

Summary
Pulmonary hypertension is associated with sudden death and is a risk factor for mortality in adult patients with sickle cell disease. The high mortality despite only mild-to-moderate increases in pulmonary vascular resistance remains an unresolved paradox. Accordingly, little is known about the cardiovascular effects of stressors, such as vaso-occlusive pain crisis (VOC) and exercise, which may acutely increase pulmonary pressures and impair right heart function. We therefore evaluated pulmonary artery pressures by echocardiogram in 25 patients with sickle cell disease in steady-state and during VOC, and by right heart catheterisation with exercise in a second cohort of 21 patients to determine whether pulmonary hypertension worsens during acute cardiopulmonary stress. TRV increased during VOC (P < 0·001), and the increased pulmonary pressures during VOC were associated with decreases in haemoglobin levels (P < 0·001), and increases in lactate dehydrogenase (P < 0·001) and plasma haemoglobin levels (P = 0·03). During exercise stress performed during cardiac catheterisation, mean pulmonary artery pressures (P < 0·001) and pulmonary vascular resistance increased (P < 0·001) in all subjects. These data suggest that acute elevations in pulmonary pressures during VOC or exercise may contribute to morbidity and mortality in patients with sickle cell disease.
Keywords: sickle cell disease, vaso-occlusive crisis, pulmonary hypertension, nitric oxide, haemolysis
Pulmonary hypertension is increasingly recognised as a complication of sickle cell disease. Despite mild to moderate increases in pulmonary arterial pressures, the 2-year mortality associated with the disease approaches 50% in adult patients. (Castro et al, 2003). We recently demonstrated that in adult patients with sickle cell disease, even mild pulmonary hypertension [defined by a tricuspid regurgitant jet velocity (TRV) ≥2·5 m/s] is a major independent risk factor for death (relative risk, 10·1; 95%confidence interval, 2·7–44), suggesting that these mild to moderate increases in pulmonary pressures are poorly tolerated (Gladwin et al, 2004). Sudden or unexplained death is a well-described phenomenon in patients with sickle cell disease, both in steady state and during vaso-occlusive crisis, occurring in 12–26% of all deaths in the Co-operative Study of Sickle Cell Disease (Charache, 1994; Platt et al, 1994).
Acute increases in pulmonary pressures during stress in these patients with severe anaemia and mild to moderate pulmonary hypertension could result in cardiovascular collapse and potentially explain some of these events. Vasoocclusive pain crisis is associated with pathological changes, such as worsening anaemia, enhanced red cell adhesion, vasoconstriction and hypoxemia, which could certainly promote or exacerbate pulmonary hypertension in patients with sickle cell disease. Similarly, during physical exertion, patients with pulmonary vascular diseases become more symptomatic, mainly because of the inability of their right ventricles to overcome the increased right ventricular afterload, and are unable to adequately increase cardiac output for the demands of exercise.
Based on these observations, we hypothesised that vasoocclusive crisis and exercise are associated with worsening pulmonary hypertension in patients with sickle cell disease. To test this hypothesis, we evaluated the effects of vaso-occlusive pain crises and exercise on pulmonary artery pressures, as well as the blood laboratory parameters associated with vasoocclusive pain crisis and changes in pulmonary pressures.
Methods
Patient characteristics
All patients were enrolled in a National Heart Lung and Blood Institute-approved human subjects protocol and all subjects provided written, informed consent. All patients had sickle cell disease (SS, SC or Sβ°-thal phenotypes). The study included two subgroups of patients. For the study of the effects of vaso-occlusive crises on pulmonary hypertension, patients were evaluated at steady-state and during hospitalisation with vaso-occlusive pain crises. For the study of the effects of exercise on pulmonary pressures, a separate subgroup of patients previously diagnosed with pulmonary hypertension by Doppler echocardiography in steady state were evaluated during right heart catheterisation. All patients evaluated had no history or echocardiographic evidence of left ventricular dysfunction or significant valvular disease. Vaso-occlusive pain crisis was defined as an acute pain episode resulting in hospitalisation. Treatment strategies for episodes vaso-occlusive crisis included intravenous hydration, the use of supplemental oxygen to maintain an oxygen saturation measured by pulse oximetry >92% and intravenous opioids administered either intermittently or via patient-controlled analgesia in all patients. Echocardiograms, as well as laboratory profiles, were performed during steady-state and during the first 24 h of admission for vaso-occlusive crises. Crisis data for all patients was obtained within 3 months of baseline echocardiographic assessments, with the exception of one patient whose baseline was obtained 1 year and 4 months before hospitalisation. Six patients had two separate crisis episodes. Data for these patients was analysed based on all individual episodes.
