a year, clinical responses were only observed in 2 of 15 patients. Combining gene therapy with other immunotherapies such as vaccination or chemotherapy may improve the antitumor efficacy.
4.4 Other therapies for improving T cell function
Enhancing co-stimulation during activation may improve priming of TAA-specific T cells. Vaccination with viruses encoding the co-stimulatory molecules CD80, ICAM-1, and LFA-3 (also known as TRICOM) in combination with TAA improves antitumor activity [121, 122]. Enhancing co-stimulation not only increases the number of antigen-specific T cells, but also elicits higher avidity T cells as determined by dissociation of MHC tetramers and tumor lysis assays. Furthermore, the TRICOM vaccine elicits more memory T cells which also have increased peptide sensitivity [123]. However, enhanced co-stimulation also leads to the deletion of T cells. CD28-signaling pathways lead to either activation-induced cell death or activation of both immature and peripheral T cells depending on the strength of TCR triggering [32, 124]. CD28 ligation and weak TCR binding results in the enhancement of T cell activation while CD28 ligation combined with strong TCR binding results in antigen-induced apoptosis. Further understanding of these opposing roles of co-stimulation may explain why some tumor immunotherapies induce ineffective T cell responses.
Some therapies that incorporate chemotherapy or irradiation also improve the functional avidity of the antitumor T cell response [65, 120]. One mechanism for this improved immunity is the depletion of regulatory T cells. As mentioned above, vaccination with cells expressing HER-2/neu in combination with chemotherapy prevents tumor growth in neu-N transgenic mice [95]. In this study the chemotherapy led to selective depletion of Tregs resulting in activation of a subset of high-affinity neu-specific CD8+ T cells. Irradiation and chemotherapy can also enhance the activation of tumor-specific T cells by improving the expression and cross-presentation of tumor antigens on both tumors and the surrounding stroma [125, 126]. T cell therapies, such as ACT, may benefit from this increased antigen presentation.
5. Discussion
Analyses of the T cell response to tumor antigens have demonstrated that tumor growth still occurs despite large numbers of tumor-reactive T cells. T cells must overcome a number of obstacles including tumor-induced immune suppression, cellular heterogeneity, and antigen loss from the tumor. This poor reactivity of T cells for TAA also results from central and peripheral tolerance mechanisms that delete or inactivate T cells with high avidity for tumor antigens. The remaining low-avidity T cells do not recognize the endogenous levels of tumor antigen. As a result the focus of research has turned towards developing therapies that enhance the activation and functional avidity of tumor-specific T cells.
The selective expansion of high-avidity T cells ex vivo is well established and ACT with these cells has shown potential. However, this therapy is labor intensive and the costs may be too great for general clinical use. Nevertheless, these studies have demonstrated that there is a subset of T cells within the TAA-specific T cell repertoire with sufficient avidity to recognize the levels of antigen expressed on the surface of tumors. Enhancing the survival and expansion of high-avidity T cells in vivo will improve the design of future T cell therapies. In some cases selecting for tumor reactive T cells requires enhancing the strength of the stimulating antigen. However, studies to date suggest that selective expansion of high-avidity T cells from the full repertoire may be more complicated. For example, vaccination with peptide mimotopes elicits T cells with both strong and weak reactivity to the endogenous tumor antigen. Understanding why, in some cases, vaccines elicit T cells with low functional avidity for TAA is crucial. Further insight into the mechanism behind peripheral deletion and functional inactivation of tumor-specific T cells will likely contribute to the answer. Other therapies such as lymphodepletion and optimization of prime-boost schedules that select for high-avidity memory T cells are also promising. Overall, the shift towards strategies aimed at improving the frequency and the functional avidity of tumor-specific T cells will likely be crucial for improving clinical efficacy of current immunotherapies.
Acknowledgements
We thank Dr. John Cohen, Kimberly Jordan, and Charles Kemmler for critical reading of this manuscript. The authors apologize to those investigators whose research was not cited due to space limitations. The authors were supported by R01 CA109560 and the Cancer Research Institute Predoctoral Emphasis Pathway in Tumor Immunology Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Prehn RT, Main JM. Immunity to methylcholanthrene-induced sarcomas. J Natl Cancer Inst. 1957;18(6):769–78. [PubMed] [Google Scholar]
- [2].Gross L. Intradermal immunization of C3H mice against a sarcoma that originated in an animal of the same line. Cancer Res. 1943;(3):326–33. [Google Scholar]
- [3].Nagorsen D, Scheibenbogen C, Marincola FM, Letsch A, Keilholz U. Natural T cell immunity against cancer. Clin Cancer Res. 2003;9(12):4296–303. [PubMed] [Google Scholar]
- [4].Clemente CG, Mihm MC, Jr., Bufalino R, Zurrida S, Collini P, Cascinelli N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 1996;77(7):1303–10. doi: 10.1002/(SICI)1097-0142(19960401)77:7<1303::AID-CNCR12>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- [5].Mihm MC, Jr., Clemente CG, Cascinelli N. Tumor infiltrating lymphocytes in lymph node melanoma metastases: a histopathologic prognostic indicator and an expression of local immune response. Lab Invest. 1996;74(1):43–7. [PubMed] [Google Scholar]
