

Abstract
By fusing the CaMKII inhibitory peptide AIP to GFP, we constructed a specific and effective CaMKII inhibitor, GFP-AIP. Expression of GFP-AIP and/or dominant-inhibitory CaMKIV in cultured neonatal rat spiral ganglion neurons (SGNs) shows that CaMKII and CaMKIV act additively and in parallel, to mediate the prosurvival effect of depolarization. Depolarization or expression of constitutively-active CaMKII functionally inactivates Bad, indicating that this is one means by which CaMKII promotes neuronal survival. CaMKIV, but not CaMKII, requires CREB to promote SGN survival, consistent with the exclusively nuclear localization of CaMKIV and indicating that the principal prosurvival function of CaMKIV is activation of CREB. Consistent with this, a constitutively-active CREB construct that provides a high level of CREB activity promotes SGN survival, although low levels of CREB activity did not do so. Also, in apoptotic SGNs, activation of CREB by depolarization is disabled, presumably as part of a cellular commitment to apoptosis.
Introduction
Neurons are distinctive in that they are electrically active, spiral ganglion (cochlear) neurons (SGNs) being particularly active. As is the case for neurons generally, membrane electrical activity or depolarization is an effective survival-promoting stimulus for SGNs, both in vitro (Hegarty et al., 1997) and in vivo (Hartshorn et al., 1991; Leake et al., 1991; Leake et al., 1992; Lousteau, 1987; Lustig et al., 1994; Wong-Riley et al., 1981). The intracellular signaling that links membrane depolarization to survival has been investigated and multiple signaling pathways are involved (Hansen et al., 2001; Vaillant et al., 1999), although there may be variability among neurons with regard to the relative importance of different pathways. A common element among these intracellular signaling pathways is that they are initiated by entry of Ca2+ ion into the cytosol via voltage-gated Ca2+ channels (VGCCs). In experiments in which neurons are chronically depolarized by elevating extracellular K+, Ca2+ entry via L-type VGCCs is required (Collins and Lile, 1989; Collins et al., 1991; Franklin et al., 1995; Galli et al., 1995; Gallo et al., 1987; Hegarty et al., 1997; Koike et al., 1989), although entry through other VGCCs, e.g., N-type, may be more important when neurons are stimulated by patterned electrical activity (Brosenitsch and Katz, 2001).
Ca2+ entry promotes neuronal survival via divergent Ca2+-dependent intracellular signaling pathways including both Ca2+/calmodulin-dependent protein kinase (CaMK) and cAMP-dependent protein kinase (PKA) (Bok et al., 2003; Hansen et al., 2003; Hansen et al., 2001). That CaMK activity promotes neuronal survival is shown by the use of constitutively active CaMKII (Hansen et al., 2003) or constitutively active CaMKIV mutants (Hansen et al., 2003; See et al., 2001). Both promote neuronal survival, indicating that either CaMKII or CaMKIV activity is sufficient, at least in part. As these kinases are Ca2+-activated, it is likely that they contribute to the survival-promoting effect of depolarization. This is supported by studies using CaMK inhibitors. A requirement for CaMKIV in the survival-promoting effect of depolarization has been shown through the use of a kinase-inactive dominant-negative mutant (See et al., 2001). Pharmacological inhibitors have been used in depolarization-dependent survival paradigms to ascertain a requirement for CaMKII (Hack et al., 1993; Hansen et al., 2003; Ikegami and Koike, 2000; Vaillant et al., 1999) but the agents used are not selective. The CaMK inhibitor KN-62 and related compounds block CaMKII and CaMKIV (Enslen et al., 1994; Tokumitsu et al., 1990); the calmodulin antagonist calmidazolium blocks activation generally of calmodulin-dependent enzymes (Gietzen et al., 1982; Gietzen et al., 1981; Louis et al., 1983).
Here we ask whether CaMKII and CaMKIV have distinct roles in mediating promotion of neuronal survival by depolarization and we probe the requirement for CREB for actions of CaMKII and CaMKIV. We developed a specific inhibitor of CaMKII, termed GFP-AIP, that can be expressed in neurons after transfection and can be targeted to different subcellular locations. By using GFP-AIP in combination with a dominant-negative (kinase null) CaMKIV mutant, we show here that CaMKII and CaMKIV make independent distinct contributions to depolarization-dependent neuronal survival.
We further asked about the extent to which CaMKII and CaMKIV recruit transcriptional and post-translational mechanisms to promote survival. As a representative transcriptional mechanism, we investigated the cAMP/Ca2+ Response Element Binding (CREB) protein. CaMKs are among those protein kinases capable of phosphorylating CREB on serine-133 (Enslen et al., 1994; Enslen et al., 1995; Matthews et al., 1994; Sun et al., 1994). Others include PKA (Gonzalez and Montminy, 1989; Sun et al., 1992), Rsk2 (Bohm et al., 1995; Swanson et al., 1999), MSK1 (Simon et al., 2004). This allows CREB to recruit the co-activator CREB Binding Protein (CBP) and activate transcription (Cardinaux et al., 2000; Chrivia et al., 1993; Kwok et al., 1994). In a further step important for transcription, CBP is itself phosphorylated by CaMKIV in response to depolarization (Impey et al., 2002). CREB has been shown to mediate the survival-promoting effect of neurotrophins (Bonni et al., 1999; Dawson and Ginty, 2002; Lonze et al., 2002; Riccio et al., 1999) but not of PKA (Bok et al., 2003). As a representative post-translational modification, we investigated functional inactivation of Bad. Neurotrophins promote neuronal survival in part by recruiting ERK and Akt protein kinases that phosphorylate Bad so as to prevent its proapoptotic function.
CaMKII does not promote survival via CREB but is capable of functionally inactivating the proapoptotic regulator Bad. CaMKIV is primarily nuclear and promotes survival by activating CREB, although recruitment of CBP by CREB may not be sufficient. Moreover, activation of CREB by depolarization is disabled in apoptotic cells, which removes a potential block to cell death.
Results
Generation and testing of GFP-AIP, a selective CaMKII inhibitor
We and others (Hack et al., 1993; Hansen et al., 2001; Ikegami and Koike, 2000; Vaillant et al., 2002) have previously implicated CaMKII activity in the mechanism by which depolarization promotes neuronal survival. However, these studies relied on the use of a CaMK inhibitor KN-62 (Tokumitsu et al., 1990), which also inhibits CaMKIV (Enslen et al., 1994; Tokumitsu et al., 1990) and possibly other calmodulin-dependent enzymes. Because CaMKIV can also function as a prosurvival signal (See et al., 2001), more specific inhibitors are necessary to determine the relative contribution of CaMKII and CaMKIV to neuronal survival.
Autocamtide-2-related inhibitory peptide (AIP, Ishida et al., 1995) is a non-phosphorylatable analog of autocamtide-2, a CaMKII specific substrate. AIP is a potent and highly specific CaMKII inhibitor (Ishida et al., 1995; Ishida et al., 1998; Zou and Cline, 1999) as it binds specifically to the catalytic domain of CaMKII, but cannot be phosphorylated and released. AIP is typically microinjected into cells but can be expressed from a transgene if fused to an appropriate stabilizer protein (Zou and Cline, 1999). To generate a fluorescent AIP-based CaMKII inhibitor we fused an oligonucleotide encoding AIP, separated by a 22 amino acid linker, to the C-terminus of the green fluorescent protein (GFP) coding sequence to generate a construct termed GFP-AIP (Figure 1A, see Methods for details). Our previous attempts to construct an inhibitor by fusing the AIP coding sequence to sequences coding small epitope tags failed, apparently because the “miniproteins” were not stably expressed (data not shown). Also, chimeras containing GFP and AIP with shorter linkers were unsuccessful as inhibitors.
Figure 1.
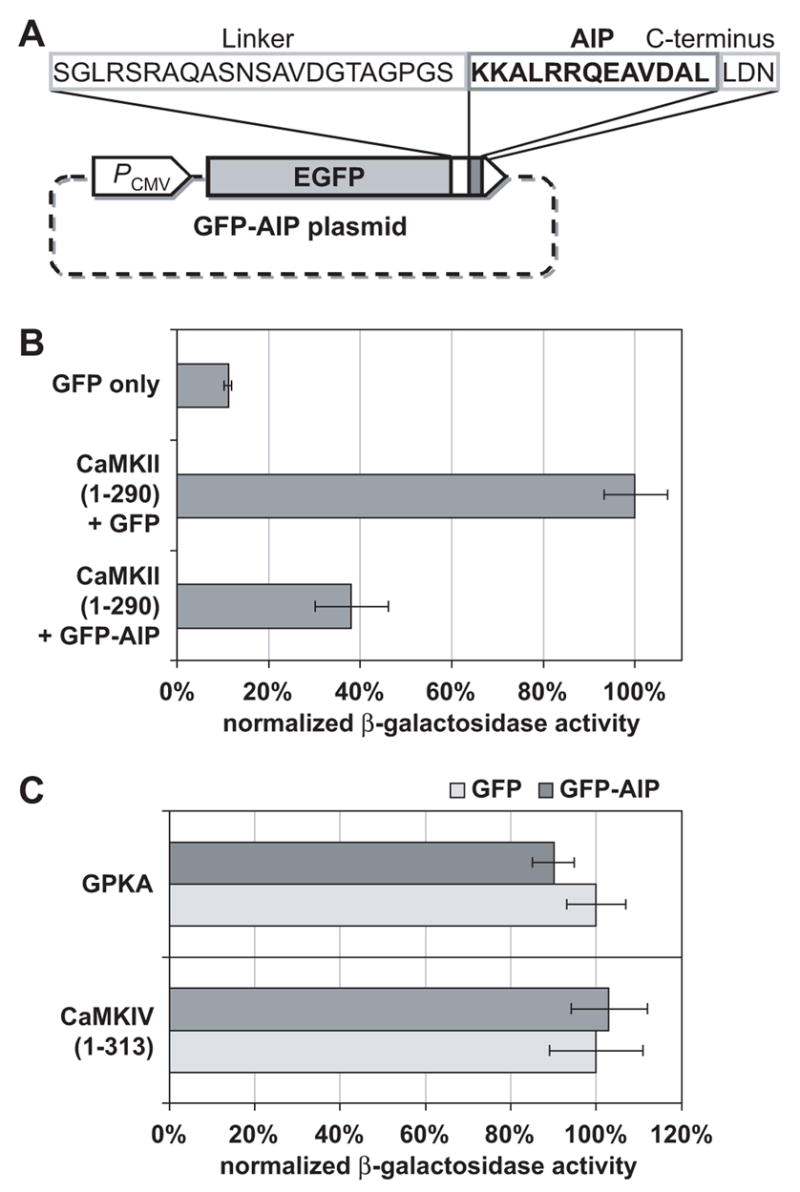
(A) As described in Methods, the CaMKII inhibitor GFP-AIP was constructed by inserting an oligonucleotide encoding the AIP sequence 3’ to GFP. A CMV promoter (PCMV) drives expression of a chimeric protein consisting of GFP fused to AIP by a 22 amino acid linker and a three amino acid C-terminal sequence. (B) The CaMKII inhibitor GFP-AIP inhibits activation of a CRE-LacZ reporter by the constitutively-active truncated mutant CaMKII, CaMKII(1-290). PC12 cells were cotransfected with CRE-LacZ reporter and plasmids encoding proteins of interest. CRE-LacZ constituted 20% (2 μg) of total plasmid, CaMKII(1-290), if present, constituted 10% (1 μg), GFP-AIP, if present, constituted 50% (5 μg), and the remainder was GFP. β-galactosidase was assayed as described in Methods and normalized to activity in cells transfected with CaMKII(1-290) + GFP, which was arbitrarily set to 100%. Error bars in this and all subsequent graphs show standard deviation. (C) GFP-AIP inhibits neither CaMKIV nor cAMP-dependent protein kinase (PKA). As in B, β–galactosidase activity in PC12 cells transfected with GFP and activated kinase was arbitrarily assigned a value of 100% and activity in PC12 cells transfected with GFP-AIP and kinase was normalized to it. CaMKIV(1-313) is a truncated constitutively-active mutant CaMKIV construct and GPKA is a GFP-tagged PKA catalytic subunit. Plasmids used were as in B, with CaMKIV(1-313) or PKA catalytic subunit replacing CaMKII(1-290).