Plasma haemoglobin enzyme-linked immunosorbent assay (ELISA)
Plasma haemoglobin was measured on dilutions of patients’ plasma using an ELISA according to the manufacturer’s directions (Bethyl Laboratories, Inc., Montgomery, TX, USA) as previously described (Wang et al, 2004).
Echocardiography
Transthoracic echocardiography was performed in all patients using the Acuson Sequoia (Siemens-Acuson Inc., Mountainview, CA, USA) and Sonos 5500 (Philips Inc., Andover, MA, USA). Tricuspid regurgitation was assessed as previously described (Gladwin et al, 2004). Pulmonary artery systolic pressure (PASP) was calculated from the TRV and the estimated central venous pressure (CVP) using a modified Bernoulli equation: PASP = 4(TRV)² + CVP. Echocardiograms were read without knowledge of patients’ clinical status.
Right heart catheterisation with supine upper extremity exercise
Right heart catheterisation with supine upper extremity exercise was performed in 21 patients as previously described (Groves & Badesch, 2004). Briefly, patients performed supine straight-arm-raising exercise using 2-pound hand weights while rapidly raising the upper extremities for 5 min or until fatigue. Haemodynamic parameters were assessed at rest and at peak exercise.
Statistical analysis
Significant differences between baseline and crisis or rest and exercise were evaluated by paired student’s t-test or analysis of variance (anova) with a Bonferoni multiple comparisons test. The calculations were performed using graphpad prism. (GraphPad Software Inc., San Diego, CA, USA). A P-value < 0·05 was considered statistically significant.
Results
Effects of vaso-occlusive crisis on TRV
Twenty-six patients (mean age 34 ± 2·5 years, 13 female, 23 HbSS, 2 HbSC and 1 HbSbβ°-thal) were evaluated in steady-state and during vaso-occlusive pain crisis in the crisis cohort. Six patients had two separate crisis episodes and we report the repeated episodes, totalling 32 events. In four patients echocardiograms could not be obtained during crisis, thus echocardiographic data was reported for 28 events. Compared with steady-state values, vaso-occlusive pain crisis was associated with a decrease in haemoglobin (Hb g/l: steady-state 93 ± 2·0; crisis 83 ± 2·0; P < 0·001) and increase in white blood cell count (WBC 109/l: steady-state 8·9 ± 0·5: crisis 11·9 ± 0·8; P = 0·001) lactate dehydrogenase (LDH U/l: steady-state 363 ± 22; crisis 423 ± 38; P < 0·001), aspartate aminotransferase (AST U/l: steady-state 44 ± 3; crisis 51 ± 4; P = 0·029), and direct bilirubin levels (direct bilirubin µmol/l: steady-state 6·8 ± 1·7; crisis 8·5 ± 1·7, P = 0·02) (Table I). In conjunction with the worsening anaemia and increased LDH, TRV was elevated during vaso-occlusive crisis (TRV m/s: steady-state 2·4 ± 0·07; crisis 2·9 ± 0·07; P < 0·001) (Fig 1). This represents a crisis-associated increase in echocardiographically estimated PASPs from 30 ± 1·0 mm Hg to 39 ± 1·7 mm Hg (P < 0·001). TRV significantly decreased to levels close to baseline in nineteen patients who had echocardiograms repeated 2·4 ± 0·7 months after crisis resolution (P < 0·001) (Fig 1).
Table I.
Mean laboratory parameters of study subjects enroled in the crisis cohort.
| Steady-state | Crisis | P | |
|---|---|---|---|
| Haemoglobin (g/l) | 93 ± 2·0 | 83 ± 2·0 | <0·001 |
| White blood cell count (109/l) | 8·9 ± 0·5 | 11·9 ± 0·8 | 0·001 |
| Platelet count (109/l) | 374 ± 25 | 349 ± 27 | 0·3 |
| Absolute reticulocyte count (109/l) | 212 ± 19 | 220 ± 17 | 0·6 |
| Lactate dehydrogenase (U/l) | 363 ± 22 | 423 ± 28 | <0·001 |
| Aspartate aminotransferase (U/l) | 44 ± 3 | 51 ± 4 | 0·026 |
| Alanine aminotransferase (U/l) | 29 ± 2 | 30 ± 2 | 0·8 |
| Total bilirubin (µmol/l) | 49·5 ± 5·1 | 58·1 ± 6·8 | 0·09 |
| Direct bilirubin (µmol/l) | 6·8 ± 1·7 | 8·5 ± 1·7 | 0·02 |
| Alkaline phosphatase (U/l) | 106 ± 9 | 105 ± 8 | 0·9 |
| Creatinine (µmol/l) | 61·8 ± 2·6 | 64·5 ± 3·5 | 0·2 |
Data represents mean ± SE.