- [6].Clark WH, Jr., Elder DE, Guerry Dt, Braitman LE, Trock BJ, Schultz D, et al. Model predicting survival in stage I melanoma based on tumor progression. J Natl Cancer Inst. 1989;81(24):1893–904. doi: 10.1093/jnci/81.24.1893. [DOI] [PubMed] [Google Scholar]
- [7].Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–13. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- [8].Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58(16):3491–4. [PubMed] [Google Scholar]
- [9].Schumacher K, Haensch W, Roefzaad C, Schlag PM. Prognostic significance of activated CD8(+) T cell infiltrations within esophageal carcinomas. Cancer Res. 2001;61(10):3932–6. [PubMed] [Google Scholar]
- [10].Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–4. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- [11].Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9(10):1269–74. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- [12].Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6(4):295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- [13].Derynck R, Jarrett JA, Chen EY, Eaton DH, Bell JR, Assoian RK, et al. Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells. Nature. 1985;316(6030):701–5. doi: 10.1038/316701a0. [DOI] [PubMed] [Google Scholar]
- [14].Kruger-Krasagakes S, Krasagakis K, Garbe C, Schmitt E, Huls C, Blankenstein T, et al. Expression of interleukin 10 in human melanoma. Br J Cancer. 1994;70(6):1182–5. doi: 10.1038/bjc.1994.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J, et al. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev. 2006;213:131–45. doi: 10.1111/j.1600-065X.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- [16].Alexander-Miller MA, Leggatt GR, Berzofsky JA. Selective expansion of high- or low-avidity cytotoxic T lymphocytes and efficacy for adoptive immunotherapy. Proc Natl Acad Sci U S A. 1996;93(9):4102–7. doi: 10.1073/pnas.93.9.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gallimore A, Dumrese T, Hengartner H, Zinkernagel RM, Rammensee HG. Protective immunity does not correlate with the hierarchy of virus-specific cytotoxic T cell responses to naturally processed peptides. J Exp Med. 1998;187(10):1647–57. doi: 10.1084/jem.187.10.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rubio V, Stuge TB, Singh N, Betts MR, Weber JS, Roederer M, et al. Ex vivo identification, isolation and analysis of tumor-cytolytic T cells. Nat Med. 2003;9(11):1377–82. doi: 10.1038/nm942. [DOI] [PubMed] [Google Scholar]
- [19].Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 1995;375(6527):148–51. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- [20].Kalergis AM, Boucheron N, Doucey MA, Palmieri E, Goyarts EC, Vegh Z, et al. Efficient T cell activation requires an optimal dwell-time of interaction between the TCR and the pMHC complex. Nat Immunol. 2001;2(3):229–34. doi: 10.1038/85286. [DOI] [PubMed] [Google Scholar]
- [21].McKeithan TW. Kinetic proofreading in T-cell receptor signal transduction. Proc Natl Acad Sci U S A. 1995;92(11):5042–6. doi: 10.1073/pnas.92.11.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wulfing C, Rabinowitz JD, Beeson C, Sjaastad MD, McConnell HM, Davis MM. Kinetics and extent of T cell activation as measured with the calcium signal. J Exp Med. 1997;185(10):1815–25. doi: 10.1084/jem.185.10.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rabinowitz JD, Beeson C, Lyons DS, Davis MM, McConnell HM. Kinetic discrimination in T-cell activation. Proc Natl Acad Sci U S A. 1996;93(4):1401–5. doi: 10.1073/pnas.93.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Valitutti S, Muller S, Dessing M, Lanzavecchia A. Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J Exp Med. 1996;183(4):1917–21. doi: 10.1084/jem.183.4.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Alexander-Miller MA, Leggatt GR, Sarin A, Berzofsky JA. Role of antigen, CD8, and cytotoxic T lymphocyte (CTL) avidity in high dose antigen induction of apoptosis of effector CTL. J Exp Med. 1996;184(2):485–92. doi: 10.1084/jem.184.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Holler PD, Kranz DM. Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity. 2003;18(2):255–64. doi: 10.1016/s1074-7613(03)00019-0. [DOI] [PubMed] [Google Scholar]
- [27].Rosette C, Werlen G, Daniels MA, Holman PO, Alam SM, Travers PJ, et al. The impact of duration versus extent of TCR occupancy on T cell activation: a revision of the kinetic proofreading model. Immunity. 2001;15(1):59–70. doi: 10.1016/s1074-7613(01)00173-x. [DOI] [PubMed] [Google Scholar]
- [28].Kersh GJ, Allen PM. Structural basis for T cell recognition of altered peptide ligands: a single T cell receptor can productively recognize a large continuum of related ligands. J Exp Med. 1996;184(4):1259–68. doi: 10.1084/jem.184.4.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Matsui K, Boniface JJ, Steffner P, Reay PA, Davis MM. Kinetics of T-cell receptor binding to peptide/I-Ek complexes: correlation of the dissociation rate with T-cell responsiveness. Proc Natl Acad Sci U S A. 1994;91(26):12862–6. doi: 10.1073/pnas.91.26.12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Slansky JE, Rattis FM, Boyd LF, Fahmy T, Jaffee EM, Schneck JP, et al. Enhanced antigen-specific antitumor immunity with altered peptide ligands that stabilize the MHC-peptide-TCR complex. Immunity. 2000;13(4):529–38. doi: 10.1016/s1074-7613(00)00052-2. [DOI] [PubMed] [Google Scholar]