To confirm the effectiveness of GFP-AIP as a CaMKII inhibitor in cells, we used a reporter assay. Because we could not obtain a number of transfected SGNs sufficient for a biochemical reporter assay, we instead used a neuronal cell line, PC12 cells. A truncated constitutively-active mutant CaMKII (CaMKII(1-290)) (Sun et al., 1994), which we have already shown to be active in promoting SGN survival (Hansen et al., 2003), was cotransfected into PC12 cells with a Ca2+/cAMP Response Element (CRE)-LacZ reporter. CaMKII(1-290) induced increased expression of β-galactosidase (Figure 1B). This action of CaMKII was strongly inhibited by cotransfection of GFP-AIP (Figure 1B). To confirm the specificity of GFP-AIP, we performed similar reporter assays using other protein kinases to induce CRE-dependent transcription. The ability of CaMKIV to induce increased reporter expression was unaffected by GFP-AIP (Figure 1C). Furthermore, cAMP-dependent protein kinase (PKA), another kinase that may be recruited by membrane depolarization to promote neuronal survival (Bok et al., 2003; Hansen et al., 2001; Hanson et al., 1998; Meyer-Franke et al., 1995), was not inhibited by GFP-AIP (Figure 1C).
CaMKII is necessary, in part, for the prosurvival effect of depolarization
To determine expression and localization of CaMKII, cultured SGNs were immunolabeled with an antibody detecting CaMKII and with isoform-specific antibodies detecting CaMKIIα and CaMKIIβ. Representive examples are shown in Figures 2A–E. CaMKII immunoreactivity was largely cytoplasmic in most neurons (Figure 2A) but some neurons also had nuclear CaMKII immunoreactivity, as has been noted previously in, for example, cerebellar granule neurons (Takeuchi et al., 2002; Takeuchi et al., 1999). The nuclear CaMKII immunoreactivity appears to be attributable mainly to CaMKIIα, which was primarily nuclear in 25% of SGNs (Figure 2B) but distributed in the cytoplasm and nucleus in 75% of SGNs (Figure 2C). CaMKIIβ immunoreactivity was primarily cytoplasmic (Figures 2D,2E). Depolarization did not affect CaMKII distribution (data not shown).
Figure 2. (A–E) CaMKII localization in SGNs.
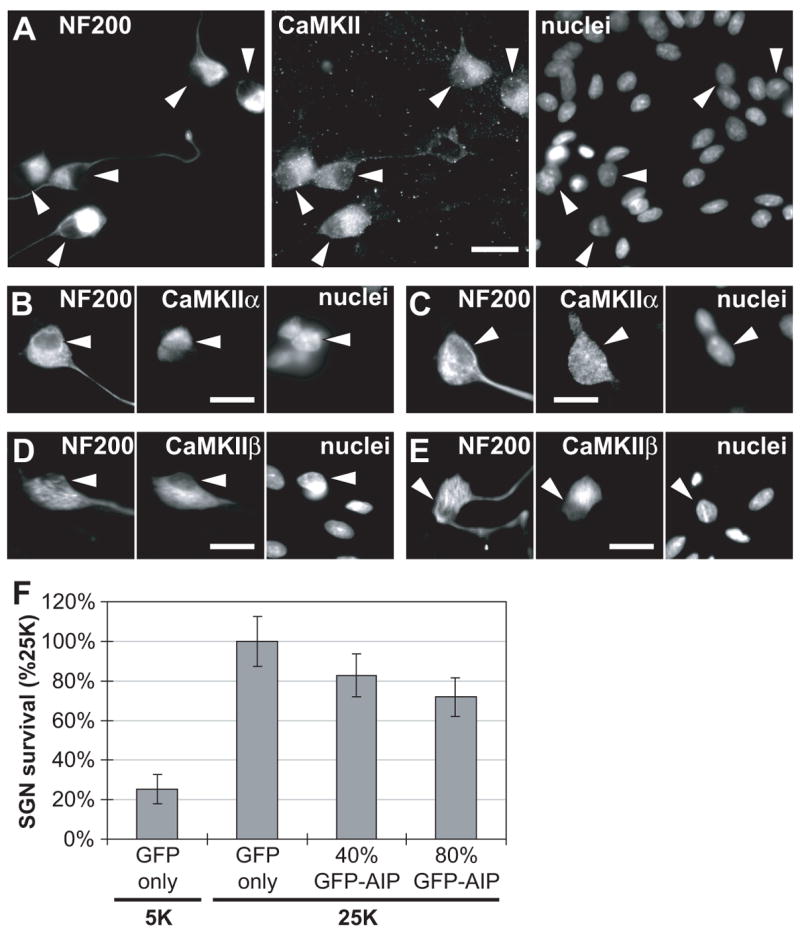
Immunofluorescence was used to detect NF200 and CaMKII in spiral ganglion cultures (48 h cultures) as described in Methods. Neuronal cytoplasm was labeled with anti-NF200 and all nuclei in the cultures (neuronal and non-neuronal cells) were labeled with Hoechst 33342. In each set of images, the positions of neuronal nuclei are indicated by arrowheads, which are at identical positions in each image in the set. Scale bar = 20 μm. (A) A pan-CaMKII antibody was used to label CaMKII. (B & C) An anti-CaMKIIα antibody was used in these two representative examples of the two different subcellular distributions of CaMKIIα that we observed in cultured SGNs. (D & E) An anti-CaMKIIβ antibody was used in these two representative examples of the observed subcellular distribution of CaMKIIβ. (F) GFP-AIP inhibits, in a dose-dependent manner, the ability of depolarization to promote SGN survival. SGNs were maintained for 96 h in either control nondepolarizing (5K) or depolarizing (25K) medium following transfection with either GFP plasmids alone or a mixture of GFP and GFP-AIP plasmids. GFP-AIP plasmid constituted 40% (4 μg) or 80% (8 μg) of the total, as indicated. The number of transfected SGNs surviving at 96 h was determined as described in Methods. The reduction in SGN survival in 25K was significant (P = 0.01 for 40% GFP-AIP; P = 0.0003 for 80% GFP-AIP) relative to GFP only.
We next used GFP-AIP to determine whether CaMKII contributes to the ability of depolarization to promote neuronal survival. Neonatal rat spiral ganglion neurons (SGNs) were depolarized in vitro by increasing extracellular [K+] from control 5.4 mM (5K) to 25 mM (25K), which we have previously shown to be an effective survival-promoting stimulus (Hansen et al., 2001; Hegarty et al., 1997). Transfection of GFP-AIP (2:3 mixture of GFP-AIP and GFP plasmids) significantly (P = 0.01) reduced the prosurvival effect of 25K (Figure 2F) and this was further significantly (P = 0.0003) reduced by increasing the proportion of GFP-AIP plasmid from 40% to 80% of the total (Figure 2F). However, inhibition of SGN survival is not complete even using this amount of GFP-AIP. Because we can’t further increase the level of GFP-AIP in the neurons, we can’t determine whether the incomplete inhibition of survival is because CaMKII is incompletely inhibited or because CaMKII does not act alone to promote survival. Nevertheless, we can conclude that, because GFP-AIP reduced the ability of depolarization to promote SGN survival in a dose-dependent manner, CaMKII activity must be necessary, at least in part, for the survival-promoting effect of depolarization.
Because CaMKII isoforms are detected in both cytoplasm and nucleus, we directed GFP-AIP to either compartment to determine the extent to which nuclear or cytoplasmic CaMKII activity contributes to survival. For this purpose, a oligonucleotide encoding either a nuclear localization signal (nls) or a nuclear export signal (nes) (Bok et al., 2003) was inserted in the GFP-AIP sequence. To maintain the inhibitory function of these constructs, the nes was placed at the NH2-terminal and the nls in the linker between the GFP and AIP sequences. As shown in Fig. 3A, inclusion of nes and nls targeting signals, respectively, restricts GFP-AIP to the nucleus or excludes GFP-AIP from the nucleus. A significant decrease in SGN survival in 25K was detected following expression of either GFP-nls-AIP or GFP-nes-AIP (Fig. 3B) suggesting that both nuclear and cytoplasmic CaMKII mediate part of the survival-promoting effect of depolarization.
Figure 3. Targeted inhibition of CaMKII reveals nuclear and cytoplasmic roles.
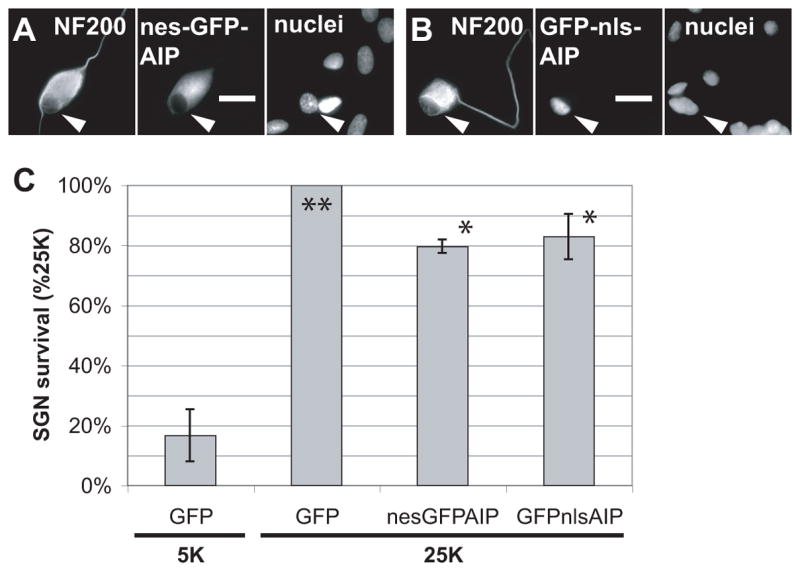
GFP-AIP was either excluded from the nucleus (A) by addition of a nuclear exclusion signal (nes) or restricted to the nucleus (B) by addition of a nuclear localization signal (nls). In A and B, the location of the GFP-AIP is shown by GFP fluorescence. Neuronal cytoplasm was labeled with anti-NF200 and all nuclei in the cultures (neuronal and non-neuronal cells) were labeled with Hoechst 33342. In A and B, the positions of the neuronal nucleus is indicated by an arrowhead, which is at an identical position in each image in the set. Scale bar = 20 μm. (C) Cytoplasmic (nes) or nuclear (nls) GFP-AIP significantly (*P < 0.01) inhibited the ability of depolarization to promote SGN survival, relative to GFP(**). Cytoplasmic or nuclear GFP-AIP were not significantly different from each other with respect to survival and were significantly different (P < 0.0001) from the 5K condition. SGNs were maintained for 96 h in either control nondepolarizing (5K) or depolarizing (25K) medium following transfection with GFP, nes-GFP-AIP, or GFP-nls-AIP plasmids, as in Figure 2F.
CaMKII functionally inactivates the proapoptotic regulator Bad
While, the nuclear role of CaMKII suggests an effect on transcriptional regulation, the primarily cytoplasmic localization of CaMKII suggests that CaMKII promotes SGN survival also by phosphorylating, directly or indirectly, cytoplasmic proteins involved in regulation of apoptosis. The pro-apoptotic regulator Bad has been shown to be phosphorylated by several prosurvival signals, which thus inactivate the proapoptotic function of Bad (Downward, 1999). The small number of SGNs in the cultures and the presence of non-neuronal cells preclude assay of Bad phosphorylation in SGNs. We could, however, apply a more direct test of the ability of CaMKII to functionally inactivate Bad by taking advantage of the ability of overexpressed wild-type Bad to induce apoptosis in SGNs maintained in culture medium that marginally promotes survival (Bok et al., 2003). SGNs rapidly undergo cell death in serum-free 5K culture medium with no trophic additions (Hegarty et al., 1997). Addition of 1% FBS slowed cell death, allowing the effect of wild-type Bad overexpression to become apparent (Figures 3,4). This was most apparent 48 h after transfer to the marginal condition. Increasing the FBS concentration to ≥5% prevented SGN death regardless of Bad overexpression (not shown). We could thus use suppression of the Bad-induced increase in SGN apoptosis by various agents as a measure of the ability of these agents to functionally inactivate Bad proapoptotic signaling.
Figure 4. Both depolarization and expression of constitutively-active CaMKII rescue SGNs from apoptosis caused by Bad overexpression.