Fig 1.
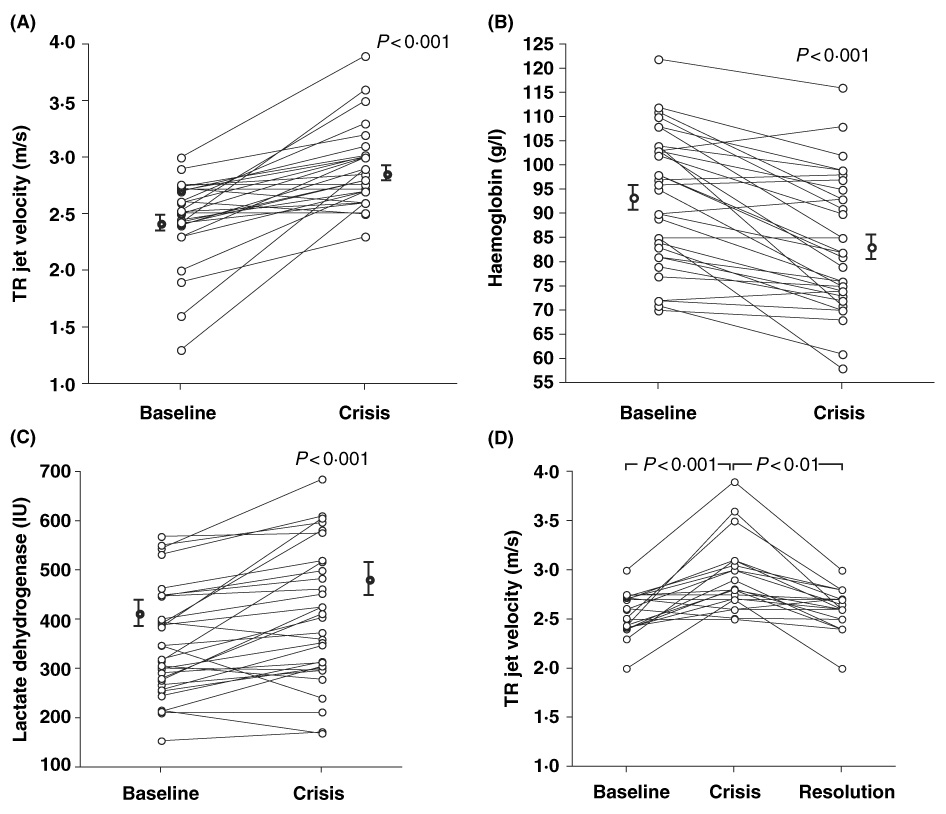
Vaso-occlusive crisis is associated with (A) decrease in haemoglobin and (B) increase in lactate dehydrogenase levels. The increase in haemolytic rate is associated with (C) worsening pulmonary hypertension that improves with (D) resolution of crisis (n = 19). All comparisons were significant by paired t-test.
Previous studies have shown that in patients with sickle cell disease, LDH is a good surrogate marker of intravascular haemolysis (Neely et al, 1969; Naumann et al, 1971; Kato et al, 2006). To gain insight into the role of haemolysis in the elevations in LDH levels associated with vaso-occlusive pain crisis, we measured plasma haemoglobin levels in a subgroup of 17 patients included in this cohort during steady state and crisis. Consistent with an increase in haemolytic rate, vaso-occlusive pain crisis was associated with an increase in plasma haemoglobin (plasma haemoglobin µmol/l: steady-state 13 ± 3; crisis 28 ± 7; P = 0·03). There was, however, only a weak and non-significant negative correlation between TRV and Hb level when steady-state and vaso-occlusive crisis data were combined (R = −0·20, P = 0·13) and there was no association when steady-state (R = −0·11, P = 0·5) and vaso-occlusive crisis (R = −0·06, P = 0·7) were analysed separately. When comparing steady-state with crisis values, there was no correlation between change in TRV and change in Hb level in all patients (R = −0·07, P = 0·7) or in those with (R = −0·21, P = 0·3) or without (R = 0·01, P = 0·98) crisis associated decrements in Hb level. In addition, during vaso-occlusive crisis TRV significantly increased in patients with (P < 0·001) or without crisis associated decrements in Hb level (P = 0·017). Taken together, these findings suggest that in this relatively small sample of patients, worsening of haemolytic anaemia is not the sole contributor to worsening pulmonary hypertension.