- [31].McMahan RH, McWilliams JA, Jordan KR, Dow SW, Wilson DB, Slansky JE. Relating TCR-peptide-MHC affinity to immunogenicity for the design of tumor vaccines. J Clin Invest. 2006;116(9):2543–51. doi: 10.1172/JCI26936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yu XZ, Martin PJ, Anasetti C. CD28 signal enhances apoptosis of CD8 T cells after strong TCR ligation. J Immunol. 2003;170(6):3002–6. doi: 10.4049/jimmunol.170.6.3002. [DOI] [PubMed] [Google Scholar]
- [33].Ueno T, Tomiyama H, Fujiwara M, Oka S, Takiguchi M. Functionally impaired HIV-specific CD8 T cells show high affinity TCR-ligand interactions. J Immunol. 2004;173(9):5451–7. doi: 10.4049/jimmunol.173.9.5451. [DOI] [PubMed] [Google Scholar]
- [34].Sykulev Y, Vugmeyster Y, Brunmark A, Ploegh HL, Eisen HN. Peptide antagonism and T cell receptor interactions with peptide-MHC complexes. Immunity. 1998;9(4):475–83. doi: 10.1016/s1074-7613(00)80631-7. [DOI] [PubMed] [Google Scholar]
- [35].Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, et al. Ligand recognition by alpha beta T cell receptors. Annu Rev Immunol. 1998;16:523–44. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- [36].Crawford F, Kozono H, White J, Marrack P, Kappler J. Detection of antigen-specific T cells with multivalent soluble class II MHC covalent peptide complexes. Immunity. 1998;8(6):675–82. doi: 10.1016/s1074-7613(00)80572-5. [DOI] [PubMed] [Google Scholar]
- [37].Yee C, Savage PA, Lee PP, Davis MM, Greenberg PD. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J Immunol. 1999;162(4):2227–34. [PubMed] [Google Scholar]
- [38].de Visser KE, Cordaro TA, Kessels HW, Tirion FH, Schumacher TN, Kruisbeek AM. Low-avidity self-specific T cells display a pronounced expansion defect that can be overcome by altered peptide ligands. J Immunol. 2001;167(7):3818–28. doi: 10.4049/jimmunol.167.7.3818. [DOI] [PubMed] [Google Scholar]
- [39].Derby MA, Wang J, Margulies DH, Berzofsky JA. Two intermediate-avidity cytotoxic T lymphocyte clones with a disparity between functional avidity and MHC tetramer staining. Int Immunol. 2001;13(6):817–24. doi: 10.1093/intimm/13.6.817. [DOI] [PubMed] [Google Scholar]
- [40].Dutoit V, Guillaume P, Cerottini JC, Romero P, Valmori D. Dissecting TCR-MHC/peptide complex interactions with A2/peptide multimers incorporating tumor antigen peptide variants: crucial role of interaction kinetics on functional outcomes. Eur J Immunol. 2002;32(11):3285–93. doi: 10.1002/1521-4141(200211)32:11<3285::AID-IMMU3285>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- [41].Bullock TN, Mullins DW, Colella TA, Engelhard VH. Manipulation of avidity to improve effectiveness of adoptively transferred CD8(+) T cells for melanoma immunotherapy in human MHC class I-transgenic mice. J Immunol. 2001;167(10):5824–31. doi: 10.4049/jimmunol.167.10.5824. [DOI] [PubMed] [Google Scholar]
- [42].Dutoit V, Rubio-Godoy V, Doucey MA, Batard P, Lienard D, Rimoldi D, et al. Functional avidity of tumor antigen-specific CTL recognition directly correlates with the stability of MHC/peptide multimer binding to TCR. J Immunol. 2002;168(3):1167–71. doi: 10.4049/jimmunol.168.3.1167. [DOI] [PubMed] [Google Scholar]
- [43].Garcia KC, Scott CA, Brunmark A, Carbone FR, Peterson PA, Wilson IA, et al. CD8 enhances formation of stable T-cell receptor/MHC class I molecule complexes. Nature. 1996;384(6609):577–81. doi: 10.1038/384577a0. [DOI] [PubMed] [Google Scholar]
- [44].Luescher IF, Vivier E, Layer A, Mahiou J, Godeau F, Malissen B, et al. CD8 modulation of T-cell antigen receptor-ligand interactions on living cytotoxic T lymphocytes. Nature. 1995;373(6512):353–6. doi: 10.1038/373353a0. [DOI] [PubMed] [Google Scholar]
- [45].Daniels MA, Jameson SC. Critical role for CD8 in T cell receptor binding and activation by peptide/major histocompatibility complex multimers. J Exp Med. 2000;191(2):335–46. doi: 10.1084/jem.191.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Xu XN, Purbhoo MA, Chen N, Mongkolsapaya J, Cox JH, Meier UC, et al. A novel approach to antigen-specific deletion of CTL with minimal cellular activation using alpha3 domain mutants of MHC class I/peptide complex. Immunity. 2001;14(5):591–602. doi: 10.1016/s1074-7613(01)00133-9. [DOI] [PubMed] [Google Scholar]
- [47].Potter TA, Rajan TV, Dick RF, 2nd, Bluestone JA. Substitution at residue 227 of H-2 class I molecules abrogates recognition by CD8-dependent, but not CD8-independent, cytotoxic T lymphocytes. Nature. 1989;337(6202):73–5. doi: 10.1038/337073a0. [DOI] [PubMed] [Google Scholar]
- [48].Choi EM, Chen JL, Wooldridge L, Salio M, Lissina A, Lissin N, et al. High avidity antigen-specific CTL identified by CD8-independent tetramer staining. J Immunol. 2003;171(10):5116–23. doi: 10.4049/jimmunol.171.10.5116. [DOI] [PubMed] [Google Scholar]
- [49].Dustin ML, Springer TA. T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature. 1989;341(6243):619–24. doi: 10.1038/341619a0. [DOI] [PubMed] [Google Scholar]
- [50].Hahn WC, Rosenstein Y, Calvo V, Burakoff SJ, Bierer BE. A distinct cytoplasmic domain of CD2 regulates ligand avidity and T-cell responsiveness to antigen. Proc Natl Acad Sci U S A. 1992;89(15):7179–83. doi: 10.1073/pnas.89.15.7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Koneru M, Monu N, Schaer D, Barletta J, Frey AB. Defective adhesion in tumor infiltrating CD8+ T cells. J Immunol. 2006;176(10):6103–11. doi: 10.4049/jimmunol.176.10.6103. [DOI] [PubMed] [Google Scholar]
- [52].Fahmy TM, Bieler JG, Edidin M, Schneck JP. Increased TCR avidity after T cell activation: a mechanism for sensing low-density antigen. Immunity. 2001;14(2):135–43. [PubMed] [Google Scholar]