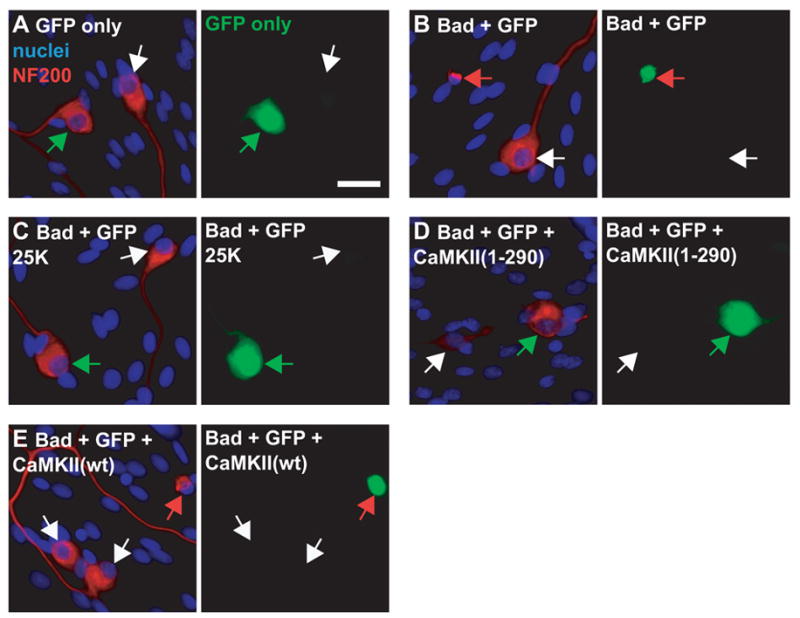
SGNs were transfected with combinations of expression plasmids including GFP (A–E), Bad (B–E), constitutively active mutant CaMKII(1-290) (D) or wild-type CaMKII (wt) (E). In the combinations, GFP plasmid was 20%; Bad, if present, was 20%; and CaMKII, if present, was 60% of the total amount of plasmid. Empty vector (pcDNA3) was used to replace plasmids not included in a particular combination. Eighteen hours after transfection the cultures were switched to 5K (A,B,D,E) or 25K (C) culture medium, all containing 1% serum. After a further 48 h the cultures were fixed and stained to detect NF200 and nuclei, as in Figure 2. Transfected cells were identified by green GFP fluorescence shown in the right panel of each pair of images. Neurons were identified by NF200 immunoreactivity (red) and nuclei were stained with Hoechst 33342 (blue), shown in the left image of each pair. Arrows point to neuronal nuclei and indicate identical positions in each image pair of images. The images were chosen so that each shows a typical transfected and a typical untransfected neuron adjacent to each other in the culture. White arrows indicate untransfected neurons in all conditions. Red arrows indicate transfected neurons that are apoptotic (identified by their condensed nuclei and collapsed cytoplasm) (B,E). Green arrows indicate transfected nonapoptotic neurons (A,C,D). Scale bar = 20 μm.
Cultures were stained, as in Figure 2, to label the nuclei and neurofilaments. This allowed identification of the neurons as well as identification of apoptotic cells, which were evident by their pyknotic nuclei and collapsed neurofilament network. Typical examples are shown in Figure 4. Transfected cells were identified by cotransfecting GFP, which alone did not cause increased apoptosis. Nuclear and cytoskeletal condensation were apparent in Bad-transfected SGNs but uncommon in untransfected cells in the same culture. In contrast, increased apoptosis was not observed in Bad-transfected SGNs maintained in depolarizing 25K medium, confirming that depolarization can functionally inactivate Bad. Similarly, increased apoptosis was not observed in SGNs cotransfected with Bad and CaMKII(1-290), a truncated constitutively-active mutant CaMKII that we have previously shown to promote SGN survival (Hansen et al., 2003). Thus, expression of CaMKII(1-290) prevents apoptosis caused by Bad overexpression, suggesting that activated CaMKII can functionally inactivate Bad. Cotransfection with wild-type CaMKII had no effect on Bad overexpression-induced apoptosis.
These data are quantified in Figure 5. Transfection of wild-type Bad increased the percentage of apoptotic SGNs from ≈30% to ≈70% for cultures maintained in 5K + 1% FBS. This increase in apoptosis was almost entirely reversed by depolarization (25K) or by transfection of constitutively-active mutant, but not wild-type, CaMKII. Thus, depolarization and CaMKII activity result in functional inactivation of Bad, implying that Bad inactivation is a means by which depolarization and CaMKII promote SGN survival. While CaMKII mediates at least part of the survival-promoting effect of depolarization, it is unlikely that CaMKII is the sole means by which depolarization can functionally inactivate Bad. We have previously shown (Bok et al., 2003) that PKA overexpression also rescues SGNs from Bad overexpression.
Figure 5. Quantitation of apoptotic SGNs confirms that depolarization or constitutively-active CaMKII rescue SGNs from apoptosis caused by Bad overexpression.
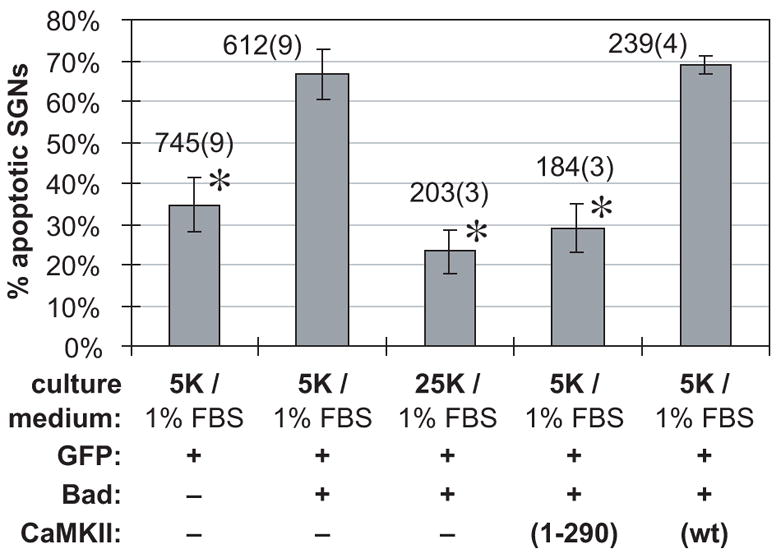
Expression of Bad caused a significant increase in the number of apoptotic SGNs. This increase, in turn, was prevented by depolarization or by transfection of constitutively-active (CaMKII(1-290)), but not wild-type (wt), CaMKII. Random fields were selected and all SGNs in the field scored. Apoptotic SGNs were identified on the basis of pyknotic nuclei and collapsed cytoplasm in cultures transfected, treated, fixed and stained as in Figure 4. The graph shows apoptotic SGNs, as a percentage of the total number scored, in each experimental condition. The culture condition (1% FBS + 5K or 25K) and the plasmids transfected are indicated. GFP plasmid was 20%; Bad, if present, was 20%; and CaMKII, if present, was 60% of the total amount of plasmid; the remainder was empty pcDNA3 vector. Each column shows the mean and standard deviation of at least three separate experiments, each performed in duplicate. The total number of SGNs counted is shown above each bar; the number in parentheses is the number of separate experiments pooled to obtain this number of SGNs. The values marked with an asterisk are not significantly different from each other but are all significantly different (P < 0.00002) from values with no asterisk.
CaMKIV is necessary, in part, for the prosurvival effect of depolarization
Overexpression of a catalytically inactive CaMKIV mutant, CaMKIV(K75E) has been previously shown to act as a dominant-inhibitory inhibitor of endogenous CaMKIV (Ahn et al., 1999; Finkbeiner et al., 1997; Ho et al., 1996; Miranti et al., 1995; Shieh et al., 1998). We therefore constructed this mutant, as described in Methods, in order to inhibit CaMKIV in SGNs. SGN survival promoted by depolarization was significantly reduced by transfection of CaMKIV(K75E) (Figure 6), indicating that CaMKIV activity is necessary, at least in part, for the survival-promoting effect of depolarization in SGN. Transfection of a 4:1 mixture of CaMKIV(K75E) to GFP plasmids strongly and significantly (P = 0.0002) reduced SGN survival. Halving the amount of CaMKIV(K75E) plasmid to 40% (2:2:1 mixture of CaMKIV(K75E) : pcDNA3 empty vector : GFP plasmids) resulted in a reduction in survival nearly as great as that caused by 80% CaMKIV(K75E) and not significantly different from it (P = 0.15).
Figure 6. Dominant-inhibitory CaMKIV, CaMKIV(K75E), inhibits the ability of depolarization to promote SGN survival.
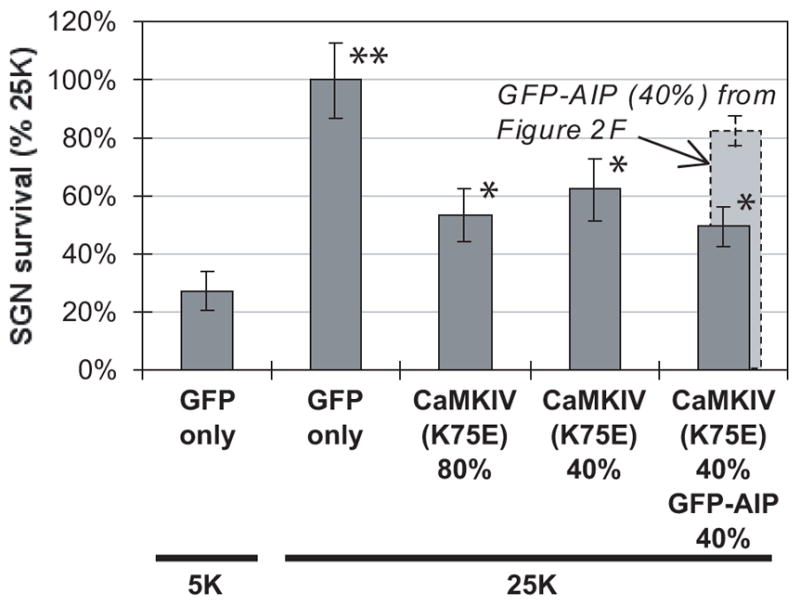
SGNs were maintained for 96 h in either control nondepolarizing (5K) or depolarizing (25K) medium following transfection with either GFP plasmids alone, a mixture of GFP and CaMKIV(K75E) plasmids, or a 1:1 mixture of CaMKIV(K75E) and GFP-AIP plasmids. CaMKIV(K75E) plasmid constituted 40% (4 μg) or 80% (8 μg) of the total, as indicated and GFP-AIP was 40% (4 μg). The number of transfected SGNs surviving at 96 h was determined as described in Methods. Values marked with a single asterisk were not significantly different from each other but were significantly different from conditions marked with no asterisk or double asterisks. The reduction in SGN survival in 25K was significant (P < 0.00005) for SGNs transfected with any of the inhibitory constructs, relative to SGNs transfected with GFP only. The survival of SGNs transfected with a combination of 40% CaMKIV(K75E) and 40% GFP-AIP was significantly reduced (P < 0.04) relative to 40% CaMKIV(K75E) alone or 40% GFP-AIP alone (from Figure 2F, shown in light gray).
The small difference in inhibition of survival between 80% and 40% CaMKIV(K75E) suggests that inhibition by 80% is complete or nearly so and that the remaining depolarization-dependent survival is due to signaling other than CaMKIV. This can be accounted for, at least in part, by CaMKII signaling. As shown in Figure 6, partial inhibition of CaMKIV combined with partial inhibition of CaMKII — achieved by cotransfection of CaMKIV(K75E) and GFP-AIP — resulted in inhibition of depolarization-dependent survival significantly greater than that due to either inhibitor alone at that level, implying that CaMKII and CaMKIV independently mediate part of the prosurvival effect of depolarization.
CREB is required for CaMKIV, but not CaMKII, prosurvival signaling
CaMKIV immunoreactivity is primarily expressed in neurons in the culture and is primarily nuclear (Figure 7A). As a nuclear protein kinase, CaMKIV can phosphorylate and activate CREB, which has been shown to be a prosurvival transcription factor (Bonni et al., 1999; Finkbeiner, 2000; Riccio et al., 1999). We therefore tested the possibility that CREB mediates the prosurvival effect of CaMKIV. A dominant negative CREB mutant, CREBm1 (CREB-Ser133Ala), was used to inhibit CREB function (Struthers et al., 1991). To verify that CREBm1 inhibits the ability of CaMKIV to activate CRE-dependent transcription, we cotransfected the CRE-LacZ reporter into PC12 cells with a constitutively-active truncated mutant CaMKIV, CaMKIV(1-313) (ref. Sun et al., 1994), in the presence or absence of CREBm1. As shown in Figure 7B, the increased reporter expression induced by CaMKIV(1-313) is inhibited by CREBm1.
Figure 7.