Effects of exercise on pulmonary arterial pressures
We evaluated pulmonary hemodynamics at rest and during upper extremity exercise to a mean peak heart rate of 60 ± 2%predicted in 21 patients (mean age 47 ± 2·4 years, 12 female, 21 HbSS) undergoing right heart catheterisation. Submaximal exercise increased right atrial pressure (rest 7 ± 0·7 mmHg, exercise 11 ± 0·9 mmHg, P < 0·001), mean pulmonary artery pressure (rest 31 ± 2 mmHg, exercise 47 ± 3 mmHg, P < 0·001), pulmonary vascular resistance (rest 190 ± 30 dyn.sec/cm5, exercise 290 ± 32 dyn.sec/cm5, P < 0·001), decreased mixed-venous oxygen saturation (rest 69 ± 2%, exercise 62 ± 2%, P < 0·001), and did not change pulmonary capillary wedge pressure (rest 15 ± 1 mmHg, exercise 16 ± 1 mmHg, P = 0·09) (Fig 2). Furthermore, exercise-induced pulmonary hypertension developed in three of six patients with normal pulmonary pressures.
Fig 2.
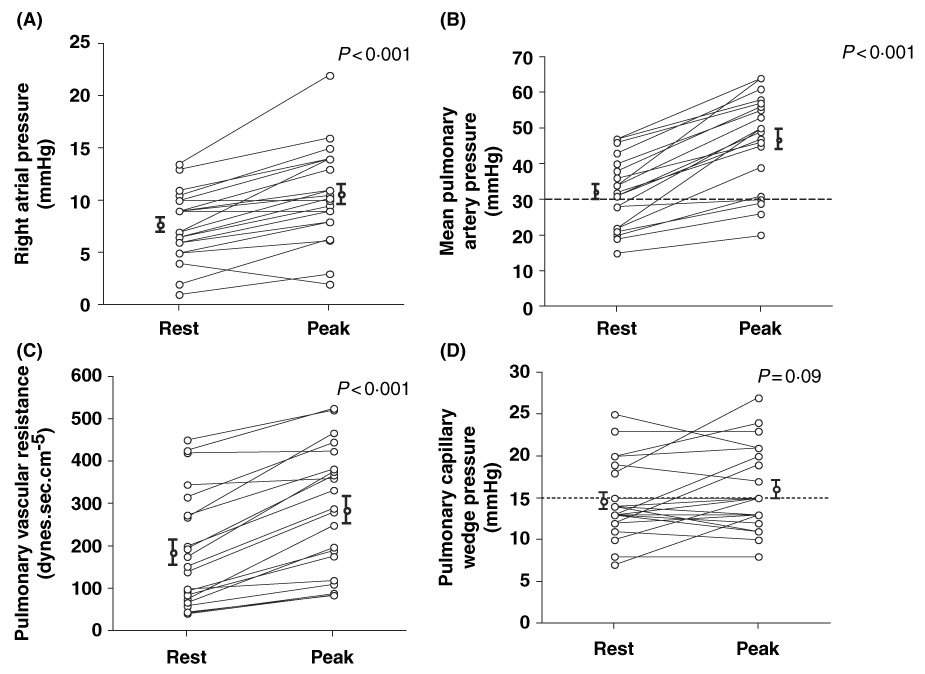
Effects of exercise in pulmonary hemodynamics in patients with sickle cell disease. Upper extremity exercise increases (A) right atrial pressure, (B) mean pulmonary artery pressure, (C) pulmonary vascular resistance, and did not change (D) pulmonary capillary wedge pressure (n = 21, all comparisons by paired t-test).
An abnormal wedge pressure during exercise occurred in two of 15 patients with normal resting values. In patients with abnormal baseline wedge pressure, exercise did not result in a significant increase from resting values (rest 21 ± 1 mmHg, exercise 22 ± 1 mmHg, P = 0·6). These data suggest that in the majority of patients, exercise-induced diastolic dysfunction does not seem to be a major contributor to elevations in pulmonary artery pressures and exercise intolerance.