- [53].Cawthon AG, Alexander-Miller MA. Optimal colocalization of TCR and CD8 as a novel mechanism for the control of functional avidity. J Immunol. 2002;169(7):3492–8. doi: 10.4049/jimmunol.169.7.3492. [DOI] [PubMed] [Google Scholar]
- [54].Slifka MK, Whitton JL. Functional avidity maturation of CD8(+) T cells without selection of higher affinity TCR. Nat Immunol. 2001;2(8):711–7. doi: 10.1038/90650. [DOI] [PubMed] [Google Scholar]
- [55].Koneru M, Schaer D, Monu N, Ayala A, Frey AB. Defective proximal TCR signaling inhibits CD8+ tumor-infiltrating lymphocyte lytic function. J Immunol. 2005;174(4):1830–40. doi: 10.4049/jimmunol.174.4.1830. [DOI] [PubMed] [Google Scholar]
- [56].Beaudenon S, Kremsdorf D, Croissant O, Jablonska S, Wain-Hobson S, Orth G. A novel type of human papillomavirus associated with genital neoplasias. Nature. 1986;321(6067):246–9. doi: 10.1038/321246a0. [DOI] [PubMed] [Google Scholar]
- [57].List AF, Greco FA, Vogler LB. Lymphoproliferative diseases in immunocompromised hosts: the role of Epstein-Barr virus. J Clin Oncol. 1987;5(10):1673–89. doi: 10.1200/JCO.1987.5.10.1673. [DOI] [PubMed] [Google Scholar]
- [58].Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266(5192):1865–9. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- [59].Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet. 1981;2(8256):1129–33. doi: 10.1016/s0140-6736(81)90585-7. [DOI] [PubMed] [Google Scholar]
- [60].Tsukuma H, Hiyama T, Tanaka S, Nakao M, Yabuuchi T, Kitamura T, et al. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N Engl J Med. 1993;328(25):1797–801. doi: 10.1056/NEJM199306243282501. [DOI] [PubMed] [Google Scholar]
- [61].Koutsky LA, Ault KA, Wheeler CM, Brown DR, Barr E, Alvarez FB, et al. A controlled trial of a human papillomavirus type 16 vaccine. N Engl J Med. 2002;347(21):1645–51. doi: 10.1056/NEJMoa020586. [DOI] [PubMed] [Google Scholar]
- [62].Chang MH, Chen CJ, Lai MS, Hsu HM, Wu TC, Kong MS, et al. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N Engl J Med. 1997;336(26):1855–9. doi: 10.1056/NEJM199706263362602. [DOI] [PubMed] [Google Scholar]
- [63].Novellino L, Castelli C, Parmiani G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother. 2005;54(3):187–207. doi: 10.1007/s00262-004-0560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- [65].Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198(4):569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Dudley ME, Roopenian DC. Loss of a unique tumor antigen by cytotoxic T lymphocyte immunoselection from a 3-methylcholanthrene-induced mouse sarcoma reveals secondary unique and shared antigens. J Exp Med. 1996;184(2):441–7. doi: 10.1084/jem.184.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Colella TA, Bullock TN, Russell LB, Mullins DW, Overwijk WW, Luckey CJ, et al. Self-tolerance to the murine homologue of a tyrosinase-derived melanoma antigen: implications for tumor immunotherapy. J Exp Med. 2000;191(7):1221–32. doi: 10.1084/jem.191.7.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kappler JW, Roehm N, Marrack P. T cell tolerance by clonal elimination in the thymus. Cell. 1987;49(2):273–80. doi: 10.1016/0092-8674(87)90568-x. [DOI] [PubMed] [Google Scholar]
- [69].Kisielow P, Bluthmann H, Staerz UD, Steinmetz M, von Boehmer H. Tolerance in T-cell-receptor transgenic mice involves deletion of nonmature CD4+8+ thymocytes. Nature. 1988;333(6175):742–6. doi: 10.1038/333742a0. [DOI] [PubMed] [Google Scholar]
- [70].Blackman M, Kappler J, Marrack P. The role of the T cell receptor in positive and negative selection of developing T cells. Science. 1990;248(4961):1335–41. doi: 10.1126/science.1972592. [DOI] [PubMed] [Google Scholar]
- [71].Theobald M, Biggs J, Hernandez J, Lustgarten J, Labadie C, Sherman LA. Tolerance to p53 by A2.1-restricted cytotoxic T lymphocytes. J Exp Med. 1997;185(5):833–41. doi: 10.1084/jem.185.5.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Anderson AC, Nicholson LB, Legge KL, Turchin V, Zaghouani H, Kuchroo VK. High frequency of autoreactive myelin proteolipid protein-specific T cells in the periphery of naive mice: mechanisms of selection of the self-reactive repertoire. J Exp Med. 2000;191(5):761–70. doi: 10.1084/jem.191.5.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Klein L, Klugmann M, Nave KA, Tuohy VK, Kyewski B. Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat Med. 2000;6(1):56–61. doi: 10.1038/71540. [DOI] [PubMed] [Google Scholar]
- [74].Yu Z, Theoret MR, Touloukian CE, Surman DR, Garman SC, Feigenbaum L, et al. Poor immunogenicity of a self/tumor antigen derives from peptide-MHC-I instability and is independent of tolerance. J Clin Invest. 2004;114(4):551–9. doi: 10.1172/JCI21695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Gross DA, Graff-Dubois S, Opolon P, Cornet S, Alves P, Bennaceur-Griscelli A, et al. High vaccination efficiency of low-affinity epitopes in antitumor immunotherapy. J Clin Invest. 2004;113(3):425–33. doi: 10.1172/JCI19418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].de Visser KE, Cordaro TA, Kioussis D, Haanen JB, Schumacher TN, Kruisbeek AM. Tracing and characterization of the low-avidity self-specific T cell repertoire. Eur J Immunol. 2000;30(5):1458–68. doi: 10.1002/(SICI)1521-4141(200005)30:5<1458::AID-IMMU1458>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- [77].Zehn D, Bevan MJ. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity. 2006;25(2):261–70. doi: 10.1016/j.immuni.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zippelius A, Pittet MJ, Batard P, Rufer N, de Smedt M, Guillaume P, et al. Thymic selection generates a large T cell pool recognizing a self-peptide in humans. J Exp Med. 2002;195(4):485–94. doi: 10.1084/jem.20011658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–4. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lyman MA, Aung S, Biggs JA, Sherman LA. A spontaneously arising pancreatic tumor does not promote the differentiation of naive CD8+ T lymphocytes into effector CTL. J Immunol. 2004;172(11):6558–67. doi: 10.4049/jimmunol.172.11.6558. [DOI] [PubMed] [Google Scholar]