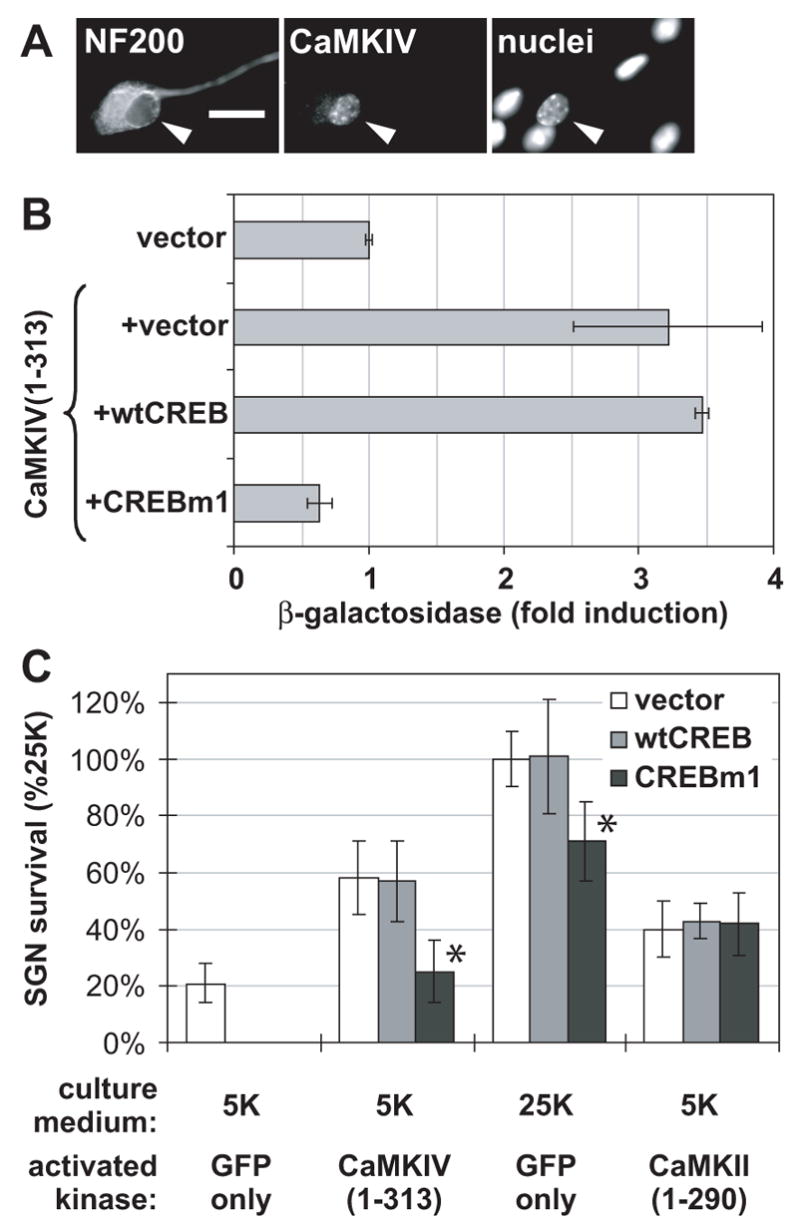
(A) CaMKIV is primarily nuclear in SGNs. This set of images of a spiral ganglion culture shows anti-CaMKIV immunofluorescence , anti-NF200 immunofluorescence to label neuronal cytoplasm, and Hoechst 33342 fluorescence to label the nuclei of all cells. The positions of neuronal nuclei are indicated with arrowheads, which indicate identical positions in the images in the set. (B) Dominant-inhibitory CREB, CREBm1, blocks activation of CRE-dependent transcription by CaMKIV. PC12 cells were transfected with CRE-LacZ reporter and GFP (each constituting 20% of the total plasmid), in combination with plasmids encoding CaMKIV(1-313) and CREBm1, as indicated (each constituting 40% of total plasmid when present). If necessary, pcDNA3 was added to make the total amount of plasmid 10 μg. β-galactosidase was assayed as described in Methods and normalized to activity in cells transfected with CaMKIV(1-313), which was arbitrarily set to 100%. Error bars show standard deviation. (C) Dominant-inhibitory CREB inhibits the ability of CaMKIV and of depolarization, but not of CaMKII, to promote SGN survival. SGNs were maintained for 96 h in either control nondepolarizing (5K) or depolarizing (25K) medium following transfection. In all cases, GFP was 20% of the mixture; activated kinase, CaMKIV(1-313) or CaMKII(1-290), if present, constituted 40% of the mixture; CREB, wild-type (wt) or dominant-inhibitory (CREBm1), if present, constituted 40% of the mixture; the remainder of the plasmid in the mixture was pcDNA3 vector. The number of transfected SGNs surviving at 96 h was determined as described in Methods. Relative to wtCREB or empty vector, CREBm1 significantly (*P < 0.001) reduced SGN survival due to CaMKIV(1-313) to the same level as survival in the control 5K condition. Also, CREBm1 significantly (*P < 0.01) reduced SGN survival due to depolarization (25K). However, CREBm1 had no significant effect on SGN survival due to CaMKII(1-290).
Cotransfection of CREBm1, but not wild-type CREB, with CaMKIV(1-313), eliminated the prosurvival effect of CaMKIV(1-313) on SGNs (Figure 7C). This indicates that CaMKIV requires CREB for its prosurvival effect. In contrast, the survival-promoting effect of constitutively-active CaMKII(1-290) was unaffected by CREBm1 (Figure 7C), indicating a lack of requirement for CREB and consistent with recruitment of cytosolic or other prosurvival effectors.
Because the ability of depolarization to promote survival appears to depend only in part on CaMKIV signaling, we would expect that CREBm1 would only partially block the prosurvival effect of depolarization. Indeed, SGNs transfected with CREBm1 and maintained in 25K medium showed reduced survival relative to neurons transfected with wild-type CREB or empty vector plasmids but survival of CREBm1-transfected SGNs was greater than survival under nondepolarizing 5K conditions (Figure 7C). This indicates that CREB-mediated transcription plays a role in depolarization-mediated SGN survival but that other signaling pathways, which are independent of CREB, also contribute. Presumably, these include CaMKII and PKA (Bok et al., 2003).
The constitutively-active CREB construct VCREB, but not the constitutively-active CREBDIEDML, promotes SGN survival
We next determined the extent to which CREB-mediated transcription is sufficient to promote SGN survival. After phosphorylation by CaMKIV or other kinases on Ser133, CREB is able to recruit the coactivator CREB Binding Protein (CBP), which activates transcription directly (Vo and Goodman, 2001). CBP itself can be phosphorylated by CaMKIV, which potentiates its efficacy as a transactivator (Impey et al., 2002). The sterol-responsive element binding protein contains an amino acid motif DIEDML that allows it to constitutively recruit CBP; phosphorylation is not necessary (Cardinaux et al., 2000). In the CREB mutant CREBDIEDML, this DIEDML motif replaces the corresponding six amino acid sequence including Ser133 in the CREB transactivation domain and confers the ability to constitutively recruit CBP (Cardinaux et al., 2000). In VCREB, another constitutively-active CREB construct, the CREB transactivation domain of CREB is replaced with that of a viral transcription factor VP16 (Tao et al., 1998). This allows direct transcriptional activation, bypassing the requirement for CBP.
The relative ability of these constitutively-active CREB constructs to activate CRE-dependent transcription is shown in Figure 8A. As above, PC12 cells were used for these assays. β-galactosidase expressed from a CRE-LacZ reporter was induced over six-fold by VCREB. Induction by CREBDIEDML was significantly less, although greater than two-fold. This difference may be due to the lack of CBP phosphorylation after its recruitment by CREBDIEDML and consequent lack of full activation of the transactivator complex, while VCREB acts as a strong transactivator independently of CBP.
Figure 8.
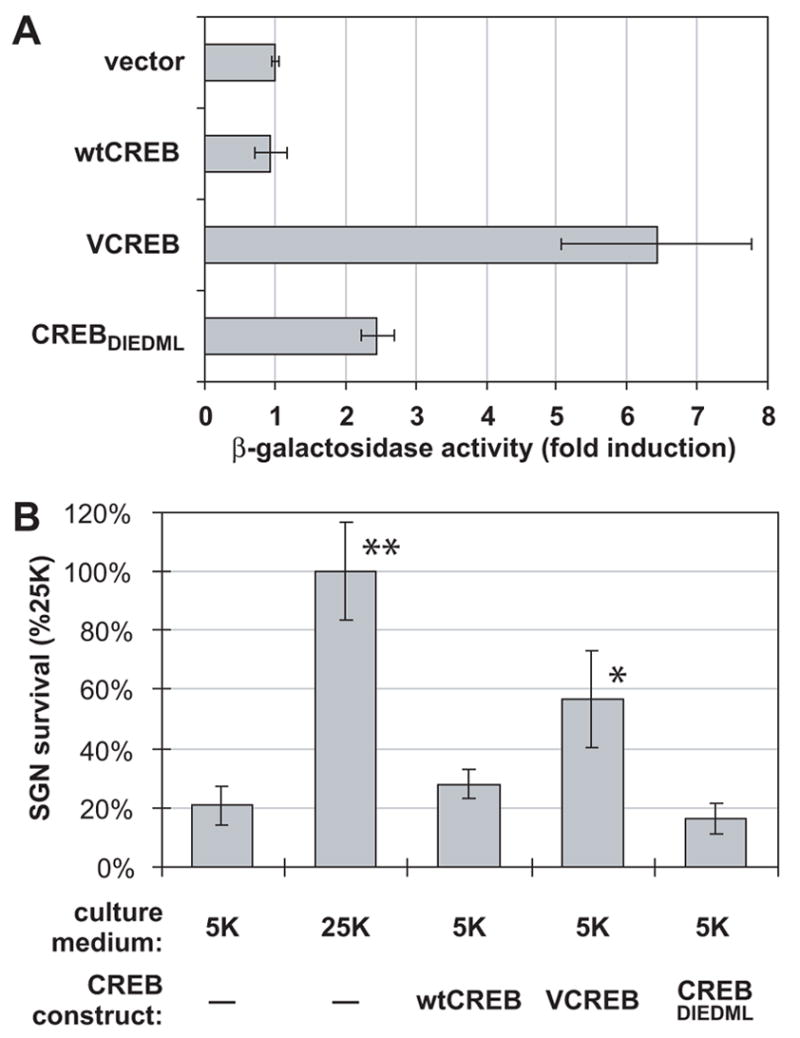
(A) VCREB is a stronger activator of CRE-dependent transcription than is CREBDIEDML in PC12 cells. PC12 cells were cotransfected with CRE-LacZ reporter and GFP (each constituting 20% of total plasmid in mixture) in combination with CREB constructs, wild-type (wt) CREB, VCREB or CREBDIEDML, which constituted 50% of total plasmid. The remainder of the plasmid in the mixture was empty pcDNA3 vector. β-galactosidase was assayed as described in Methods and normalized to activity in cells transfected with reporter and empty pcDNA3 vector, which was arbitrarily set to 1. (B) VCREB, but not CREBDIEDML, promotes SGN survival. SGNs were maintained for 96 h in either control nondepolarizing (5K) or depolarizing (25K) medium following transfection with either GFP plasmids alone or a mixture of GFP and CREB plasmids, wtCREB, VCREB or CREBDIEDML, as indicated. GFP plasmid constituted 20% (2 μg) of the total; CREB plasmids constituted 80% of the total, if present, otherwise 80% of the plasmid was pcDNA3 vector. The number of transfected SGNs surviving at 96 h was determined as described in Methods. The increase in SGN survival over 5K was significant only for 25K and for VCREB-transfected cells (*P < 0.001). The 25K and VCREB transfection conditions were significantly different from each other (**P < 0.001).
After confirming that VCREB and CREBDIEDML are constitutive activators of CRE-dependent transcription, we asked whether such transcriptional activation was sufficient to promote SGN survival. SGNs maintained in 5K were transfected with constitutively-active CREB constructs or wild-type CREB. Of these, only VCREB enhanced SGN survival; wild-type CREB and CREBDIEDML did not (Figure 8B). This shows that CRE-dependent transcription is sufficient to promote neuronal survival provided that the level is high enough. The level or quality of transcriptional activation induced by CREBDIEDML is apparently insufficient.
CREB activation is disabled during apoptosis
Given the significance of the CaMKIV-CREB pathway in prosurvival signaling, we next asked whether it was inactivated after withdrawal of trophic support or during the apoptotic process. SGNs were deprived of trophic support for 24 h to induce apoptosis in a subset of the neurons. The cultures were then briefly (15 min) exposed to depolarizing (25K) medium to acutely activate CREB. We used immunocytochemistry to detect phosphorylated CREB (pCREB) immunofluorescence in individual SGNs, which could additionally be classified as apoptotic or nonapoptotic by the criteria discussed above. Typical examples are shown in Figure 9A.
