

Abstract
Metabotropic glutamate receptors are expressed throughout the nervous system, but their function as well as their ability to promote neuronal survival rests heavily upon the intracellular mechanisms governed by this family of G-proteins. In this regard, we examined one of the primary pathways that can oversee cell survival, namely protein kinase B (Akt1), and its functional integration with some of its substrates that may work in concert with group I metabotropic glutamate receptor (mGluRI) activation to protect primary hippocampal neurons during oxidative stress. We demonstrate that neuroprotection against free radical injury through mGluRI activation with DHPG requires the activation of Akt1, since loss of Akt1 activity assessed through its GSK-3α/β substrate by pharmacological blockade of the phosphatidylinositide-3-kinase pathway or the gene silencing of Akt1 expression prevents neuronal protection during mGluRI activation. Closely coupled to the robust neuroprotection by mGluRI activation are the inhibitory phosphorylation and prevention of caspase 3 cleavage of the Forkhead transcription factor FOXO3a, the down-regulation of Bim expression, and the protection of β-catenin by Akt1 against phosphorylation and degradation to promote its translocation from the cytoplasm to the nucleus and allow it to assist with a “pro-survival” cellular program. Further insight into the cellular mechanisms that determine neuronal protection by the metabotropic glutamate system will foster the successful therapeutic development of mGluRs for neurodegenerative disorders.
Keywords: Akt, β-catenin, Bim, forkhead, FOXO3a, glycogen synthase kinase-3β, ischemia, metabotropic glutamate receptor, nitric oxide, protein kinase B, apoptosis, oxidative stress, gene silencing, hippocampal neurons, phosphatidylinositide-3-kinase, siRNA
INTRODUCTION
Metabotropic glutamate receptors are expressed throughout the mammalian nervous system and participate in a variety of functions that include the processing of cognitive, sensory, motor, and olfactory information. In hippocampal neurons, group I metabotropic receptors (mGluRIs) are involved in normal physiologic functions that modulate calcium currents (Dietrich, D, et al., 1997, Maiese, K, et al., 1999, Paschen, W, 2004, Weber, JT, 2004) and direct memory imprinting through both long-term potentiation and long-term depression (Chen, J, et al., 2000, Manahan-Vaughan, D, 1997). The mGluRIs also play a significant protective role in a broad range of disease states that range from the prevention of the degeneration of motor neurons in amyotrophic lateral sclerosis patients (Valerio, A, et al., 2002), the facilitation of dopamine release from nigrostriatal terminals during Parkinson's disease (Campusano, JM, et al., 2002, Shimazoe, T, et al., 2002), and the inhibition of toxic amyloid formation and deposition in the brain (Lee, RK, et al., 1995, Ulus, IH and Wurtman, RJ, 1997).
At the cellular level, mGluRI activation can block neuronal injury associated with apoptosis (Baskys, A, et al., 2005, Blandini, F, et al., 2004, Chong, ZZ, et al., 2005a, Lin, SH and Maiese, K, 2001), modulate endonuclease activation (Vincent, AM, et al., 1999), and maintain cellular membrane asymmetry to avert the phagocytic demise of neurons during inflammatory cell activation (Chong, ZZ, et al., 2005a, Vincent, AM and Maiese, K, 2000). Protection by mGluRI occurs at or below the level of free radical generation and oxidative stress (Maiese, K, et al., 1996, Sagara, Y and Schubert, D, 1998, Vincent, AM, et al., 1997). Yet, the pathways that determine neuronal protection, especially in ischemic sensitive hippocampal neurons, are complex in nature and require elucidation for the successful development of therapeutic targets that employ the metabotropic system (Baskys, A, et al., 2005, Maiese, K, et al., 2005a).
Given these objectives, the cellular pathways that are regulated by protein kinase B, also known as Akt, become a prime consideration to uncover the pathways responsible for mGluRI neuroprotection. Akt is a serine/threonine kinase with three family members (Akt1, Akt2, and Akt3) that are ubiquitously expressed in mammals. Of the three family members, Akt1 is highly expressed in brain and is a central modulator for cell survival (Chong, ZZ, et al., 2005b) that can prevent cell injury during ischemic insults and free radical exposure (Chong, ZZ, et al., 2002, Maiese, K, et al., 2005b). The phosphorylation of two major residues, Thr308 and Ser473, are needed for the activation of Akt1, but it is the phosphorylation of Ser473 that is considered to be a critical component for its complete activation (Bellacosa, A, et al., 1998).