Discussion
A seminal question that has arisen from the study of pulmonary hypertension in patients with sickle cell disease is how can the mild elevations in mean pulmonary artery pressure and pulmonary vascular resistance seen in these patients be associated with an increased risk of sudden death? (Gladwin et al, 2004; Hassoun & Krishnan, 2004; Klings & Farber, 2004). Sudden death typically results from progressive right heart failure and hemodynamic collapse in patients with other forms of pulmonary arterial hypertension. However, unlike patients with idiopathic pulmonary arterial hypertension, who are young, have no co-morbid organ dysfunction, and do not have severe anaemia, patients with sickle cell disease and pulmonary hypertension have multi-organ complications and severe anaemia. We argue that patients with severe haemolytic anaemia chronically live at the limits of cardiopulmonary compensation and do not survive to see elevations of pulmonary hypertension and vascular resistance to a level similar to those of patients with other forms of pulmonary arterial hypertension. A similar symptomatic presentation and high associated mortality was observed in pregnant women with moderate pulmonary hypertension coupled with the physiological anaemia and high cardiac output state of pregnancy (Weiss & Hess, 2000).
Importantly, this anaemia and requirement for high cardiac output worsens during vaso-occlusive crisis. Our data suggest that patients with sickle cell disease and underlying chronic pulmonary hypertension (which is often unrecognised) develop more severe anaemia and acute exacerbation of pulmonary hypertension with acute crisis. It seems likely that this exacerbation could contribute to the sudden death sometimes seen during vaso-occlusive pain crisis (Parfrey et al, 1985). In addition, in patients with sickle cell disease, exercise is associated with marked worsening of pulmonary hypertension, a finding that probably contributes to the presence of functional limitation and worse prognosis in these individuals.
Pulmonary arterial hypertension is an increasingly recognised complication of chronic hereditary and acquired haemolytic anaemias, including sickle cell disease, (Sutton et al, 1994; Castro et al, 2003; Morris et al, 2003; Gladwin et al, 2004) thalassaemia intermedia and major, (Du et al, 1997; Aessopos et al, 2001) paroxysmal nocturnal haemoglobinuria, (Heller et al, 1992) hereditary spherocytosis and stomatocytosis, (Verresen et al, 1991; Jardine & Laing, 2004) microangiopathic haemolytic anaemias (Jubelirer, 1991; Labrune et al, 1999) and pyruvate kinase deficiency, (Chou & DeLoughery, 2001) suggesting the existence of a syndrome of haemolysis-associated endothelial dysfunction (Kaul et al, 2004; Minneci et al, 2005; Nolan et al, 2005; Rother et al, 2005; Kato et al, 2006), caused by the release of erythrocyte haemoglobin, which scavenges nitric oxide (Reiter et al, 2002), and erythrocyte arginase, which metabolises l-arginine, the substrate for nitric oxide synthesis (Morris et al, 2003; Schnog et al, 2004) (Morris et al, 2005). Cell-free plasma haemoglobin destroys nitric oxide at a rate 500–1000 fold faster than intra-erythrocytic haemoglobin (Gladwin et al, 2003a; Schechter & Gladwin, 2003). Plasma haemoglobin and oxygen free radical-mediated consumption of nitric oxide produce a state of resistance to nitric oxide detected by blood flow physiology studies in patients with sickle cell disease (Aslan et al, 2001; Reiter et al, 2002; Eberhardt et al, 2003; Gladwin et al, 2003b; Reiter & Gladwin, 2003). It is clear, however, that other factors, such as in situ thrombosis, pulmonary thromboembolic disease, iron overload and liver dysfunction contribute to the pathogenesis of pulmonary hypertension in sickle cell disease.
Here we have shown that vaso-occlusive crisis is frequently associated with worsening haemolytic anaemia, which is supported by increased LDH, AST and plasma haemoglobin levels. We have recently confirmed and extended the observation that intravascular haemolysis is the predominant source of increased steady state LDH levels in patients with sickle cell disease (Neely et al, 1969; Naumann et al, 1971; Adhikary et al, 1986; Kato et al, 2006). In addition, a substantial body of evidence suggests that acute vaso-occlusive crisis is associated with intensification of haemolysis (Neely et al, 1969; Naumann et al, 1971; Ballas & Marcolina, 2006). Ballas and Marcolina (2006), using radioisotopic labelling techniques, definitively demonstrated, in vivo, in patients with sickle cell disease that the red cell survival time of 35·4 d during steady state declined to only 16·6 d during vaso-occlusive crisis. These data were further supported by a parallel decline from 17·0 to 7·5 in the ratio of erythrocyte haemoglobin to reticulocyte haemoglobin, an indirect marker of erythrocyte survival. Accelerated release of haemoglobin and arginase into blood plasma during crisis-associated hyperhaemolysis would be expected to further impair nitric oxide bioavailability with consequent acute worsening of the endothelial dysfunction seen in patients with sickle cell disease (Reiter et al, 2002). In fact, worsening of forearm flow-mediated dilation has been observed in patients with sickle cell disease during vaso-occlusive crisis, although the difference in this small, probably underpowered study was not statistically significant (Blum et al, 2005). These mechanisms may contribute to the significant crisis-associated worsening and subsequent improvement of pulmonary hypertension observed after resolution of vaso-occlusive pain crisis. However, we cannot conclusively rule out the possibility that the drop in haemoglobin and lack of increase in reticulocyte count in these patients might be due to sequestration of sickle erythrocytes and reticulocytes to activated and adhesive endothelium, and that adhesion/vaso-occlusion might contribute to acute increases in pulmonary pressures. In fact, these and other mechanisms, such as hypoxia, increase in vasoconstrictive mediators, such as endothelin-1 (Hammerman et al, 1997; Rybicki & Benjamin, 1998; Ergul et al, 2004), or sympathetic nervous system activation, are contributors to the end result of a potentially clinically significant exacerbation of pulmonary hypertension during vaso-occlusive crisis.