Figure 9. Depolarization fails to induce CREB phosphorylation in apoptotic SGNs.
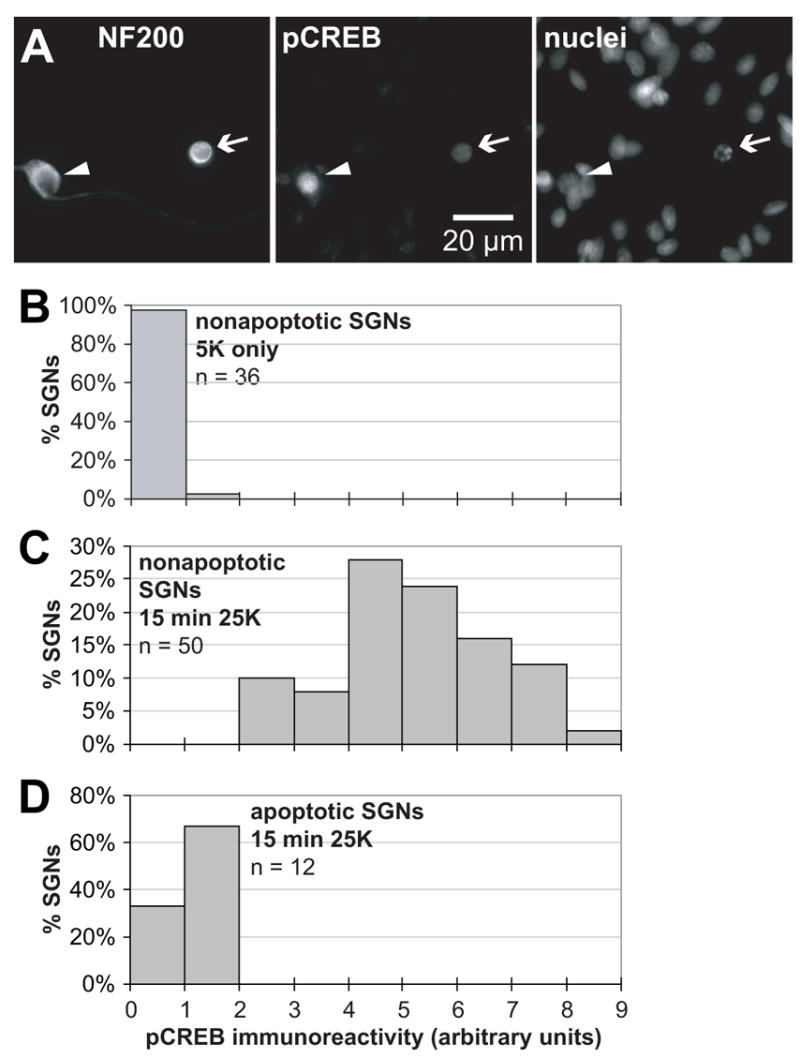
SGN cultures were deprived of trophic support by maintenance in serum-free nondepolarizing medium (5K) for 24 h. (A) The culture was exposed to depolarizing (25K) medium for 15 min. The cells were fixed and an antibody to phosphorylated CREB (pCREB) was used to detect pCREB immunofluorescence. NF200 and nuclei were labeled, and apoptotic cells identified, as in Figure 3. An apoptotic (arrow) and nearby nonapoptotic SGN (arrowhead) are shown. The apoptotic SGN, evident by its pyknotic nucleus and collapsed cytoplasm shows a reduced acute increase in pCREB immunofluorescence, relative to the nonapoptotic SGN, in response to a 15 min exposure to 25K. (B) Quantitation of pCREB immunofluorescence in nonapoptotic SGNs deprived of trophic support for 24 h was performed using NIH Image as described in Methods. The number of SGNs exhibiting various levels of pCREB immunofluorescence is plotted in the histogram. N = the number of SGNs scored. (C) Quantitation of pCREB immunofluorescence in nonapoptotic SGNs deprived of trophic support as in B but exposed to 25K for 15 min . (D) Quantitation of pCREB immunofluorescence in apoptotic SGNs deprived of trophic support but exposed to 25K for 15 min as in C.
Images like those in Figure 9A were used to quantify the acute increase in pCREB immunofluorescence in apoptotic and nonapoptotic SGNs, as described in Methods. We first showed that pCREB immunofluorescence was indeed reduced in SGNs deprived of trophic support and not exposed to 25K (Figure 9B). Among such neurons, we observed that non-apoptotic SGNs acutely exposed to 25K exhibited a large increase in pCREB immunofluorescence (Figure 9C) comparable to that observed in control SGNs exposed to 25K without prolonged deprivation (Bok et al., 2003). This indicates that even in SGN cultures deprived of trophic support sufficiently long to initiate cell death, the intracellular signaling pathway leading to CREB was still present and available to be activated in those SGNs that had not yet become apoptotic. In contrast, in those SGNs that had become apoptotic, CREB could no longer be acutely activated by 15 min exposure to 25K (Figure 9D). Thus, among the changes in intracellular signaling characteristic of apoptosis, is also a disabling of the CREB prosurvival pathway. This is consistent with observations of See et al. (2001) that CaMKIV is degraded in a caspase-dependent manner in cerebellar granule neurons (CGNs) deprived of trophic support, associated with a loss of pCREB immunofluorescence. It is also consistent with our observation that readdition of trophic factors to SGNs deprived of trophic support for 24 h fails to rescue them from cell death (D. Fairfield and S.H. Green, unpublished observation).
Discussion
We describe here a novel reagent GFP-AIP, which is a specific and effective CaMKII inhibitor. We have used it here to verify a requirement for CaMKII in prosurvival signaling resulting from membrane depolarization. Moreover, it is a versatile reagent that may be useful in investigating CaMKII actions in a variety of physiological contexts. Because this protein can be restricted to specific intracellular locations by addition of appropriate targeting signals, it can be used to assess the locus of CaMKII action in cellular regulation. The presence of GFP (or other fluorescent protein) in the chimera allows confirmation of expression and appropriate subcellular targeting. By using GFP-AIP and dominant-inhibitory CaMKIV, separately and in combination, we show that depolarization uses both CaMK pathways, in parallel, to promote neuronal survival. CaMKIV, but not CaMKII, requires CREB to promote survival, consistent with the exclusively nuclear localization of CaMKIV.
CaMKII
Because of its primarily cytoplasmic subcellular localization, we suggest that CaMKII functions to promote survival by targeting cytoplasmic effectors. This is underscored, in particular, by our observation that CaMKII promotes SGN survival, at least in part, by functionally inactivating Bad. The ability of Bad to move from the cytoplasm, where it is sequestered by 14-3-3 protein, to the mitochondria, where it can carry out its proapoptotic function, is regulated by phosphorylation (Downward, 1999). Thus, Bad plays a central role in the regulation of apoptosis by intracellular signaling, with at least four different protein kinase systems phosphorylating at least four different sites on Bad — PI-3-OH kinase -protein kinase B (PKB/Akt) on Ser136 (Datta et al., 1999; del Peso et al., 1997; Fang et al., 1999), ERK-Rsk on Ser112 (Bonni et al., 1999; Fang et al., 1999; Scheid et al., 1999; Tan et al., 1999), cAMP-dependent protein kinase (PKA) on Ser 112 and/or Ser155 (Harada et al., 1999; Lizcano et al., 2000; Virdee et al., 2000), and Cdc2 on Ser128 (Konishi et al., 2002). Here, we provide evidence that CaMKII also regulates apoptosis by inactivating Bad, but the mechanism by which this is accomplished is likely to be complex. One phosphorylation site on Bad, Ser170 (Dramsi et al., 2002), is a potential CaMKII target, based on the deduced CaMKII target consensus (White et al., 1998), raising the possibility that CaMKII phosphorylates Bad directly. However, coexpression of Bad and CaMKII(1-290) in PC12 cells (Bok, Li, and Green, unpublished observations) results in Bad hyperphosphorylation, including phosphorylation of Ser112, which is unlikely to be a CaMKII site. This implies an indirect pathway for Bad phosphorylation by CaMKII. PKB appears to be recruited by another calmodulin-dependent kinase, CaMK kinase, to promote survival (Yano et al., 1998) and the PI-3-OH kinase - PKB pathway has been implicated in the prosurvival effect of depolarization in some neurons (Ikegami and Koike, 2000; Vaillant et al., 1999). It is therefore possible that CaMKII recruits PKB. However, involvement of PKB in promotion of survival by depolarization appears to be via a pathway distinct from CaMKII (Hansen et al., 2001; Ikegami and Koike, 2000; Vaillant et al., 1999). Moreover, neither PKB nor ERK signaling appears to be required for the prosurvival effect of depolarization in SGNs (Hansen et al., 2001). Thus, the mechanism by which CaMKII inactivates Bad may involve multiple signaling pathways and may differ among cell types.
In addition to Bad inactivation, targets of CaMKII other than Bad can contribute to its prosurvival effect. CaMKII suppresses nuclear translocation of histone deacetylase, thereby promoting neuronal survival (Bolger and Yao, 2005; Linseman et al., 2003). In the nucleus, the kinesin family motor protein KIF4 releases the prosurvival factor PARP-1 in a CaMKII dependent manner to promote neuronal survival (Midorikawa et al., 2006). Also, CaMKII has been shown to activate the prosurvival transcriptional regulator NF-κB in T lymphocytes (Hughes et al., 2001) and in neurons (Meffert et al., 2003). Because dominant-negative CREB constructs do not reduce the prosurvival effect of CaMKII, it is unlikely that CREB is the nuclear target of CaMKII.
These studies reveal a prosurvival role for CaMKII, in particular, a role in mediating the prosurvival effects of membrane depolarization. Very strong depolarization compromises neuronal survival and this excitotoxic effect may also be mediated, at least in part, by CaMKII: inhibition of CaMKII can be protective against excitotoxic insults (Hajimohammadreza et al., 1995; Takano et al., 2003). Presumably, the degree of CaMKII activation or signaling pathways activated in parallel distinguish survival and death outcomes of CaMKII activity.
CaMKIV and CREB
We further show that depolarization also promotes survival by recruiting a nuclear pathway involving CaMKIV and CREB. This is supported by the observations that dominant-inhibitory CaMKIV and dominant-inhibitory CREB both reduce the ability of depolarization to promote survival and dominant-inhibitory CREB blocks the ability of CaMKIV to promote survival. If CaMKIV promotes survival primarily via CREB then we would expect constitutively-active CREB to be sufficient to promote survival to a degree. Indeed, this is the case for the VP16-CREB fusion (VCREB) used in this and in other (Bonni et al., 1999; Riccio et al., 1999) studies. However, here we also used another constitutively-active CREB mutant, CREBDIEDML, and found that it failed to support SGN survival. One explanation for this result is that the level of transcriptional activation afforded by CREBDIEDML is insufficient to promote survival while the higher level (Figure 8A) afforded by VCREB is sufficient, at least in part.
An alternative possibility is that recruitment of CBP by CREB is necessary but is not sufficient for promotion of survival via CREB-dependent gene expression. CREBDIEDML while constitutively recruiting CBP does not promote survival. CaMKIV phosphorylates CREB on Ser133, allowing it recruit CBP, and, additionally, phosphorylates CBP-Ser301 (Impey et al., 2002). This can account for observations that, at least in some contexts, recruitment of CBP by CREB is not sufficient for transcriptional activation by CaMKIV (Chawla et al., 1998; Hardingham et al., 1999; Hu et al., 1999; Impey et al., 2002). this could also account for our observations here, given that VCREB, which circumvents the need for CBP, promotes SGN survival. While potentially interesting, this hypothesis is speculative, in view of studies showing that, in some contexts, recruitment of CBP by CREB is indeed sufficient for transcription (Cardinaux et al., 2000; Du et al., 2000). Possibly, CBP phosphorylation by CaMKIV is necessary for transcription of genes involved in neuronal survival but not in all other contexts.
This latter hypothesis could also account for a previous observation that CREB is entirely dispensable for the survival-promoting action of PKA in SGNs (Bok et al., 2003) even though, like CaMKIV, PKA phosphorylates CREB on Ser133 in SGNs (Bok et al., 2003). Possibly, PKA is unable to phosphorylate CBP in a manner that allows transcription of genes required for neuronal survival. PKA has been reported to phosphorylate CBP on Ser1772 in a manner important for Pit-1-mediated transcription (Xu et al., 1998) but not for CREB-mediated transcription (Zanger et al., 1999). This possible distinction between CaMKIV and PKA could be responsible for the ability of the former but not the latter to promote survival via CREB. A possible difficulty with this hypothesis is that a CBP N-terminal fragment (1-460) can also show cAMP responsiveness in some cell types (e.g., PC12 but not F9 or COS) (Swope et al., 1996), indicating that CBP regulation by PKA is complex and cell type-specific.