Interestingly, Akt1 does not function in isolation to foster cell survival, but relies upon an intimate relationship with several of its substrates. In particular, Akt1 can inhibit the activity of the “pro-apoptotic” Forkhead family member FOXO3a through preferential phosphorylation of FOXO3a at the residue of Ser253 (Brunet, A, et al., 1999). In addition to the direct post-translational modulation by Akt1, activity of FOXO3a also is controlled through a parallel and potential regulatory loop pathway that requires degradation by caspase 3 (Chong, ZZ, et al., 2004b), since FOXO3a has been shown to be a substrate for caspase 3-like proteases at the consensus sequence DELD304A (Charvet, C, et al., 2003). Downstream in this cascade is the BH3-only family member Bim that mediates cellular injury through Bax-mediated cytochrome c release from mitochondria, but is ultimately under the regulation of FOXO3a (Gilley, J, et al., 2003).
In addition to FOXO3a, Akt1 is closely aligned with the control of another critical “pro-survival” protein, namely β-catenin, by modulating the activity of glycogen synthase kinase-3β (GSK-3β). Without the influence of Akt1 to phosphorylate and inactivate GSK-3β, GSK-3β activity can promote cell injury (Crowder, RJ and Freeman, RS, 2000), increase caspase 3 activity (Koh, SH, et al., 2003), and is involved in neurodegenerative disorders, such as Alzheimer's disease (Chong, ZZ, et al., 2005e). More recently, the mGluR system has been shown to prevent amyloid toxicity in a hippocampal slice preparation through inhibition of GSK-3β activity (Liu, F, et al., 2005). Interestingly, GSK-3β phosphorylates β-catenin to lead to the degradation of β-catenin through ubiquination (Yamamoto, H, et al., 1999). In many cell systems, inhibition of cell injury can be regulated through the stabilization and activation of β-catenin while loss of β-catenin increases vulnerability to apoptotic injury (Chen, S, et al., 2001, Li, F, et al., 2006, Wu, WB, et al., 2003).
Here we illustrate a number of novel pathways for neuronal protection by mGluRIs that focus upon Akt1, but depend upon the concerted regulation of key substrates of Akt1. In primary hippocampal neurons, protection by mGluRI activation during free radical nitric oxide (NO) exposure requires the activation of Akt1, since loss of Akt1 activity by pharmacological blockade of the phosphatidylinositide-3-kinase (PI 3-K) pathway or the gene silencing of Akt1 expression prevents neuronal protection during mGluRI activation. The neuroprotective capacity of mGluRI activation is governed at subsequent levels that employ the inhibitory phosphorylation of FOXO3a, maintain the integrity of phosphorylated FOXO3a, and prevent Bim expression. Intimately linked to mGluRI protection is the preservation of β-catenin by Akt1 against phosphorylation and degradation to promote its translocation from the cytoplasm to the nucleus and allow it to block cell injury programs.
MATERIALS AND METHODS
Primary Hippocampal Neuronal Cultures
Briefly per our prior protocols (Chong, ZZ, et al., 2006, Chong, ZZ, et al., 2004b), the hippocampi were obtained from E-19 Sprague-Dawley rat pups and incubated in Hanks' balanced salt solution (HBBS) supplemented with 1 mM sodium pyruvate and 10 mM HEPES buffer solution (Invitrogen, Carlsbad, CA). The neurons were isolated by trituration with 10 repetitions and then centrifuged for 2 minutes at 200 g. The cells were washed in growth medium (Leibovitz's L-15 medium, Invitrogen, Carlsbad, CA) containing 6% sterile rat serum (ICN, Aurora, OH), 150 mM NaHCO3, 2.25 mg/ml of transferrin, 2.5 μg/ml of insulin, 10 nM progesterone, 90 μM putrescine, 15 nM selenium, 35 mM glucose, 1 mM L-glutamine, penicillin and streptomycin (50 μg/ml), and vitamins. The dissociated neurons were plated at a density of ∼1.5 ×103 cells/mm2 in 35 mm polylysine/laminin-coated plates and were maintained in growth medium at 37 °C in a humidified atmosphere of 5% CO2 and 95% room air for 10-14 days.
Experimental Treatments
NO administration was performed by replacing the culture media with media containing 6-(2-hydroxy-1-methyl-2-nitrosohydrazino)-N-methyl-1-hexanamine (NOC-9, 300 μM) (Calbiochem, San Diego, CA) or sodium nitroprusside (SNP, 300 μM) (Sigma, St. Louis, MO) per the experimental paradigm. More than one NO generator was used as a control to demonstrate that cells were responding to NO rather than to other by-products of these agents (Maiese, K and Vincent, AM, 2000). Data for the two NO donors was combined since no significant differences in cell injury were present among the agents. The mGluRI agonist (S)-3,5-dihydroxyphenylglycine (DHPG) was applied to neuronal cultures 1 hour prior to NO exposure and the treatment was continuous. For phosphatidylinositide-3-kinase (PI 3-K) inhibition, wortmannin (W) (Calbiochem, La Jolla, CA) or LY294002 (Tocris, Ellisville, MO) were added directly to the cultures 1 hour prior to NO application and treatment for PI 3-K inhibition was continuous.