Taken together with the effects of exercise on pulmonary hypertension, these data provide potential insights into the pathophysiology of sudden death in patients with sickle cell disease. Despite the uncertainty about the direct role of pulmonary hypertension as a cause of mortality in patients with sickle cell disease (Hassoun & Krishnan, 2004; Klings & Farber, 2004), it is possible that acute worsening of pulmonary hypertension either induced by vaso-occlusive pain crisis or by exertion could result in acute right ventricular failure and dysrhythmia in severely anaemic patients with mild-to-moderate pulmonary hypertension who are already at the limits of their cardiopulmonary compensation.
In conclusion, our results suggest that acute elevations in pulmonary arterial pressures during crisis or exercise may contribute to morbidity and mortality in patients with sickle cell disease, especially in those patients with pre-existing pulmonary hypertension and that pulmonary hypertension screening in these patients should be performed in steady state. In addition, exercise testing may improve the sensitivity of right heart catheterisation studies to detect pulmonary hypertension in patients with sickle cell disease. Clinicians should be aware of this phenomenon when evaluating patients with sickle cell disease during vaso-occlusive crisis or in those with suspected pulmonary hypertension.
Acknowledgement
The study was funded by the Intramural Research Division of the National Institutes of Health, Bethesda, MD.
References
- Adhikary PK, Hara S, Dwivedi C, Davis JW, Weaver C, Pavuluri SR. Vaso-occlusive crisis episodes in sickle cell disease. Journal of Medicine. 1986;17:227–240. [PubMed] [Google Scholar]
- Aessopos A, Farmakis D, Karagiorga M, Voskaridou E, Loutradi A, Hatziliami A, Joussef J, Rombos J, Loukopoulos D. Cardiac involvement in thalassemia intermedia: a multicenter study. Blood. 2001;97:3411–3416. doi: 10.1182/blood.v97.11.3411. [DOI] [PubMed] [Google Scholar]
- Aslan M, Ryan TM, Adler B, Townes TM, Parks DA, Thompson JA, Tousson A, Gladwin MT, Patel RP, Tarpey MM, Batinic-Haberle I, White CR, Freeman BA. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Procedings of the National Academy of Sciences of the United States of America. 2001;98:15215–15220. doi: 10.1073/pnas.221292098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas SK, Marcolina MJ. Hyperhemolysis during the evolution of uncomplicated acute painful episodes in patients with sickle cell anemia. Transfusion. 2006;46:105–110. doi: 10.1111/j.1537-2995.2006.00679.x. [DOI] [PubMed] [Google Scholar]
- Blum A, Yeganeh S, Peleg A, Vigder F, Kryuger K, Khatib A, Khazim K, Dauerman H. Endothelial function in patients with sickle cell anemia during and after sickle cell crises. Journal of Thrombosis and Thrombolysis. 2005;19:83–86. doi: 10.1007/s11239-005-1377-7. [DOI] [PubMed] [Google Scholar]
- Castro O, Hoque M, Brown BD. Pulmonary hypertension in sickle cell disease: cardiac catheterization results and survival. Blood. 2003;101:1257–1261. doi: 10.1182/blood-2002-03-0948. [DOI] [PubMed] [Google Scholar]
- Charache S. Sickle cells and sudden death. Journal of Laboratory and Clinical Medicine. 1994;124:473–474. [PubMed] [Google Scholar]
- Chou R, DeLoughery TG. Recurrent thromboembolic disease following splenectomy for pyruvate kinase deficiency. American Journal of Hematology. 2001;67:197–199. doi: 10.1002/ajh.1107. [DOI] [PubMed] [Google Scholar]
- Du ZD, Roguin N, Milgram E, Saab K, Koren A. Pulmonary hypertension in patients with thalassemia major. American Heart Journal. 1997;134:532–537. doi: 10.1016/s0002-8703(97)70091-7. [DOI] [PubMed] [Google Scholar]
- Eberhardt RT, McMahon L, Duffy SJ, Steinberg MH, Perrine SP, Loscalzo J, Coffman JD, Vita JA. Sickle cell anemia is associated with reduced nitric oxide bioactivity in peripheral conduit and resistance vessels. American Journal of Hematology. 2003;74:104–111. doi: 10.1002/ajh.10387. [DOI] [PubMed] [Google Scholar]