CaMKIV/CREB-dependent expression of certain genes has been previously implicated in neuronal survival. In some neurons this involves up-regulation of BDNF synthesis (Shieh and Ghosh, 1999; Shieh et al., 1998; Tao et al., 1998), which enables autocrine or paracrine BDNF support of survival (Acheson et al., 1995; Davies and Wright, 1995; Ghosh et al., 1994). However, this is unlikely to be the case for SGNs in which BDNF expression, while CREB-dependent, appears not to be up-regulated by depolarization or by activated CaMKIV (Zha et al., 2001). Another candidate target gene is the antiapoptotic regulator Bcl-2, expression of which is CREB-dependent (Wilson et al., 1996) and is up-regulated by neurotrophic or neuroprotective signaling (Mabuchi et al., 2001; Pugazhenthi et al., 2000; Riccio et al., 1999).
See et al. (2001) have reported that CaMKIV is degraded in a caspase-dependent manner in cerebellar granule neurons (CGNs) deprived of trophic support, associated with a loss of pCREB immunoreactivity. Also, CaMKIV and pCREB immunoreactivity are lost in cortical neurons in which cell death has been induced by treatment with anti-amyloid precursor protein antibody (Mbebi et al., 2002), although in this case caspase activity is not required. Here, we show loss of the CREB activation in apoptotic, but not in nonapoptotic, SGNs after withdrawal of trophic support. These observations suggest that neuronal apoptosis, induced by at least two different means, is accompanied by a disabling of a key prosurvival signaling pathway: the CaMKIV-CREB pathway.
Depolarization recruits at least three signaling systems that independently and additively promote survival acting in distinct subcellular compartments on distinct substrates
The observation that simultaneous blockade of CaMKII and CaMKIV reduces survival to a greater degree than blocking either alone suggests that these kinase systems act independently in parallel to mediate the prosurvival effect of depolarization. We have previously shown (Bok et al., 2003; Hansen et al., 2001) that depolarization recruits a cAMP-dependent pathway, in addition to CaMKs, to promote SGN survival. Moreover, PKA acts in the cytoplasm and not via CREB to promote survival (Bok et al., 2003). Thus, depolarization uses at least three distinct Ca2+-dependent signaling pathways that act in parallel and in distinct intracellular compartments to promote SGN survival. CaMKIV acts in the nucleus, via CREB, to prevent cell death. In parallel with CaMKIV actions in the nucleus to control the amount of apoptotic regulators, PKA (Bok et al., 2003) and probably also CaMKII, acting in the cytoplasm, prevent cell death by post-translational modification of apoptotic regulators, e.g., Bad.
Materials and methods
Spiral ganglion neuron culture and transfection
Our basic culture medium consisted of high-glucose (4.5 mg/ml) Dulbecco’s Modified Eagle’s Medium (DMEM), 0.1 mg/ml penicillin, 0.1 mg/ml streptomycin, N2 supplement, 10 μg/ml insulin and is designated herein as ‘5K’ because in it [K+] = 5.4 mM. We also used a depolarizing medium (25K) in which Na+ was replaced with equimolar K+ to raise [K+] to 25 mM while maintaining osmolarity. Prior to and during transfection, the cultures were maintained in 25K to which 5% fetal bovine serum was added (“25K+S”). The DMEM, antibiotics, and serum were obtained from the University of Iowa Diabetes and Endocrinology Research Core and the other supplements were from Sigma (St. Louis, MO) or Invitrogen (Carlsbad, CA).
Dissociated cultures of spiral ganglion neurons (SGNs) were prepared from post-natal day 4–5 rats and maintained by a modification of the methods described previously (Bok et al., 2003; Hansen et al., 2003). Briefly, rat cochleae were removed from the temporal bone and placed in ice-cold phosphate buffered saline (PBS). Each spiral ganglion was isolated from the cochlea by sequential removal of the bony cochlear capsule, the spiral ligament, and the organ of Corti, leaving the spiral ganglion within the modiolus. These were collected in Hank’s Balanced Salt Solution (HBSS) on ice, then transferred to Ca2+/Mg2+-free HBSS with 0.1% trypsin and 0.1% collagenase at 37°C for 20 min to enzymatically dissociate the cells. After three washes with culture medium, a further mechanical dissociation was performed by trituration using a mechanical pipettor with 1 ml pipette tips. The dissociated cells were plated in 25K+S. Neurons were plated in tissue culture dishes (Falcon or Corning, purchased from Fisher Scientific) or slides (Lab-Tek, Rochester, NY) previously coated sequentially with polyornithine (Sigma, 0.1 mg/ml in 10 mM borate buffer, pH 8.4) for 1 hr at room temperature (RT) followed by laminin (Invitrogen, 20 μg/ml in HBSS) overnight at 4°C.
Transfection was performed when the neurons had firmly attached to the substrate, about 6 h after plating. We used a calcium phosphate-based protocol modified from that of Gabellini et al. (Bonni et al., 1999; Riccio et al., 1999) as we have previously described (Zha et al., 2001). Briefly, plasmids of interest were mixed with 1.25 M CaCl2 solution and then sterile deionized water was added to bring the CaCl2 concentration to 0.25 M. An equal volume of 2x depolarizing HEPES buffer (50 mM HEPES + 220 mM NaCl + 1.5 mM Na2HPO4 + 60 mM KCl, pH 7.1) was added slowly and with agitation. The DNA/Ca2+/PO4 mixture was left for 20 min at RT to allow precipitates to form and then added to the culture medium (25K+S) on the cells at a 1:10 (v/v) ratio, the final concentration of DNA being 10 μg/ml, which we determined to be optimal. After 6 h, the culture medium containing DNA mixture was washed once with DMEM and replaced with 25K medium. Typically, 10% of the SGNs were transfected. In some cases multiple plasmids were co-transfected. In experiments performed using two plasmids that both encode a detectable product, we found that >95% of the transfected cells expressed both gene products (not shown). Because the transfection efficiency is highly dependent on the DNA concentration, when transfecting mixtures of different plasmids the total amount of plasmid was always 10 μg/ml and the ratio of the plasmids varied as described for each figure.
Immunocytochemistry
After culture for the times indicated, the cells were fixed for 15 min with 4% paraformaldehyde in PBS, washed with PBS, incubated with blocking buffer (PBS + 2% BSA + 5% normal goat serum + 0.1% NaN3) for 1 h at 37°, then with primary antibodies in blocking buffer for 1 h at 37° or overnight at 4°, then with fluorescently-labeled secondary antibodies in blocking buffer for 1 h at 37°. After washing with PBS, the nuclei were stained with Hoechst 33342 (10 μg/ml in PBS) for 15 min, washed again with PBS and viewed with a Leica Leitz DMR microscope equipped for fluorescence optics. The images were captured using a Photometrics CH250 cooled CCD camera with IPLab Spectrum software (Signal Analytics Corp., Vienna, VA). The images were prepared for publication using Adobe Photoshop and Illustrator.
The primary antibodies used in this study were rabbit anti-phosphoCREB antibody that specifically detects CREB phosphorylated at Ser133 (Upstate Biotech, Waltham, MA, 1:1000 dilution), rabbit anti-pan-CaMKII antibody (Transduction Labs, Lexington, KY, 1:200), mouse anti-CaMKIV antibody (Transduction Labs, 1:200), and mouse or rabbit anti-neurofilament 200 kDa isoform (NF200) antibody (Sigma, 1:400 dilution) to identify neurons. Anti-CaMKIIα and anti-CaMKIIβ antibodies were generously provided by Dr. Johannes Hell (Dept. Pharmacology, University of Iowa). Secondary antibodies used were Alexa Fluor® 488 or 568 goat anti-rabbit IgG and Alexa Fluor® 350 or 568 goat anti-mouse IgG antibodies (Molecular Probes, Eugene, OR, 1:400 dilution).
NIH image on a Macintosh computer or the equivalent Image J software on a Windows computer was used to quantify phosphoCREB immunofluorescence. In the program, a circular selection was made just inside the boundary of each neuronal nucleus and the intensity of the phosphoCREB immunofluorescence determined as the average pixel density within the circle. Background fluorescence was determined as the average pixel density within a circle of equal diameter just outside of the nucleus and this background was subtracted from the value obtained for nuclear fluorescence. The scale used was arbitrary but linear and consistent among all experiments.
Assay of neuronal survival/apoptosis
For survival assays, replicate wells were prepared for each plasmid or combination of plasmids transfected. Two wells were fixed 18 h after the start of transfection, by which time expression of the introduced genes was already apparent, and were used to establish the transfection efficiency of that combination of plasmids. The remaining wells (in separate multiwell plates) were washed 3x with PBS and then maintained for 96 h under 5K or 25K condition. The cells were then fixed, permeabilized, and stained with anti-NF200 antibody, as described above. All transfected (GFP-expressing) neurons (NF200-positive) in each well were counted to assess survival. This number was corrected for differing transfection efficiencies among different combinations of plasmids. To allow for comparison among experiments, the number of SGNs transfected with GFP only and maintained in 25K was arbitrarily set to 100%, to which relative survival in other experimental conditions was normalized.
For apoptosis assays, SGNs were cotransfected with combinations of plasmids including wild type Bad, GFP, and wild type or truncated constitutively active CaMKII. After recovery from transfection, the cultures were maintained for 48 h in 5K + 1% fetal bovine serum, fixed and stained with anti-NF200 antibody and Hoechst 33342. SGN apoptosis was established by nuclear pyknosis and collapse of the cytoskeleton.
Statistically significant differences among conditions are indicated in the figures and figure legends. Statistical comparisons among the multiple conditions in each experiment were done with Tukey's Studentized Range (HSD) Test, using SAS System software. Pairwise comparisons were made using Student’s t-test.
β-galactosidase assay
PC12 cell cultures in 60 mm dishes were transfected with a CRE-LacZ reporter plasmid, constructed as described below, in combination with plasmids encoding proteins of interest and/or control GFP (to facilitate identification of transfected cells and to allow a constant amount of DNA in all transfections). 48 h after transfection the cells were washed with serum-free culture medium and replated in serum-free medium in 35 mm dishes. To control for small differences in transfection efficiency among different combinations of plasmids, duplicate aliquots of transfected cells were separately fixed at the same time that the β-galactosidase assays were performed (12 after replating in serum-free medium) and the number of transfected (GFP-positive) cells counted. The level of β–galactosidase expressed in each condition was normalized to this number of transfectants.
To assay β–galactosidase activity, the cells in 35 mm dishes were scraped off in 400 μL 0.4% NP40 in PBS. 200 μL of the cell lysates were mixed with 800 μL of Z-buffer (0.1 M PBS, pH 7.5, 2 mM MgSO4, 10 mM KCl, 40 mM β-mercaptoethanol) and 160 μL of o-Nitrophenyl β-D-galactopyranoside (4 mg/ml). The mixtures were incubated at 37° until color developed and the reactions were terminated by addition of 240 μL of 1 M Na2CO3. β-galactosidase activity was determined by measuring OD420.
Expression constructs
Enhanced green fluorescence protein-C1 (EGFP-C1) was purchased from Clontech. Expression plasmids of wild type and truncated, constitutively active forms of CaMKII and CaMKIV were from Dr. Rich Maurer and have been described in Sun et al. (Sun et al., 1994). The wild type BAD expression vector was provided by Dr. Gabriel Nuñez (del Peso et al., 1997). VCREB and CREBDIEDML were obtained from Dr. Richard Goodman and are all under control of the same CMV promoter.
CREBm1 and wild-type CREB were obtained from Dr. Marc Montminy (Montminy et al., 1990) and subcloned by us into pcDNA3 (Invitrogen), so were also under control of the CMV promoter. GFP-AIP was constructed by inserting oligonucleotides encoding autocamtide-2 related inhibitory peptide (AIP; KKALRRQEAVDAL) into EGFP-C1 vector between BamHI and XbaI. Oligonucleotides used were 5’-GCGGGATCCAAGAAGGCCCTGAGGCGCCAGGAGGCCGTGGACGCCCTCTAGACGC–3’ and 5’-GCGTCTAGAGGGCGTCCACGGCCTCCTGGCGCCTCAGGGCCTTCTTGGATCCCGC-3’. This eliminated the XbaI site, facilitating detection of the insert.