Assessment of Akt Kinase Activity
Per our prior work (Chong, ZZ, et al., 2004a), Akt1 activity was determined by using a commercially available nonradioactive Akt1 kinase assay kit with GSK-3β fusion protein. Cells were lysed in ice with 150 μl of lysis buffer containing 1% Triton X-100, 10% glycerol, 137 mM NaCl, 20 mM Tris-HCl (pH 7.5), 2 μg/ml aprotinin, 2 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride, 20 mM NaF, 1 mM Na2PPi, and 1 mM Na3VO4. Equal amounts of lysates (200 μg) were pre-cleared by centrifugation and pre-absorbed with protein A-protein G (1:1) agarose slurry. Immunoprecipitation was carried out over night using the immobilized anti-Akt1G1 monoclonal antibody (Cell Signaling Technology, Beverly, MA) cross-linked to agarose. Immunoprecipitates were washed three times with lysis buffer and twice with Akt kinase buffer (20 mM HEPES, pH 7.4, 10 mM MgCl2, 10 mM MnCl2). Kinase assays were performed for 30 min at 30°C under continuous agitation in kinase buffer containing 200 μM ATP and 1 μg of GSK-3 fusion protein according to the manufacturer's instructions (Cell Signaling Technology, Beverly, MA). Samples were analyzed by Western blot analysis using 12.5% SDSpolyacrylamide gel and rabbit antibody against p-GSK-3α/β (Cell Signaling Technology, Beverly, MA). Data for the kinase activity are expressed as percentage of control activity.
Neuronal cell Survival
Neuronal injury was determined by bright field microscopy using a 0.4% trypan blue dye exclusion method 24 hours following NO per our previous protocols (Chong, ZZ, et al., 2003, Chong, ZZ, et al., 2005c). Mean survival was determined by counting 8 non-overlapping fields with each containing approximately 10-20 cells (viable + non-viable).
Phosphorylation of FOXO3a and β-catenin, Expression of Bim
Cells were homogenized and following protein determination, each sample (50 μg/lane) was then subjected to 7.5% (FOXO3a, β-catenin) or 12.5% (Bim) SDS-polyacrylamide gel electrophoresis. After transfer, the membranes were incubated with a goat polyclonal antibody against total FOXO3a (1: 100), a goat polyclonal antibody against phosphorylated FOXO3a (p-FOXO3a, Ser253, 1: 100) (Santa Cruz Biotechnologies, Santa Cruz, CA), a rabbit antibody against Bim (1: 100) (Calbiochem, San Diego, CA), or a rabbit antibody against phosphorylated-β-catenin (1:1000) (p-β-catenin, Ser33/37/Thr41, Cell Signaling, Beverly, MA). After washing, the membranes were incubated with a horse-radish peroxidase (HRP) conjugated secondary antibody (goat anti-rabbit IgG, 1:1500 or rabbit anti-goat IgG, 1:2000) (Pierce, Rockford, IL). The antibody-reactive bands were revealed by chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ).
Immunocytochemistry for β-catenin
Cells were fixed for either single or double staining with 4% paraformaldehyde blocked with 1.5% normal horse serum. For specific staining of β-catenin, cells were incubated with rabbit anti-β-catenin (1:100, Cell Signaling Technology, Beverly, MA), then with biotinylated anti-rabbit IgG (1:50) followed by Texas Red streptavidin (1:50). Cells were washed in PBS. The cells were then stained with DAPI (Sigma, St. Louis, MA) for nuclear identification. Fluorescence imaging used the wavelengths of 565 nm (red) and 400 nm (DAPI).
Statistical Analysis
For each experiment, the mean and standard error were determined. Statistical differences between groups were assessed by means of analysis of variance (ANOVA) from 4 to 6 replicate experiments with the post-hoc Student's t-test. Statistical significance was considered at p<0.05.
RESULTS
mGluRIs Significantly Increase Akt1 Activity During Free Radical Exposure
Direct assessment of Akt1 activity was performed through its specific GSK-3α/β substrate measured through the expression of phosphorylated (p)-GSK-3α/β (Fig. 1A and 1B) 6 hours following NO exposure. DHPG (750 μM) was applied to neuronal cultures 1 hour prior to NO exposure (NOC-9, 300 μM). In Fig. 1A and 1B, both NO and DHPG independently increased the activity of the p-GSK-3α/β substrate, but DHPG, either alone or during NO exposure, enhanced the activity of Akt1 and the expression of p-GSK-3α/β to a greater degree than application of NO alone. This increased expression of p-GSK-3α/β activity was blocked by the inhibitors of PI 3-K wortmannin (W, 500 nM), which forms a covalent link with the lysine residue of PI 3-K (Wymann, MP, et al., 1996), and LY294002 (LY, 10 μM), which reversibly competes for ATP binding (Vlahos, CJ, et al., 1994) (Fig. 1A).