- Ergul S, Brunson CY, Hutchinson J, Tawfik A, Kutlar A, Webb RC, Ergul A. Vasoactive factors in sickle cell disease: in vitro evidence for endothelin-1-mediated vasoconstriction. American Journal of Hematology. 2004;76:245–251. doi: 10.1002/ajh.20107. [DOI] [PubMed] [Google Scholar]
- Gladwin MT, Lancaster JR, Jr, Freeman BA, Schechter AN. Nitric oxide’s reactions with hemoglobin: a view through the SNO-storm. Nature Medicine. 2003a;9:496–500. doi: 10.1038/nm0503-496. [DOI] [PubMed] [Google Scholar]
- Gladwin MT, Schechter AN, Ognibene FP, Coles WA, Reiter CD, Schenke WH, Csako G, Waclawiw MA, Panza JA, Cannon RO., III Divergent nitric oxide bioavailability in men and women with sickle cell disease. Circulation. 2003b;107:271–278. doi: 10.1161/01.cir.0000044943.12533.a8. [DOI] [PubMed] [Google Scholar]
- Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, Brown B, Coles WA, Nichols JS, Ernst I, Hunter LA, Blackwelder WC, Schechter AN, Rodgers GP, Castro O, Ognibene FP. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. New England Journal of Medicine. 2004;350:886–895. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- Groves BM, Badesch D. Cardiac catheterization of patients with pulmonary hypertension. In: Peacock AJ, Rubin LJ, editors. Pulmonary Circulation: Disease and their Treatment. London: Arnold; 2004. pp. 121–131. [Google Scholar]
- Hammerman SI, Kourembanas S, Conca TJ, Tucci M, Brauer M, Farber HW. Endothelin-1 production during the acute chest syndrome in sickle cell disease. American Journal of Respiratory and Critical Care Medicine. 1997;156:280–285. doi: 10.1164/ajrccm.156.1.9611085. [DOI] [PubMed] [Google Scholar]
- Hassoun PM, Krishnan JA. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. New England Journal of Medicine. 2004;350:2521–2522. author reply 2521–2522. [PubMed] [Google Scholar]
- Heller PG, Grinberg AR, Lencioni M, Molina MM, Roncoroni AJ. Pulmonary hypertension in paroxysmal nocturnal hemoglobinuria. Chest. 1992;102:642–643. doi: 10.1378/chest.102.2.642. [DOI] [PubMed] [Google Scholar]
- Jardine DL, Laing AD. Delayed pulmonary hypertension following splenectomy for congenital spherocytosis. Internal Medicine Journal. 2004;34:214–216. doi: 10.1111/j.1444-0903.2004.00580.x. [DOI] [PubMed] [Google Scholar]
- Jubelirer SJ. Primary pulmonary hypertension. Its association with microangiopathic hemolytic anemia and thrombocytopenia. Archives of Internal Medicine. 1991;151:1221–1223. doi: 10.1001/archinte.151.6.1221. [DOI] [PubMed] [Google Scholar]
- Kato GJ, McGowan V, Machado RF, Little JA, Taylor JG, VI, Morris CR, Nichols J, Wang X, Poljakovic M, Morris M, Jr, Gladwin MT. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension and death in patients with sickle cell disease. Blood. 2006;107:2279–2285. doi: 10.1182/blood-2005-06-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul DK, Liu XD, Chang HY, Nagel RL, Fabry ME. Effect of fetal hemoglobin on microvascular regulation in sickle transgenic-knockout mice. Journal of Clinical Investigation. 2004;114:1136–1145. doi: 10.1172/JCI21633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klings ES, Farber HW. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. New England Journal of Medicine. 2004;350:2521–2522. doi: 10.1056/NEJM200406103502418. author reply 2521–2522. [DOI] [PubMed] [Google Scholar]
- Labrune P, Zittoun J, Duvaltier I, Trioche P, Marquet J, Niaudet P, Odievre M. Haemolytic uraemic syndrome and pulmonary hypertension in a patient with methionine synthase deficiency. European Journal of Pediatrics. 1999;158:734–739. doi: 10.1007/s004310051190. [DOI] [PubMed] [Google Scholar]