- [81].Speiser DE, Miranda R, Zakarian A, Bachmann MF, McKall-Faienza K, Odermatt B, et al. Self antigens expressed by solid tumors Do not efficiently stimulate naive or activated T cells: implications for immunotherapy. J Exp Med. 1997;186(5):645–53. doi: 10.1084/jem.186.5.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Gervois N, Guilloux Y, Diez E, Jotereau F. Suboptimal activation of melanoma infiltrating lymphocytes (TIL) due to low avidity of TCR/MHC-tumor peptide interactions. J Exp Med. 1996;183(5):2403–7. doi: 10.1084/jem.183.5.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. 1999;5(6):677–85. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- [84].Hernandez J, Aung S, Redmond WL, Sherman LA. Phenotypic and functional analysis of CD8(+) T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J Exp Med. 2001;194(6):707–17. doi: 10.1084/jem.194.6.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Romero P, Dunbar PR, Valmori D, Pittet M, Ogg GS, Rimoldi D, et al. Ex vivo staining of metastatic lymph nodes by class I major histocompatibility complex tetramers reveals high numbers of antigen-experienced tumor-specific cytolytic T lymphocytes. J Exp Med. 1998;188(9):1641–50. doi: 10.1084/jem.188.9.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Hernandez J, Aung S, Marquardt K, Sherman LA. Uncoupling of proliferative potential and gain of effector function by CD8(+) T cells responding to self-antigens. J Exp Med. 2002;196(3):323–33. doi: 10.1084/jem.20011612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol. 2001;19:47–64. doi: 10.1146/annurev.immunol.19.1.47. [DOI] [PubMed] [Google Scholar]
- [88].Ohlen C, Kalos M, Cheng LE, Shur AC, Hong DJ, Carson BD, et al. CD8(+) T cell tolerance to a tumor-associated antigen is maintained at the level of expansion rather than effector function. J Exp Med. 2002;195(11):1407–18. doi: 10.1084/jem.20011063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Staveley-O’Carroll K, Sotomayor E, Montgomery J, Borrello I, Hwang L, Fein S, et al. Induction of antigen-specific T cell anergy: An early event in the course of tumor progression. Proc Natl Acad Sci U S A. 1998;95(3):1178–83. doi: 10.1073/pnas.95.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Nguyen LT, Elford AR, Murakami K, Garza KM, Schoenberger SP, Odermatt B, et al. Tumor growth enhances cross-presentation leading to limited T cell activation without tolerance. J Exp Med. 2002;195(4):423–35. doi: 10.1084/jem.20010032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Jones LA, Chin LT, Longo DL, Kruisbeek AM. Peripheral clonal elimination of functional T cells. Science. 1990;250(4988):1726–9. doi: 10.1126/science.2125368. [DOI] [PubMed] [Google Scholar]
- [92].Webb S, Morris C, Sprent J. Extrathymic tolerance of mature T cells: clonal elimination as a consequence of immunity. Cell. 1990;63(6):1249–56. doi: 10.1016/0092-8674(90)90420-j. [DOI] [PubMed] [Google Scholar]
- [93].Ochsenbein AF, Sierro S, Odermatt B, Pericin M, Karrer U, Hermans J, et al. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature. 2001;411(6841):1058–64. doi: 10.1038/35082583. [DOI] [PubMed] [Google Scholar]
- [94].Molldrem JJ, Lee PP, Kant S, Wieder E, Jiang W, Lu S, et al. Chronic myelogenous leukemia shapes host immunity by selective deletion of high-avidity leukemia-specific T cells. J Clin Invest. 2003;111(5):639–47. doi: 10.1172/JCI16398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Ercolini AM, Ladle BH, Manning EA, Pfannenstiel LW, Armstrong TD, Machiels JP, et al. Recruitment of latent pools of high-avidity CD8(+) T cells to the antitumor immune response. J Exp Med. 2005;201(10):1591–602. doi: 10.1084/jem.20042167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Valmori D, Dutoit V, Schnuriger V, Quiquerez AL, Pittet MJ, Guillaume P, et al. Vaccination with a Melan-A peptide selects an oligoclonal T cell population with increased functional avidity and tumor reactivity. J Immunol. 2002;168(8):4231–40. doi: 10.4049/jimmunol.168.8.4231. [DOI] [PubMed] [Google Scholar]
- [97].Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med. 1998;4(3):321–7. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Mukherji B, Chakraborty NG, Yamasaki S, Okino T, Yamase H, Sporn JR, et al. Induction of antigen-specific cytolytic T cells in situ in human melanoma by immunization with synthetic peptide-pulsed autologous antigen presenting cells. Proc Natl Acad Sci U S A. 1995;92(17):8078–82. doi: 10.1073/pnas.92.17.8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Hu X, Chakraborty NG, Sporn JR, Kurtzman SH, Ergin MT, Mukherji B. Enhancement of cytolytic T lymphocyte precursor frequency in melanoma patients following immunization with the MAGE-1 peptide loaded antigen presenting cell-based vaccine. Cancer Res. 1996;56(11):2479–83. [PubMed] [Google Scholar]
- [100].Le Gal FA, Ayyoub M, Dutoit V, Widmer V, Jager E, Cerottini JC, et al. Distinct structural TCR repertoires in naturally occurring versus vaccine-induced CD8+ T-cell responses to the tumor-specific antigen NY-ESO-1. J Immunother. 2005;28(3):252–7. doi: 10.1097/01.cji.0000161398.34701.26. [DOI] [PubMed] [Google Scholar]