Fig. (1). mGluRIs significantly increase Akt1 activity during free radical exposure, but PI 3-K blockade or gene silencing eliminates Akt1 activity.

(A and B) Neuronal protein extracts (50 μg/lane) were immunoblotted with anti-phosphorylated -GSK-3α/β (p-GSK-3α/β) following incubation with the Akt1 substrate GSK-3α/β. Exposure to DHPG (750 μg/ml) or NO (NOC-9 (shown) or SNP, 300 μM) significantly increased p-GSK-3α/β expression. Application of the inhibitors of PI 3-K wortmannin (W, 500 nM) or LY294002 (LY, 10 μM) or gene silencing of Akt1 expression were sufficient to block the expression of p-GSK-3α/β in the presence of DHPG during NO. In (A) and (B), band density was performed using the public domain NIH Image program (developed at the U.S. National Institutes of Health and available at http://rsb.info.nih.gov/nih-image/) (*P<0.01 vs. control; †P<0.01 vs. DHPG/NO or DHPG). In all cases, control = untreated neurons.
Gene Silencing Blocks Akt1 Activation by mGluRI
Since wortmannin and LY294002 function at the level of PI 3-K to modulate Akt1 activity, we next examined whether direct elimination of Akt1 expression through the specific gene silencing of Akt1 with siRNA could also prevent mGluRI activation of Akt1. Neurons were transfected with Akt1 siRNA and activity of Akt1 was observed through pGSK-3α/β substrate analysis (Fig. 1B). Gene silencing of Akt1 during administration of DHPG (750 μM) or during DHPG (750 μM) with NO exposure prevents phosphorylation of p-GSK-3α/β.
Activation of mGluRI for Neuronal Protection Requires the PI 3-K Pathway and Akt1
After exposure to NO (NOC-9 or SNP, 300 μM), neurons have membrane injury and were permeable to trypan blue dye (Fig. 2A). In contrast, application of DHPG (750 μM) at a concentration which a plateau of injury is achieved on survival curves for the mGluR agonists (Lin, SH and Maiese, K, 2001, Maiese, K, et al., 1999, Vincent, AM, et al., 1999) significantly reduced trypan blue staining (Fig. 2A). Neurons following DHPG administration increased neuronal survival during NO exposure from 38 ± 3% to 66 ± 4%. Yet, application of wortmannin (W, 500 nM) or LY294002 (LY, 10 μM) at concentrations that block activation of Akt1 during NO (Fig. 1A) significantly reduced the ability of DHPG to protect against NO toxicity, suggesting that DHPG requires activation of the PI 3-K and Akt to exert neuronal protection against free radical injury.
Fig. (2). Blockade of the PI 3-K pathway or gene silencing of Akt1 eliminates neuroprotection during mGluRI activation.
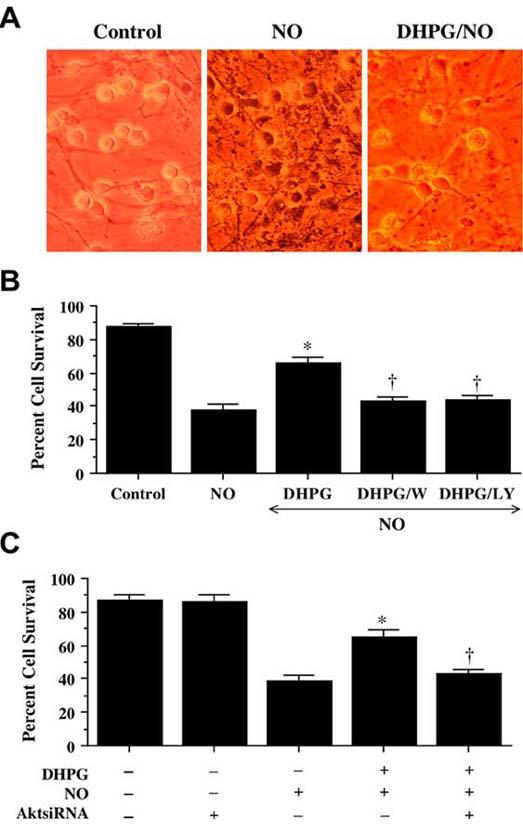
(A) Neuronal survival was assessed by trypan blue dye exclusion 24 hours following exposure to a NO donor (NOC-9 or SNP, 300 μM). Group I mGluR agonist (DHPG, 750 μM) was applied 1 hour prior to NO exposure. Representative images illustrated that cultures with NO exposure were stained with trypan blue, but DHPG significantly reduced trypan blue uptake. (B) Application of DHPG (750 μM) alone increased neuronal survival during NO exposure (NOC-9 or SNP, 300 μM), but protection was lost with application of DHPG combined with inhibitors of PI 3-K wortmannin (W, 500 nM) or LY294002 (LY, 10 μM) (*P<0.01 vs. NO; †P<0.01 vs. DHPG/NO). (C) Administration of DHPG (750 μM) alone increased significantly neuronal survival during NO exposure (NOC-9 or SNP, 300 μM), but protection was abrogated during gene silencing with Akt1 siRNA (*P<0.01 vs. NO; †P<0.01 vs. DHPG/NO). In B and C, to simplify the figures, the results for the two NO donors were combined.