- Minneci PC, Deans KJ, Zhi H, Yuen PS, Star RA, Banks SM, Schechter AN, Natanson C, Gladwin MT, Solomon SB. Hemolysis-associated endothelial dysfunction mediated by accelerated NO inactivation by decompartmentalized oxyhemoglobin. Journal of Clinical Investigation. 2005;115:3409–3414. doi: 10.1172/JCI25040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CR, Morris SM, Jr, Hagar W, Van Warmerdam J, Claster S, Kepka-Lenhart D, Machado L, Kuypers FA, Vichinsky EP. Arginine therapy: a new treatment for pulmonary hypertension in sickle cell disease? American Journal of Respiratory and Critical Care Medicine. 2003;168:63–69. doi: 10.1164/rccm.200208-967OC. [DOI] [PubMed] [Google Scholar]
- Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, Hazen SL, Vichinsky EP, Morris SM, Jr, Gladwin MT. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. Journal of the American Medical Association. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann HN, Diggs LW, Barreras L, Williams BJ. Plasma hemoglobin and hemoglobin fractions in sickle cell crisis. American Journal of Clinical Pathology. 1971;56:137–147. doi: 10.1093/ajcp/56.2.137. [DOI] [PubMed] [Google Scholar]
- Neely CL, Wajima T, Kraus AP, Diggs LW, Barreras L. Lactic acid dehydrogenase activity and plasma hemoglobin elevations in sickle cell disease. American Journal of Clinical Pathology. 1969;52:167–169. doi: 10.1093/ajcp/52.2.167. [DOI] [PubMed] [Google Scholar]
- Nolan VG, Wyszynski DF, Farrer LA, Steinberg MH. Hemolysis associated priapism in sickle cell disease. Blood. 2005;106:3264–3267. doi: 10.1182/blood-2005-04-1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfrey NA, Moore W, Hutchins GM. Is pain crisis a cause of death in sickle cell disease? American Journal of Clinical Pathology. 1985;84:209–212. doi: 10.1093/ajcp/84.2.209. [DOI] [PubMed] [Google Scholar]
- Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease. Life expectancy and risk factors for early death. New England Journal of Medicine. 1994;330:1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- Reiter CD, Gladwin MT. An emerging role for nitric oxide in sickle cell disease vascular homeostasis and therapy. Current Opinion in Hematology. 2003;10:99–107. doi: 10.1097/00062752-200303000-00001. [DOI] [PubMed] [Google Scholar]
- Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nature Medicine. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA. 2005;293:1653–1662. doi: 10.1001/jama.293.13.1653. [DOI] [PubMed] [Google Scholar]
- Rybicki AC, Benjamin LJ. Increased levels of endothelin-1 in plasma of sickle cell anemia patients. Blood. 1998;92:2594–2596. [PubMed] [Google Scholar]
- Schechter AN, Gladwin MT. Hemoglobin and the paracrine and endocrine functions of nitric oxide. New England Journal of Medicine. 2003;348:1483–1485. doi: 10.1056/NEJMcibr023045. [DOI] [PubMed] [Google Scholar]
- Schnog JJ, Jager EH, van der Dijs FP, Duits AJ, Moshage H, Muskiet FD, Muskiet FA. Evidence for a metabolic shift of arginine metabolism in sickle cell disease. Annals of Hematology. 2004;83:371–375. doi: 10.1007/s00277-004-0856-9. [DOI] [PubMed] [Google Scholar]
- Sutton LL, Castro O, Cross DJ, Spencer JE, Lewis JF. Pulmonary hypertension in sickle cell disease. American Journal of Cardiology. 1994;74:626–628. doi: 10.1016/0002-9149(94)90760-9. [DOI] [PubMed] [Google Scholar]
- Verresen D, De Backer W, Van Meerbeeck J, Neetens I, Van Marck E, Vermeire P. Spherocytosis and pulmonary hypertension coincidental occurrence or causal relationship? European Respiratory Journal. 1991;4:629–631. [PubMed] [Google Scholar]
- Wang X, Tanus-Santos JE, Reiter CD, Dejam A, Shiva S, Smith RD, Hogg N, Gladwin MT. Biological activity of nitric oxide in the plasmatic compartment. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11477–11482. doi: 10.1073/pnas.0402201101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss BM, Hess OM. Pulmonary vascular disease and pregnancy: current controversies, management strategies, and perspectives. European Heart Journal. 2000;21:104–115. doi: 10.1053/euhj.1999.1701. [DOI] [PubMed] [Google Scholar]