- [101].Chen W, Yewdell JW, Levine RL, Bennink JR. Modification of cysteine residues in vitro and in vivo affects the immunogenicity and antigenicity of major histocompatibility complex class I-restricted viral determinants. J Exp Med. 1999;189(11):1757–64. doi: 10.1084/jem.189.11.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Nishikawa H, Qian F, Tsuji T, Ritter G, Old LJ, Gnjatic S, et al. Influence of CD4+CD25+ regulatory T cells on low/high-avidity CD4+ T cells following peptide vaccination. J Immunol. 2006;176(10):6340–6. doi: 10.4049/jimmunol.176.10.6340. [DOI] [PubMed] [Google Scholar]
- [103].Dyall R, Bowne WB, Weber LW, LeMaoult J, Szabo P, Moroi Y, et al. Heteroclitic immunization induces tumor immunity. J Exp Med. 1998;188(9):1553–61. doi: 10.1084/jem.188.9.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, et al. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med. 1998;188(2):277–86. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Valmori D, Fonteneau JF, Valitutti S, Gervois N, Dunbar R, Lienard D, et al. Optimal activation of tumor-reactive T cells by selected antigenic peptide analogues. Int Immunol. 1999;11(12):1971–80. doi: 10.1093/intimm/11.12.1971. [DOI] [PubMed] [Google Scholar]
- [106].Tangri S, Ishioka GY, Huang X, Sidney J, Southwood S, Fikes J, et al. Structural features of peptide analogs of human histocompatibility leukocyte antigen class I epitopes that are more potent and immunogenic than wild-type peptide. J Exp Med. 2001;194(6):833–46. doi: 10.1084/jem.194.6.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Parkhurst MR, Salgaller ML, Southwood S, Robbins PF, Sette A, Rosenberg SA, et al. Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J Immunol. 1996;157(6):2539–48. [PubMed] [Google Scholar]
- [108].Hernandez J, Schoeder K, Blondelle SE, Pons FG, Lone YC, Simora A, et al. Antigenicity and immunogenicity of peptide analogues of a low affinity peptide of the human telomerase reverse transcriptase tumor antigen. Eur J Immunol. 2004;34(8):2331–41. doi: 10.1002/eji.200425134. [DOI] [PubMed] [Google Scholar]
- [109].Hoffmann TK, Loftus DJ, Nakano K, Maeurer MJ, Chikamatsu K, Appella E, et al. The ability of variant peptides to reverse the nonresponsiveness of T lymphocytes to the wild-type sequence p53(264-272) epitope. J Immunol. 2002;168(3):1338–47. doi: 10.4049/jimmunol.168.3.1338. [DOI] [PubMed] [Google Scholar]
- [110].Yang S, Linette GP, Longerich S, Haluska FG. Antimelanoma activity of CTL generated from peripheral blood mononuclear cells after stimulation with autologous dendritic cells pulsed with melanoma gp100 peptide G209-2M is correlated to TCR avidity. J Immunol. 2002;169(1):531–9. doi: 10.4049/jimmunol.169.1.531. [DOI] [PubMed] [Google Scholar]
- [111].Rivoltini L, Squarcina P, Loftus DJ, Castelli C, Tarsini P, Mazzocchi A, et al. A superagonist variant of peptide MART1/Melan A27-35 elicits anti-melanoma CD8+ T cells with enhanced functional characteristics: implication for more effective immunotherapy. Cancer Res. 1999;59(2):301–6. [PubMed] [Google Scholar]
- [112].Stuge TB, Holmes SP, Saharan S, Tuettenberg A, Roederer M, Weber JS, et al. Diversity and Recognition Efficiency of T Cell Responses to Cancer. Plos Med. 2004;1(2):e28. doi: 10.1371/journal.pmed.0010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Anderton SM, Radu CG, Lowrey PA, Ward ES, Wraith DC. Negative selection during the peripheral immune response to antigen. J Exp Med. 2001;193(1):1–11. doi: 10.1084/jem.193.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Rees W, Bender J, Teague TK, Kedl RM, Crawford F, Marrack P, et al. An inverse relationship between T cell receptor affinity and antigen dose during CD4(+) T cell responses in vivo and in vitro. Proc Natl Acad Sci U S A. 1999;96(17):9781–6. doi: 10.1073/pnas.96.17.9781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Zeh HJ, 3rd, Perry-Lalley D, Dudley ME, Rosenberg SA, Yang JC. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol. 1999;162(2):989–94. [PubMed] [Google Scholar]
- [116].Monsurro V, Nagorsen D, Wang E, Provenzano M, Dudley ME, Rosenberg SA, et al. Functional heterogeneity of vaccine-induced CD8(+) T cells. J Immunol. 2002;168(11):5933–42. doi: 10.4049/jimmunol.168.11.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Johnson LA, Heemskerk B, Powell DJ, Jr., Cohen CJ, Morgan RA, Dudley ME, et al. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J Immunol. 2006;177(9):6548–59. doi: 10.4049/jimmunol.177.9.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Zhao Y, Zheng Z, Robbins PF, Khong HT, Rosenberg SA, Morgan RA. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J Immunol. 2005;174(7):4415–23. doi: 10.4049/jimmunol.174.7.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Morgan RA, Dudley ME, Yu YY, Zheng Z, Robbins PF, Theoret MR, et al. High efficiency TCR gene transfer into primary human lymphocytes affords avid recognition of melanoma tumor antigen glycoprotein 100 and does not alter the recognition of autologous melanoma antigens. J Immunol. 2003;171(6):3287–95. doi: 10.4049/jimmunol.171.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Oh S, Hodge JW, Ahlers JD, Burke DS, Schlom J, Berzofsky JA. Selective induction of high avidity CTL by altering the balance of signals from APC. J Immunol. 2003;170(5):2523–30. doi: 10.4049/jimmunol.170.5.2523. [DOI] [PubMed] [Google Scholar]