Furthermore, gene silencing of Akt1 during administration of DHPG (750 μM) with NO exposure blocks the ability of mGluRI to protect neurons. As shown in Fig. 2C, NO significantly decreases survival from 87 ± 2% to 39 ± 4% (p<0.01). As expected, DHPG (750 μM) prevented cell injury assessed by trypan blue staining (Fig. 2C), but this protection was lost with gene silencing of Akt1 with survival decreasing to 43 ± 3% (p<0.01) (Fig. 2C), illustrating that activation of Akt1 is essential for DHPG to prevent free radical induced neuronal degeneration. Of note, transfection with siRNA for Akt1 was not toxic to neurons.
Activation of mGluRI Modulates Inhibitory Phosphorylation of FOXO3a and Prevents Its Destruction Though Caspase 3 Pathway
Western blot assay was performed for phosphorylated FOXO3a (p-FOXO3a) at the preferential phosphorylation site for Akt of Ser253 (Brunet, A, et al., 1999) as well as for the expression of total FOXO3a at 6 and 12 hours after NO (NOC-9, 300 μM) exposure (Fig. 3A and 3B). Exposure to NO significantly increased the expression of p-FOXO3a over a 6 hour period when compared to untreated control neurons (Fig. 3A and 3B). After 6 hours following NO exposure, expression of p-FOXO3a and total FOXO3a was lost and was similar to control levels of expression (Fig. 3A and 3B). In contrast, DHPG (750 μM) alone or in combination with NO exposure maintained the expression of p-FOXO3a and total FOXO3a over a 12 hour course, suggesting that DHPG (750 μM) can block destruction of p-FOXO3a (Fig. 3A and 3B).
Fig. (3). Activation of mGluRI results in the inhibitory phosphorylation of FOXO3a and FOXO3a integrity is maintain with DHPG or caspase 3 inhibition during NO.

(A, B, and C) Neuronal protein extracts (50 μg/lane) were immunoblotted with anti-phosphorylated-FOXO3a (p-FOXO3a, Ser253) (A, C) or anti-total FOXO3a (B) at 6 and 12 hours (h) following with 1 hour pre-treatment with DHPG (750 μM), DEVD (50 μM), LEHD (50μM), NO (NOC-9 (shown) or SNP, 300 μM) alone, or combined DHPG (750 μM) with NO. NO led to the loss of p-FOXO3a (A, C) and the loss of total FOXO3a (B) with 12 hours, but exposure to DHPG (750 μM) or DEVD (50 μM for caspase 3 inhibition maintained p-FOXO3a (A, C) and total FOXO3a (B) at 6 and 12 hours following NO. With caspase 9 inhibition (LEHD, 50 μM), minimal p-FOXO3a expression is maintained at 12 hours which may reflect downstream caspase 3 inhibition. In (A, B, and C), band density was performed using the public domain NIH Image program (developed at the U.S. National Institutes of Health and available at http://rsb.info.nih.gov/nih-image/) (*P<0.01 vs. control; †P<0.01 vs. NO 6 h). In all cases, control = untreated neurons.
Since mGluR activation has been shown to prevent the induction of caspase 3 activity during cell injury (Lin, SH and Maiese, K, 2001, Maiese, K, et al., 2005a, Maiese, K, et al., 2000) and that FOXO3a, has been shown to be a substrate for caspase 3-like proteases at the consensus sequence DELD304A (Charvet, C, et al., 2003), we examined whether inhibition of caspase 3 activity could prevent FOXO3a destruction (Fig. 3C). We employed the inhibition of caspase 9 as an alternative control since mGluRI activation also can block activity of this caspase pathway (Chong, ZZ, et al., 2005a). In Fig. 3C, western blot assay was performed for p-FOXO3a at 6 hours and 12 hours following NO (NOC-9, 300 μM) exposure during inhibition of caspase 3 and 9 - like activity. Consistent with FOXO3a containing a consensus sequence for caspase 3, inhibition of caspase 3 - like activity, prevented p-FOXO3a degradation in neurons 6 hours and 12 hours following NO exposure. In contrast, inhibition of caspase 9 - like activity produced significantly less expression of p-FOXO3a than did inhibition of caspase 3 - like activity, but the maintenance of some p-FOXO3a expression may be secondary to some reduction in caspase 3 - like activity during the blockade of caspase 9 - like activity, since the intrinsic caspase pathway is associated with the release of cytochrome c and subsequent activation of caspase 9 followed by activation of caspase 3 (Li, P, et al., 1997).