To target GFP-AIP to the nucleus or to prevent its entry into the nucleus and restrict it to the cytoplasm, a nuclear localization signal (nls) or a nuclear export signal (nes) was, respectively added to the construct as we have described for nls- and nes-containing GFP-PKA (Bok et al., 2003).
The CRE-LacZ reporter was constructed by inserting oligonucleotides having one copy of CRE enhancer sequence (TGACGTCA) at BamHI site within the multiple cloning site of the pβgal-Promoter vector (Clontech). Oligonucleotides used were 5’-GGCGGATCCAGATCTCTGACGTCAGCTCGAGGGATCCGGC-3’ and 5’-GCCGGATCCCTCGAGCTGACGTCAGAGATCTGGATCCGCC-3’. A catalytically inactive CaMKIV mutant, CaMKIV(K75E), was constructed using site-directed mutagenesis by overlap extension PCR (Ho et al., 1989). The mutated, amplified cDNA replaced the corresponding region of GFP-tagged wild type CaMKIV between BspEI and PstI. The primers used were
| Forward #1 5’-TGAGCAAAGACCCCAACGAGAAGC-3’ |
| Reverse #1 5’-CCTGTCCACTGTCTCCTTTAACACTTT-3’, |
| Forward #2 5’-AAAGTGTTAAAGGAGACAGTGGACAAG-3’ |
| Reverse #2 5’-TGCCTATTTCTCCACCTCCTCGCA-3’. |
Acknowledgments
Support for this study was from NIH grant DC02961. We thank Drs. Marlan Hansen and Stefan Strack for comments on the manuscript. We also thank Drs. Richard Goodman, Richard Maurer and Gabriel Nuñez for generously providing plasmids used in these studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Acheson A, Conover JC, Fandl JP, DeChiara TM, Russell M, Thadani A, Squinto SP, Yancopoulos GD, Lindsay RM. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374:450–453. doi: 10.1038/374450a0. [DOI] [PubMed] [Google Scholar]
- Ahn S, Ginty DD, Linden DJ. A late phase of cerebellar long-term depression requires activation of CaMKIV and CREB. Neuron. 1999;23:559–568. doi: 10.1016/s0896-6273(00)80808-9. [DOI] [PubMed] [Google Scholar]
- Bohm M, Moellmann G, Cheng E, Alvarez-Franco M, Wagner S, Sassone-Corsi P, Halaban R. Identification of p90RSK as the probable CREB-Ser133 kinase in human melanocytes. Cell Growth Differ. 1995;6:291–302. [PubMed] [Google Scholar]
- Bok J, Zha XM, Cho YS, Green SH. An extranuclear locus of cAMP-dependent protein kinase action is necessary and sufficient for promotion of spiral ganglion neuronal survival by cAMP. J Neurosci. 2003;23:777–787. doi: 10.1523/JNEUROSCI.23-03-00777.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger TA, Yao TP. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J Neurosci. 2005;25:9544–9553. doi: 10.1523/JNEUROSCI.1826-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Brosenitsch TA, Katz DM. Physiological patterns of electrical stimulation can induce neuronal gene expression by activating N-type calcium channels. J Neurosci. 2001;21:2571–2579. doi: 10.1523/JNEUROSCI.21-08-02571.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinaux JR, Notis JC, Zhang Q, Vo N, Craig JC, Fass DM, Brennan RG, Goodman RH. Recruitment of CREB binding protein is sufficient for CREB-mediated gene activation. Mol Cell Biol. 2000;20:1546–1552. doi: 10.1128/mcb.20.5.1546-1552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla S, Hardingham GE, Quinn DR, Bading H. CBP: A Signal-regulated transcriptional coactivator controlled by nuclear calcium and CaM Kinase IV. Science. 1998;281:1505–1509. doi: 10.1126/science.281.5382.1505. [DOI] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RPS, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- Collins F, Lile JD. The role of dihydropyridine-sensitive voltage gated calcium channels in potassium mediated neuronal survival. Brain Res. 1989;502:99–108. doi: 10.1016/0006-8993(89)90465-4. [DOI] [PubMed] [Google Scholar]
- Collins F, Schmidt MF, Guthrie PB, Kater SB. Sustained increase in intracellular calcium promotes neuronal survival. J Neurosci. 1991;11:2582–2587. doi: 10.1523/JNEUROSCI.11-08-02582.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Davies AM, Wright EM. Neurotrophic factors. Neurotrophin autocrine loops. Curr Biol. 1995;5:723–726. doi: 10.1016/s0960-9822(95)00144-8. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Ginty DD. CREB family transcription factors inhibit neuronal suicide. Nat Med. 2002;8:450–451. doi: 10.1038/nm0502-450. [DOI] [PubMed] [Google Scholar]
- del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nuñez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- Downward J. How BAD phosphorylation is good for survival. Nat Cell Biol. 1999;1:E33–35. doi: 10.1038/10026. [DOI] [PubMed] [Google Scholar]
- Dramsi S, Scheid MP, Maiti A, Hojabrpour P, Chen X, Schubert K, Goodlett DR, Aebersold R, Duronio V. Identification of a novel phosphorylation site, Ser-170, as a regulator of Bad pro-apoptotic activity. J Biol Chem. 2002;277:6399–6272. doi: 10.1074/jbc.M109990200. [DOI] [PubMed] [Google Scholar]
- Du K, Asahara H, Jhala US, Wagner BL, Montminy M. Characterization of a CREB gain-of-function mutant with constitutive transcriptional activity in vivo. Mol Cell Biol. 2000;20:4320–4327. doi: 10.1128/mcb.20.12.4320-4327.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enslen H, Sun P, Brickey D, Soderling SH, Klamo E, Soderling TR. Characterization of Ca2+/calmodulin-dependent protein kinase IV. Role in transcriptional regulation. J Biol Chem. 1994;269:15520–15527. [PubMed] [Google Scholar]
- Enslen H, Tokumitsu H, Soderling TR. Phosphorylation of CREB by CaM-kinase IV activated by CaM-kinase IV kinase. Biochem Biophys Res Commun. 1995;207:1038–1043. doi: 10.1006/bbrc.1995.1289. [DOI] [PubMed] [Google Scholar]
- Fang X, Yu S, Eder A, Mao M, Bast RC, Jr, Boyd D, Mills GB. Regulation of BAD phosphorylation at serine 112 by the Rasmitogen-activated protein kinase pathway. Oncogene. 1999;18:6635–6640. doi: 10.1038/sj.onc.1203076. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S. CREB couples neurotrophin signals to survival messages. Neuron. 2000;25:11–14. doi: 10.1016/s0896-6273(00)80866-1. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- Franklin JL, Sanz-Rodriguez C, Juhasz A, Deckwerth TL, Johnson EM., Jr Chronic depolarization prevents programmed death of sympathetic neurons in vitro but does not support growth: requirement for Ca2+ influx but not Trk activation. J Neurosci. 1995;15:643–664. doi: 10.1523/JNEUROSCI.15-01-00643.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli C, Meucci O, Scorziello A, Werge TM, Calissano P, Schettini G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-1 through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci. 1995;15:1172–1179. doi: 10.1523/JNEUROSCI.15-02-01172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Kingsbury A, Balazs R, Jorgensen OS. The role of depolarization in the survival and differentiation of cerebellar granule cells in culture. J Neurosci. 1987;7:2203–2213. doi: 10.1523/JNEUROSCI.07-07-02203.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- Gietzen K, Sadorf I, Bader H. A model for the regulation of the calmodulin-dependent enzymes erythrocyte Ca2+-transport ATPase and brain phosphodiesterase by activators and inhibitors. Biochem J. 1982;207:541–548. doi: 10.1042/bj2070541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietzen K, Wuthrich A, Bader H. R 24571: a new powerful inhibitor of red blood cell Ca++-transport ATPase and of calmodulin-regulated functions. Biochem Biophys Res Commun. 1981;101:418–425. doi: 10.1016/0006-291x(81)91276-6. [DOI] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–682. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- Hack N, Hidaka H, Wakefield MJ, Balazs R. Promotion of granule cell survival by high K+ or excitatory amino acid treatment and Ca2+/calmodulin-dependent protein kinase activity. Neuroscience. 1993;57:9–20. doi: 10.1016/0306-4522(93)90108-r. [DOI] [PubMed] [Google Scholar]
- Hajimohammadreza I, Probert AW, Coughenour LL, Borosky SA, Marcoux FW, Boxer PA, Wang KK. A specific inhibitor of calcium/calmodulin-dependent protein kinase-II provides neuroprotection against NMDA- and hypoxia/hypoglycemia-induced cell death. J Neurosci. 1995;15:4093–4101. doi: 10.1523/JNEUROSCI.15-05-04093.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen MR, Devaiah AK, Bok J, Zha X, Green SH. Ca2+/calmodulin-dependent protein kinases II and IV function similarly in neurotrophic signaling but differ in their effects on neurite growth in spiral ganglion neurons. J, Neurosci Res. 2003;72:169–184. doi: 10.1002/jnr.10551. [DOI] [PubMed] [Google Scholar]
- Hansen MR, Zha XM, Bok J, Green SH. Multiple distinct signal pathways, including an autocrine neurotrophic mechanism, contribute to the survival-promoting effect of depolarization on spiral ganglion neurons. J Neurosci. 2001;21:2256–2267. doi: 10.1523/JNEUROSCI.21-07-02256.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Jr, Shen S, Wiemelt AP, McMorris FA, Barres BA. Cyclic AMP elevation is sufficient to promote the survival of spinal motor neurons in vitro. J Neurosci. 1998;18:7361–7371. doi: 10.1523/JNEUROSCI.18-18-07361.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H, Becknell B, Wilm M, Mann M, Huang LJ, Taylor SS, Scott JD, Korsmeyer SJ. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol Cell. 1999;3:413–422. doi: 10.1016/s1097-2765(00)80469-4. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Chawla S, Cruzalegui FH, Bading H. Control of recruitment and transcription-activating function of CBP determines gene regulation by NMDA receptors and L-type calcium channels. Neuron. 1999;22:789–798. doi: 10.1016/s0896-6273(00)80737-0. [DOI] [PubMed] [Google Scholar]
- Hartshorn DO, Miller JM, Altschuler RA. Protective effect of electrical stimulation in the deafened guinea pig cochlea. Otolaryngology - Head & Neck Surgery. 1991;104:311–319. doi: 10.1177/019459989110400305. [DOI] [PubMed] [Google Scholar]
- Hegarty JL, Kay AR, Green SH. Trophic support of cultured spiral ganglion neurons by depolarization exceeds and is additive with that by neurotrophins or cyclic AMP, and requires elevation of [Ca2+]i within a set range. J Neurosci. 1997;17:1959–1970. doi: 10.1523/JNEUROSCI.17-06-01959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho N, Gullberg M, Chatila T. Activation protein 1-dependent transcriptional activation of interleukin 2 gene by Ca2+/calmodulin kinase type IV/Gr. J Exp Med. 1996;184:101–112. doi: 10.1084/jem.184.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Hu SC, Chrivia J, Ghosh A. Regulation of CBP-mediated transcription by neuronal calcium signaling. Neuron. 1999;22:799–808. doi: 10.1016/s0896-6273(00)80738-2. [DOI] [PubMed] [Google Scholar]
- Hughes K, Edin S, Antonsson A, Grundstrom T. Calmodulin-dependent kinase II mediates T cell receptor/CD3- and phorbol ester-induced activation of IκB Kinase. J Biol Chem. 2001;276:36008–36013. doi: 10.1074/jbc.M106125200. [DOI] [PubMed] [Google Scholar]
- Ikegami K, Koike T. Membrane depolarization-mediated survival of sympathetic neurons occurs through both phosphatidylinositol 3-kinase- and CaM kinase II-dependent pathways. Brain Res. 2000;866:218–226. doi: 10.1016/s0006-8993(00)02284-8. [DOI] [PubMed] [Google Scholar]
- Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–244. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H. A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem Biophys Res Commun. 1995;212:806–812. doi: 10.1006/bbrc.1995.2040. [DOI] [PubMed] [Google Scholar]
- Ishida A, Shigeri Y, Tatsu Y, Uegaki K, Kameshita I, Okuno S, Kitani T, Yumoto N, Fujisawa H. Critical amino acid residues of AIP, a highly specific inhibitory peptide of calmodulin-dependent protein kinase II. FEBS Lett. 1998;427:115–118. doi: 10.1016/s0014-5793(98)00405-0. [DOI] [PubMed] [Google Scholar]