- [122].Hodge JW, Chakraborty M, Kudo-Saito C, Garnett CT, Schlom J. Multiple costimulatory modalities enhance CTL avidity. J Immunol. 2005;174(10):5994–6004. doi: 10.4049/jimmunol.174.10.5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Yang S, Hodge JW, Grosenbach DW, Schlom J. Vaccines with enhanced costimulation maintain high avidity memory CTL. J Immunol. 2005;175(6):3715–23. doi: 10.4049/jimmunol.175.6.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Gao JX, Zhang H, Bai XF, Wen J, Zheng X, Liu J, et al. Perinatal blockade of b7-1 and b7-2 inhibits clonal deletion of highly pathogenic autoreactive T cells. J Exp Med. 2002;195(8):959–71. doi: 10.1084/jem.20011948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203(5):1259–71. doi: 10.1084/jem.20052494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Zhang B, Bowerman NA, Salama JK, Schmidt H, Spiotto MT, Schietinger A, et al. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med. 2007 doi: 10.1084/jem.20062056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Robbins PF, El-Gamil M, Li YF, Kawakami Y, Loftus D, Appella E, et al. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J Exp Med. 1996;183(3):1185–92. doi: 10.1084/jem.183.3.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269(5228):1281–4. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- [129].Zorn E, Hercend T. A natural cytotoxic T cell response in a spontaneously regressing human melanoma targets a neoantigen resulting from a somatic point mutation. Eur J Immunol. 1999;29(2):592–601. doi: 10.1002/(SICI)1521-4141(199902)29:02<592::AID-IMMU592>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- [130].Linard B, Bezieau S, Benlalam H, Labarriere N, Guilloux Y, Diez E, et al. A ras-mutated peptide targeted by CTL infiltrating a human melanoma lesion. J Immunol. 2002;168(9):4802–8. doi: 10.4049/jimmunol.168.9.4802. [DOI] [PubMed] [Google Scholar]
- [131].Buzyn A, Ostankovitch M, Zerbib A, Kemula M, Connan F, Varet B, et al. Peptides derived from the whole sequence of BCR-ABL bind to several class I molecules allowing specific induction of human cytotoxic T lymphocytes. Eur J Immunol. 1997;27(8):2066–72. doi: 10.1002/eji.1830270834. [DOI] [PubMed] [Google Scholar]
- [132].Yotnda P, Garcia F, Peuchmaur M, Grandchamp B, Duval M, Lemonnier F, et al. Cytotoxic T cell response against the chimeric ETV6-AML1 protein in childhood acute lymphoblastic leukemia. J Clin Invest. 1998;102(2):455–62. doi: 10.1172/JCI3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Passoni L, Scardino A, Bertazzoli C, Gallo B, Coluccia AM, Lemonnier FA, et al. ALK as a novel lymphoma-associated tumor antigen: identification of 2 HLA-A2.1-restricted CD8+ T-cell epitopes. Blood. 2002;99(6):2100–6. doi: 10.1182/blood.v99.6.2100. [DOI] [PubMed] [Google Scholar]
- [134].Ressing ME, Sette A, Brandt RM, Ruppert J, Wentworth PA, Hartman M, et al. Human CTL epitopes encoded by human papillomavirus type 16 E6 and E7 identified through in vivo and in vitro immunogenicity studies of HLA-A*0201-binding peptides. J Immunol. 1995;154(11):5934–43. [PubMed] [Google Scholar]
- [135].Gratama JW, Zutter MM, Minarovits J, Oosterveer MA, Thomas ED, Klein G, et al. Expression of Epstein-Barr virus-encoded growth-transformation-associated proteins in lymphoproliferations of bone-marrow transplant recipients. Int J Cancer. 1991;47(2):188–92. doi: 10.1002/ijc.2910470205. [DOI] [PubMed] [Google Scholar]
- [136].van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254(5038):1643–7. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- [137].Boel P, Wildmann C, Sensi ML, Brasseur R, Renauld JC, Coulie P, et al. BAGE: a new gene encoding an antigen recognized on human melanomas by cytolytic T lymphocytes. Immunity. 1995;2(2):167–75. doi: 10.1016/s1074-7613(95)80053-0. [DOI] [PubMed] [Google Scholar]
- [138].Van den Eynde B, Peeters O, De Backer O, Gaugler B, Lucas S, Boon T. A new family of genes coding for an antigen recognized by autologous cytolytic T lymphocytes on a human melanoma. J Exp Med. 1995;182(3):689–98. doi: 10.1084/jem.182.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Wang RF, Johnston SL, Zeng G, Topalian SL, Schwartzentruber DJ, Rosenberg SA. A breast and melanoma-shared tumor antigen: T cell responses to antigenic peptides translated from different open reading frames. J Immunol. 1998;161(7):3598–606. [PubMed] [Google Scholar]
- [140].Tsang KY, Zaremba S, Nieroda CA, Zhu MZ, Hamilton JM, Schlom J. Generation of human cytotoxic T cells specific for human carcinoembryonic antigen epitopes from patients immunized with recombinant vaccinia-CEA vaccine. J Natl Cancer Inst. 1995;87(13):982–90. doi: 10.1093/jnci/87.13.982. [DOI] [PubMed] [Google Scholar]
- [141].Bakker AB, Schreurs MW, de Boer AJ, Kawakami Y, Rosenberg SA, Adema GJ, et al. Melanocyte lineage-specific antigen gp100 is recognized by melanoma-derived tumor-infiltrating lymphocytes. J Exp Med. 1994;179(3):1005–9. doi: 10.1084/jem.179.3.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Rivoltini L, Topalian SL, et al. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci U S A. 1994;91(9):3515–9. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Wang W, Epler J, Salazar LG, Riddell SR. Recognition of breast cancer cells by CD8+ cytotoxic T-cell clones specific for NY-BR-1. Cancer Res. 2006;66(13):6826–33. doi: 10.1158/0008-5472.CAN-05-3529. [DOI] [PubMed] [Google Scholar]
- [144].Correale P, Walmsley K, Nieroda C, Zaremba S, Zhu M, Schlom J, et al. In vitro generation of human cytotoxic T lymphocytes specific for peptides derived from prostate-specific antigen. J Natl Cancer Inst. 1997;89(4):293–300. doi: 10.1093/jnci/89.4.293. [DOI] [PubMed] [Google Scholar]