mGluRI Activation can Prevent Bim Expression During NO Exposure
FOXO3a may lead to cell injury through the regulation of Bim. Bim is a BH3-only family member and functions upstream of Bax-mediated cytochrome c release from the mitochondria (Chong, ZZ, et al., 2005d, Chong, ZZ, et al., 2005f, Doonan, F and Cotter, TG, 2004). Over-expression of FOXO3a has been reported to enhance Bim expression during cell stress such as trophic factor withdrawal and lead to cell injury that requires Bim activity (Gilley, J, et al., 2003). In addition, FOXO3a can activate the Bim promoter through two conserved FOXO binding sites (Gilley, J, et al., 2003) and promote cell injury (Sunters, A, et al., 2003).
We therefore examined the ability of mGluRI activation to alter Bim expression. Using western blot analysis, NO (NOC-9, 300 μM) exposure significantly increased the expression of Bim over a 6 and 12 hour period when compared to control cultures (Fig. 4). Yet, DHPG (750 μM) alone or in combination with NO exposure prevented Bim expression over the same 6 and 12 hour period, suggesting that modulation of Bim by mGluRI occurs upstream through inhibitory phosphorylation of FOXO3a by mGluRI (Fig. 3A and 3B).
Fig. (4). mGluRI prevents the expression of Bim.
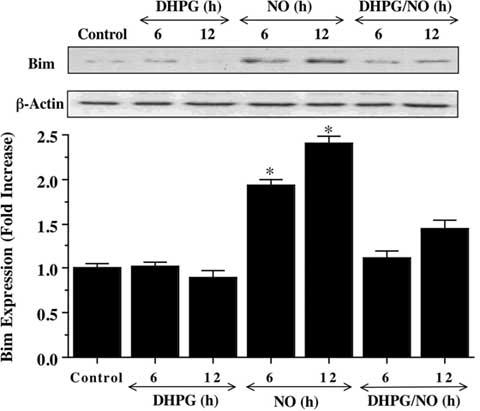
Neuronal protein extracts (50 μg/lane) were immunoblotted with anti-Bim at 6 and 12 hours (h) following with 1 hour pre-treatment with DHPG (750 μM), NO (NOC-9 (shown) or SNP, 300 μM) alone, or combined DHPG (750 μM) with NO. NO led to the rapid expression of Bim at 6 and 12 hours after NO exposure. In contrast, DHPG (750 μM) prevented Bim expression during this same time period. Band density was performed using the public domain NIH Image program (developed at the U.S. National Institutes of Health and available at http://rsb.info.nih.gov/nih-image/) (*P<0.01 vs. control). In all cases, control = untreated neurons.
Activation of mGluRI Uses a Downstream Pathway of Akt1 that Involves Maintenance of the “Anti-apoptotic” Protein β-catenin and Its Nuclear Translocation
We next examined the ability of mGluRI to modulate β-catenin phosphorylation and intracellular trafficking, since β-catenin can be indirectly modulated by the Akt1 pathway through GSK-3β. DHPG (750 μM) either alone or in conjunction with NO exposure prevented the expression of phosphorylated β-catenin (p-β-catenin) 6 hours following NO exposure. In contrast, expression of p-β-catenin was increased during NO alone or during combined DHPG (750 μM) and NO during gene silencing of Akt1, illustrating that DHPG requires Akt1 to prevent the phosphorylation of β-catenin (Fig. 5).
Fig. (5). mGluRI uses Akt1 activation to protect against phosphorylation of β-catenin during NO exposure.

Neuronal protein extracts (50 μg/lane) were immunoblotted with anti-phosphorylated-β-catenin (p-β-catenin) at 6 hours following NO (NOC-9 (shown) or SNP, 300 μM) exposure. Transfection with Akt1 siRNA significantly reduced expression of p-β-catenin during DHPG (750 μM) alone or during NO exposure. Band density was performed using the public domain NIH Image program (developed at the U.S. National Institutes of Health and available at http://rsb.info.nih.gov/nih-image/) (*P<0.01 vs. NO).
Immunofluorescent staining for β-catenin and DAPI nuclear staining were used to follow the subcellular translocation of β-catenin in neurons during DHPG (750 μM) and NO exposure (Fig. 6, 7). During NO exposure alone, significant immunofluorescent staining for β-catenin in the cytoplasm of neurons with minimal nuclear staining is present. This is evident by the ability to detect significant DAPI nuclear staining in cells during merged NO images since prominent β-catenin staining is not present in the nucleus (Fig. 6). In contrast, with administration of DHPG (750 μM) with or without NO exposure, β-catenin is translocated to the nucleus with minimal cytoplasmic staining as shown by the light nuclear regions on merged images of β-catenin and DAPI. In Fig. 7, nuclear β-catenin staining is quantitated that reveals a high proportion of β-catenin in the nucleus during DHPG (750 μM) alone or in combination with NO, suggesting that DHPG (750 μM) fosters nuclear translocation of β-catenin to allow it to initiate an anti-apoptotic program.