- Koike T, Martin DP, Johnson EM., Jr Role of Ca2+ channels in the ability of membrane depolarization to prevent neuronal death induced by trophic-factor deprivation: evidence that levels of internal Ca2+ determine nerve growth factor dependence of sympathetic ganglion cells. Proc Natl Acad Sci USA. 1989;86:6421–6425. doi: 10.1073/pnas.86.16.6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Lehtinen M, Donovan N, Bonni A. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell. 2002;9:1005–1016. doi: 10.1016/s1097-2765(02)00524-5. [DOI] [PubMed] [Google Scholar]
- Kwok RPS, Lundblad JR, Chrivia JC, Richards JP, Bächinger HP, Brennan RG, Roberts SGE, Green MR, Goodman RH. Nuclear protein CBP is a co-activator for the transcription factor CREB. Nature. 1994;370:223–226. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- Leake PA, Hradek GT, Rebscher SJ, Snyder RL. Chronic intracochlear electrical stimulation induces selective survival of spiral ganglion neurons in neonatally deafened cats. Hear Res. 1991;54:251–271. doi: 10.1016/0378-5955(91)90120-x. [DOI] [PubMed] [Google Scholar]
- Leake PA, Snyder RL, Hradek GT, Rebscher SJ. Chronic intracochlear electrical stimulation in neonatally deafened cats: effects of intensity and stimulating electrode location. Hear Res. 1992;64:99–117. doi: 10.1016/0378-5955(92)90172-j. [DOI] [PubMed] [Google Scholar]
- Linseman DA, Bartley CM, Le SS, Laessig TA, Bouchard RJ, Meintzer MK, Li M, Heidenreich KA. Inactivation of the myocyte enhancer factor-2 repressor histone deacetylase-5 by endogenous Ca2+/calmodulin-dependent kinase II promotes depolarization-mediated cerebellar granule neuron survival. J Biol Chem. 2003;278:41472–41481. doi: 10.1074/jbc.M307245200. [DOI] [PubMed] [Google Scholar]
- Lizcano JM, Morrice N, Cohen P. Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155. Biochem J. 2000;349:547–557. doi: 10.1042/0264-6021:3490547. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lonze BE, Riccio A, Cohen S, Ginty DD. Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron. 2002;34:371–385. doi: 10.1016/s0896-6273(02)00686-4. [DOI] [PubMed] [Google Scholar]
- Louis CF, Turnquist J, Jarvis B. Inhibition of calmodulin-dependent and independent cardiac sarcoplasmic reticulum activities by R24571. Cell Calcium. 1983;4:107–116. doi: 10.1016/0143-4160(83)90039-8. [DOI] [PubMed] [Google Scholar]
- Lousteau RJ. Increased spiral ganglion cell survival in electrically stimulated deafened guinea pig cochleae. Laryngoscope. 1987;97:836–842. [PubMed] [Google Scholar]
- Lustig LR, Leake PA, Snyder RL, Rebscher SJ. Changes in the cat cochlear nucleus following neonatal deafening and chronic intracochlear electrical stimulation. Hear Res. 1994;74:29–37. doi: 10.1016/0378-5955(94)90173-2. [DOI] [PubMed] [Google Scholar]
- Mabuchi T, Kitagawa K, Kuwabara K, Takasawa K, Ohtsuki T, Xia Z, Storm D, Yanagihara T, Hori M, Matsumoto M. Phosphorylation of cAMP response element-binding protein in hippocampal neurons as a protective response after exposure to glutamate in vitro and ischemia in vivo. J Neurosci. 2001;21:9204–9213. doi: 10.1523/JNEUROSCI.21-23-09204.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews RP, Guthrie CR, Wailes LM, Zhao X, Means AR, McKnight GS. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol Cell Biol. 1994;14:6107–6116. doi: 10.1128/mcb.14.9.6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbebi C, See V, Mercken L, Pradier L, Muller U, Loeffler JP. Amyloid precursor protein family-induced neuronal death is mediated by impairment of the neuroprotective calcium/calmodulin protein kinase IV-dependent signaling pathway. J Biol Chem. 2002;277:20979–20990. doi: 10.1074/jbc.M107948200. [DOI] [PubMed] [Google Scholar]
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-κB functions in synaptic signaling and behavior. Nat Neurosci. 2003;6:1072–1078. doi: 10.1038/nn1110. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- Midorikawa R, Takei Y, Hirokawa N. KIF4 motor regulates activity-dependent neuronal survival by suppressing PARP-1 enzymatic activity. Cell. 2006;125:371–383. doi: 10.1016/j.cell.2006.02.039. [DOI] [PubMed] [Google Scholar]
- Miranti CK, Ginty DD, Huang G, Chatila T, Greenberg ME. Calcium activates serum response factor-dependent transcription by a Ras- and Elk-1-independent mechanism that involves a Ca2+/calmodulin- dependent kinase. Mol Cell Biol. 1995;15:3672–3684. doi: 10.1128/mcb.15.7.3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montminy MR, Gonzalez GA, Yamamoto KY. Regulation of cAMP-inducible genes by CREB. Trends Neurosci. 1990;13:184–188. doi: 10.1016/0166-2236(90)90045-c. [DOI] [PubMed] [Google Scholar]
- Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, Reusch JE. Akt/protein kinase B up–regulates Bcl-2 expression through cAMP- response element-binding protein. J Biol Chem. 2000;275:10761–10766. doi: 10.1074/jbc.275.15.10761. [DOI] [PubMed] [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Schubert KM, Duronio V. Regulation of bad phosphorylation and association with Bcl-xL by the MAPK/Erk kinase. J Biol Chem. 1999;274:31108–31113. doi: 10.1074/jbc.274.43.31108. [DOI] [PubMed] [Google Scholar]
- See V, Boutillier AL, Bito H, Loeffler JP. Calcium/calmodulin-dependent protein kinase type IV (CaMKIV) inhibits apoptosis induced by potassium deprivation in cerebellar granule neurons. FASEB J. 2001;15:134–144. doi: 10.1096/fj.00-0106com. [DOI] [PubMed] [Google Scholar]
- Shieh PB, Ghosh A. Molecular mechanisms underlying activity-dependent regulation of BDNF expression. J Neurobiol. 1999;41:127–134. [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- Simon J, Arthur C, Fong AL, Dwyer JM, Davare M, Reese E, Obrietan K, Impey S. Mitogen- and stress-activated protein kinase 1 mediates cAMP response element-binding protein phosphorylation and activation by neurotrophins. J Neurosci. 2004;24:4324–4332. doi: 10.1523/JNEUROSCI.5227-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struthers RS, Vale WW, Arias C, Sawchenko PE, Montminy MR. Somatotroph hypoplasia and dwarfism in transgenic mice expressing a non-phosphorylatable CREB mutant. Nature. 1991;350:622–624. doi: 10.1038/350622a0. [DOI] [PubMed] [Google Scholar]
- Sun P, Enslen H, Myung PS, Maurer RA. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994;8:2527–2539. doi: 10.1101/gad.8.21.2527. [DOI] [PubMed] [Google Scholar]
- Sun P, Schoderbek WE, Maurer RA. Phosphorylation of cyclic adenosine 3',5'-monophosphate (cAMP) response element-binding protein isoforms by the cAMP-dependent protein kinase. Mol Endocrinol. 1992;6:1858–1866. doi: 10.1210/mend.6.11.1480175. [DOI] [PubMed] [Google Scholar]
- Swanson KD, Taylor LK, Haung L, Burlingame AL, Landreth GE. Transcription factor phosphorylation by pp90rsk2. Identification of Fos kinase and NGFI-B kinase I as pp90rsk2. J Biol Chem. 1999;274:3385–3395. doi: 10.1074/jbc.274.6.3385. [DOI] [PubMed] [Google Scholar]
- Swope DL, Mueller CL, Chrivia JC. CREB-binding protein activates transcription through multiple domains. J Biol Chem. 1996;271:28138–28145. doi: 10.1074/jbc.271.45.28138. [DOI] [PubMed] [Google Scholar]
- Takano H, Fukushi H, Morishima Y, Shirasaki Y. Calmodulin and calmodulin-dependent kinase II mediate neuronal cell death induced by depolarization. Brain Res. 2003;962:41–47. doi: 10.1016/s0006-8993(02)03932-x. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Fukunaga K, Miyamoto E. Activation of nuclear Ca2+/calmodulin-dependent protein kinase II and brain-derived neurotrophic factor gene expression by stimulation of dopamine D2 receptor in transfected NG108-15 cells. J Neurochem. 2002;82:316–328. doi: 10.1046/j.1471-4159.2002.00967.x. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Yamamoto H, Matsumoto K, Kimura T, Katsuragi S, Miyakawa T, Miyamoto E. Nuclear localization of the delta subunit of Ca2+/calmodulin-dependent protein kinase II in rat cerebellar granule cells. J Neurochem. 1999;72:815–825. doi: 10.1046/j.1471-4159.1999.0720815.x. [DOI] [PubMed] [Google Scholar]
- Tan Y, Ruan H, Demeter MR, Comb MJ. p90RSK blocks Bad-mediated cell death via a protein kinase C- dependent pathway. J Biol Chem. 1999;274:34859–34867. doi: 10.1074/jbc.274.49.34859. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Chijiwa T, Hagiwara M, Mizutani A, Terasawa M, Hidaka H. KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4- phenylpiperazine, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1990;265:4315–4320. [PubMed] [Google Scholar]
- Vaillant AR, Mazzoni I, Tudan C, Boudreau M, Kaplan DR, Miller FD. Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase-Akt pathway to synergistically regulate neuronal survival. J Cell Biol. 1999;146:955–966. doi: 10.1083/jcb.146.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant AR, Zanassi P, Walsh GS, Aumont A, Alonso A, Miller FD. Signaling mechanisms underlying reversible, activity-dependent dendrite formation. Neuron. 2002;34:985–998. doi: 10.1016/s0896-6273(02)00717-1. [DOI] [PubMed] [Google Scholar]
- Virdee K, Parone PA, Tolkovsky AM. Phosphorylation of the pro-apoptotic protein BAD on serine 155, a novel site, contributes to cell survival. Curr Biol. 2000;10:1151–1154. doi: 10.1016/s0960-9822(00)00702-8. [DOI] [PubMed] [Google Scholar]
- Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem. 2001;276:13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- White RR, Kwon YG, Taing M, Lawrence DS, Edelman AM. Definition of optimal substrate recognition motifs of Ca2+-calmodulin-dependent protein kinases IV and II reveals shared and distinctive features. J Biol Chem. 1998;273:3166–3172. doi: 10.1074/jbc.273.6.3166. [DOI] [PubMed] [Google Scholar]
- Wilson BE, Mochon E, Boxer LM. Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol Cell Biol. 1996;16:5546–5556. doi: 10.1128/mcb.16.10.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Riley MTT, Walsh SM, Leake-Jones PA. Maintenance of neuronal activity by electrical stimulation of unilaterally deafened cats demonstrable with cytochrome oxidase technique. Ann Otol Rhinol Laryngol. 1981;90:30–32. doi: 10.1177/00034894810902s211. [DOI] [PubMed] [Google Scholar]
- Xu L, Lavinsky RM, Dasen JS, Flynn SE, McInerney EM, Mullen TM, Heinzel T, Szeto D, Korzus E, Kurokawa R, Aggarwal AK, Rose DW, Glass CK, Rosenfeld MG. Signal-specific co-activator domain requirements for Pit-1 activation. Nature. 1998;395:301–306. doi: 10.1038/26270. [DOI] [PubMed] [Google Scholar]
- Yano S, Tokumitsu H, Soderling TR. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature. 1998;396:584–587. doi: 10.1038/25147. [DOI] [PubMed] [Google Scholar]
- Zanger K, Cohen LE, Hashimoto K, Radovick S, Wondisford FE. A novel mechanism for cyclic adenosine 3',5'-monophosphate regulation of gene expression by CREB-binding protein. Mol Endocrinol. 1999;13:268–275. doi: 10.1210/mend.13.2.0245. [DOI] [PubMed] [Google Scholar]
- Zha XM, Bishop JF, Hansen MR, Victoria L, Abbas PJ, Mouradian MM, Green SH. BDNF synthesis in spiral gangion neurons is constitutive and CREB-dependent. Hear Res. 2001;156:53–68. doi: 10.1016/s0378-5955(01)00267-2. [DOI] [PubMed] [Google Scholar]
- Zou DJ, Cline HT. Postsynaptic calcium/calmodulin-dependent protein kinase II is required to limit elaboration of presynaptic and postsynaptic neuronal arbors. J Neurosci. 1999;19:8909–8918. doi: 10.1523/JNEUROSCI.19-20-08909.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]