- [145].Wang RF, Appella E, Kawakami Y, Kang X, Rosenberg SA. Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J Exp Med. 1996;184(6):2207–16. doi: 10.1084/jem.184.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Brichard V, Van Pel A, Wolfel T, Wolfel C, De Plaen E, Lethe B, et al. The tyrosinase gene codes for an antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med. 1993;178(2):489–95. doi: 10.1084/jem.178.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Butterfield LH, Koh A, Meng W, Vollmer CM, Ribas A, Dissette V, et al. Generation of human T-cell responses to an HLA-A2.1-restricted peptide epitope derived from alpha-fetoprotein. Cancer Res. 1999;59(13):3134–42. [PubMed] [Google Scholar]
- [148].Ioannides CG, Fisk B, Fan D, Biddison WE, Wharton JT, O’Brian CA. Cytotoxic T cells isolated from ovarian malignant ascites recognize a peptide derived from the HER-2/neu proto-oncogene. Cell Immunol. 1993;151(1):225–34. doi: 10.1006/cimm.1993.1233. [DOI] [PubMed] [Google Scholar]
- [149].Vonderheide RH, Hahn WC, Schultze JL, Nadler LM. The telomerase catalytic subunit is a widely expressed tumor-associated antigen recognized by cytotoxic T lymphocytes. Immunity. 1999;10(6):673–9. doi: 10.1016/s1074-7613(00)80066-7. [DOI] [PubMed] [Google Scholar]
- [150].Domenech N, Henderson RA, Finn OJ. Identification of an HLA-A11-restricted epitope from the tandem repeat domain of the epithelial tumor antigen mucin. J Immunol. 1995;155(10):4766–74. [PubMed] [Google Scholar]
- [151].Theobald M, Biggs J, Dittmer D, Levine AJ, Sherman LA. Targeting p53 as a general tumor antigen. Proc Natl Acad Sci U S A. 1995;92(26):11993–7. doi: 10.1073/pnas.92.26.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Robbins PF, el-Gamil M, Li YF, Topalian SL, Rivoltini L, Sakaguchi K, et al. Cloning of a new gene encoding an antigen recognized by melanoma-specific HLA-A24-restricted tumor-infiltrating lymphocytes. J Immunol. 1995;154(11):5944–50. [PubMed] [Google Scholar]
- [153].Shichijo S, Nakao M, Imai Y, Takasu H, Kawamoto M, Niiya F, et al. A gene encoding antigenic peptides of human squamous cell carcinoma recognized by cytotoxic T lymphocytes. J Exp Med. 1998;187(3):277–88. doi: 10.1084/jem.187.3.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Nakao M, Shichijo S, Imaizumi T, Inoue Y, Matsunaga K, Yamada A, et al. Identification of a gene coding for a new squamous cell carcinoma antigen recognized by the CTL. J Immunol. 2000;164(5):2565–74. doi: 10.4049/jimmunol.164.5.2565. [DOI] [PubMed] [Google Scholar]
- [155].Yang D, Nakao M, Shichijo S, Sasatomi T, Takasu H, Matsumoto H, et al. Identification of a gene coding for a protein possessing shared tumor epitopes capable of inducing HLA-A24-restricted cytotoxic T lymphocytes in cancer patients. Cancer Res. 1999;59(16):4056–63. [PubMed] [Google Scholar]
- [156].Oka Y, Elisseeva OA, Tsuboi A, Ogawa H, Tamaki H, Li H, et al. Human cytotoxic T-lymphocyte responses specific for peptides of the wild-type Wilms’ tumor gene (WT1 ) product. Immunogenetics. 2000;51(2):99–107. doi: 10.1007/s002510050018. [DOI] [PubMed] [Google Scholar]
- [157].Lustgarten J, Dominguez AL, Pinilla C. Identification of cross-reactive peptides using combinatorial libraries circumvents tolerance against Her-2/neu-immunodominant epitope. J Immunol. 2006;176(3):1796–805. doi: 10.4049/jimmunol.176.3.1796. [DOI] [PubMed] [Google Scholar]
- [158].Gritzapis AD, Mahaira LG, Perez SA, Cacoullos NT, Papamichail M, Baxevanis CN. Vaccination with human HER-2/neu (435-443) CTL peptide induces effective antitumor immunity against HER-2/neu-expressing tumor cells in vivo. Cancer Res. 2006;66(10):5452–60. doi: 10.1158/0008-5472.CAN-05-4018. [DOI] [PubMed] [Google Scholar]
- [159].McWilliams JA, McGurran SM, Dow SW, Slansky JE, Kedl RM. A modified tyrosinase-related protein 2 epitope generates high-affinity tumor-specific T cells but does not mediate therapeutic efficacy in an intradermal tumor model. J Immunol. 2006;177(1):155–61. doi: 10.4049/jimmunol.177.1.155. [DOI] [PubMed] [Google Scholar]
- [160].Smith JW, 2nd, Walker EB, Fox BA, Haley D, Wisner KP, Doran T, et al. Adjuvant immunization of HLA-A2-positive melanoma patients with a modified gp100 peptide induces peptide-specific CD8+ T-cell responses. J Clin Oncol. 2003;21(8):1562–73. doi: 10.1200/JCO.2003.09.020. [DOI] [PubMed] [Google Scholar]
- [161].Lee KH, Wang E, Nielsen MB, Wunderlich J, Migueles S, Connors M, et al. Increased vaccine-specific T cell frequency after peptide-based vaccination correlates with increased susceptibility to in vitro stimulation but does not lead to tumor regression. J Immunol. 1999;163(11):6292–300. [PubMed] [Google Scholar]
- [162].Lee P, Wang F, Kuniyoshi J, Rubio V, Stuges T, Groshen S, et al. Effects of interleukin-12 on the immune response to a multipeptide vaccine for resected metastatic melanoma. J Clin Oncol. 2001;19(18):3836–47. doi: 10.1200/JCO.2001.19.18.3836. [DOI] [PubMed] [Google Scholar]
- [163].Lau R, Wang F, Jeffery G, Marty V, Kuniyoshi J, Bade E, et al. Phase I trial of intravenous peptide-pulsed dendritic cells in patients with metastatic melanoma. J Immunother. 2001;24(1):66–78. doi: 10.1097/00002371-200101000-00008. [DOI] [PubMed] [Google Scholar]
- [164].Weber J, Sondak VK, Scotland R, Phillip R, Wang F, Rubio V, et al. Granulocyte-macrophage-colony-stimulating factor added to a multipeptide vaccine for resected Stage II melanoma. Cancer. 2003;97(1):186–200. doi: 10.1002/cncr.11045. [DOI] [PubMed] [Google Scholar]
- [165].Fong L, Hou Y, Rivas A, Benike C, Yuen A, Fisher GA, et al. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc Natl Acad Sci U S A. 2001;98(15):8809–14. doi: 10.1073/pnas.141226398. [DOI] [PMC free article] [PubMed] [Google Scholar]