Fig. (6). Activation of mGluRIs fosters the intracellular trafficking of β-catenin from the cytoplasm to the nucleus.
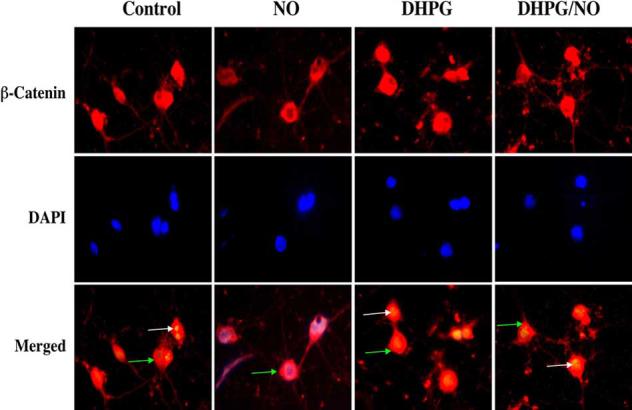
DHPG (750 μM) or combined DHPG (750 μM) with NO (NOC-9 (shown) or SNP, 300 μM) was followed at 6 hours with immunofluorescent staining for β-catenin (Texas-red). Nuclei of neurons were counterstained with DAPI. In merged images, cells with DHPG alone or combined DHPG and NO with white arrows show neuronal nuclei with strong β-catenin staining (red) and green arrows show neuronal cytoplasm with decreased β-catenin staining in contrast to cells with NO alone with green arrow demonstrating minimal β-catenin nuclear staining (yellow), illustrating the ability of DHPG to promote nuclear transfer of β-catenin.
Fig. (7). Quantitation of nuclear translocation for β-catenin during mGluRI activation.
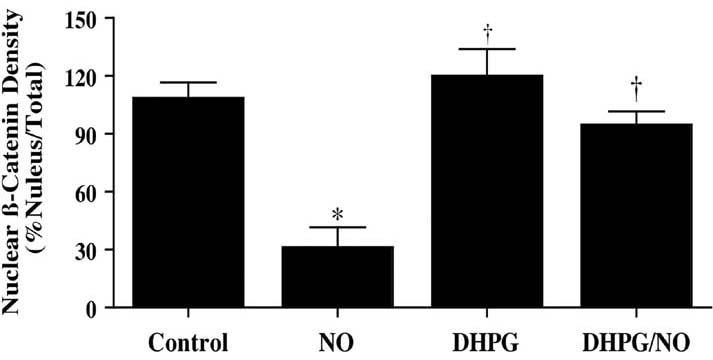
DHPG (750 μM) promotes β-catenin translocation from the cytoplasm to the nucleus during (NOC-9 (shown) or SNP, 300 μM). Intensity of β-catenin nuclear staining was performed using the public domain NIH Image program and control = untreated neurons (*P<0.01 vs. Control; †P<0.01 vs. NO).
DISCUSSION
The mGluR system plays a large role in a number disorders that can range from psychiatric disease to neurodegenerative disorders. For example, targeting the mGluR system for the treatment of bipolar disease has been proposed since these receptors may modulate signal transduction during affective disorders (Quiroz, JA, et al., 2004). Although some work suggests that the mGluR system may be detrimental such as during mGluRI activation and lead to prolonged epileptiform discharges (Wong, RK, et al., 2005), other studies support a protective role for mGluRs. During progressive disease states, such as amyotrophic lateral sclerosis, the mGluRIa receptor may offer endogenous cellular protection, since surviving motor neurons from the spinal cord of amyotrophic lateral sclerosis patients maintain mGluRIa at levels comparable to that from controls (Valerio, A, et al., 2002). If one also considers other neurodegenerative disorders, such as Alzheimer's disease that is characterized by extracellular plaques of amyloid-β peptide aggregates and intracellular neurofibrillary tangles (Chong, ZZ, et al., 2005e, Panchal, M, et al., 2004), a loss of metabotropic binding sites has been reported in patients with Alzheimer's disease (Dewar, D, et al., 1991). In addition, desensitization of mGluRIs in the frontal cortex of Alzheimer's disease patients have been described that correlate with disease progression (Albasanz, JL, et al., 2005) and the activation of mGluRIs have been shown to prevent amyloid deposition (Lee, RK, et al., 1995, Ulus, IH and Wurtman, RJ, 1997). Given the significant potential of the mGluRI system to modulate disease throughout the nervous system (Chong, ZZ, et al., 2005d, Maiese, K, et al., 2005a), understanding the cellular mechanisms controlled by mGluRIs may bring us closer to effectively employing the metabotropic system for therapeutic intervention.
The mGluRIs appear to rely heavily upon protein kinase B (Akt1) for the protection of neurons during free radical injury. The Akt1 pathway functions as a central modulator for cell survival (Chong, ZZ, et al., 2005b) and has been shown to prevent injury during ischemic injury and free radical exposure (Chong, ZZ, et al., 2002, Maiese, K, et al., 2005b). We demonstrate that activation of mGluRIs through DHPG increases the activation of Akt1 to a significantly greater degree than NO alone, suggesting that DHPG relies upon Akt1 activation to protect neurons. Yet, blockade of the PI 3-K pathway with application of wortmannin (W, 500 nM) or LY294002 (LY, 10 μM) at concentrations that prevent activation of Akt1 during NO exposure (Fig. 1A) or transfection of neurons with Akt1 siRNA to silence Akt1 expression prevents DHPG from protecting neurons during free radical injury (Fig. 2B, 2C).
Downstream from Akt1 in its cellular pathways are the transcription factors of the Forkhead family that are involved in several cellular processes such as cellular injury (Gilley, J, et al., 2003), cell cycle progression (Schmidt, M, et al., 2002), oxidative stress (Kops, GJ, et al., 2002), and the longevity of an organism (Taub, J, et al., 1999). Following its activation, Akt can phosphorylate the Forkhead family members resulting in their retention in the cytosol of cells and the subsequent blockade of their transcriptional activities that can prevent cell injury (Chong, ZZ, et al., 2005d). Our present work identifies a novel modulation by the mGluRI system that regulates the Forkhead family member FOXO3a to foster protection during oxidative stress (Fig. 3). We show that NO exposure can increase the phosphorylation of FOXO3a within 6 hours, but over the course of a 12 hour period the expression of phosphorylated and total FOXO3a is lost, suggesting its destruction. The subsequent proteolysis of phosphorylated and total FOXO3a results in amino-terminal (Nt) fragments that can precipitate cellular injury (Charvet, C, et al., 2003). In contrast, activation of the mGluRIs maintains inhibitory phosphorylation of FOXO3a over both a 6 and 12 hour period to block FOXO3a activation. Furthermore, prevention of phosphorylated and total FOXO3a degradation by mGluR1 activation may assist with neuronal protection by inhibiting the formation of Nt fragments.
Interestingly, the proteolytic processing of FOXO3a is linked to the induction of caspase 3 activity. FOXO3a has been shown to be a substrate for caspase 3-like proteases at the consensus sequence DELD304A (Charvet, C, et al., 2003). We show that blockade of caspase 3 - like activity prevents the destruction of phosphorylated FOXO3a in neurons during NO exposure. Yet, inhibition of caspase 9 - like activity does not offer a similar level of prevention for the degradation of phosphorylated FOXO3a. These results would be expected since FOXO3a has a consensus sequence only for caspase 3, but some protection by caspase 9 inhibition against FOXO3a degradation would be expected since caspase 3 activation is dependent upon initial caspase 9 activation (Li, P, et al., 1997). These results correlate well with prior reports that illustrate mGluRIs prevent caspase 3 and caspase 9 activity (Lin, SH and Maiese, K, 2001, Maiese, K, et al., 2005a, Maiese, K, et al., 2000), suggesting that the mGluRIs are able to block the degradation of FOXO3a as a result of the inhibition of these caspases.
Bim, a BH3-only family member, also is closely involved with the induction of cellular injury through the Bax-mediated release of cytochrome c, but the control of Bim may require the activity of FOXO3a (Gilley, J, et al., 2003). We illustrate that neuronal protection by mGluRI activation may require a parallel pathway that not only inhibits FOXO3a activity, but also may require the prevention of Bim expression and its subsequent activation. We illustrate that NO exposure can significantly increase Bim expression over a 6 and 12 hour period when compared to control cultures (Fig. 4). In contrast, activation of mGluRI with DHPG alone or in combination with NO exposure prevented Bim expression over this same time period, suggesting mGluRI controls both FOXO3a and Bim activity, but that control of Bim may stem from FOXO3a inhibition.
In addition to the Akt1 substrate FOXO3a, we also examined the ability of mGluRI and Akt1 to modulate the phosphorylation of β-catenin and its intracellular translocation from the cytoplasm to the cell nucleus. Akt1 can block activation of GSK-3β to prevent cell injury (Crowder, RJ and Freeman, RS, 2000), caspase 3 activity (Koh, SH, et al., 2003), and neurodegenerative disease (Chong, ZZ, et al., 2005e). The detrimental effects of GSK-3β activation may be executed through the phosphorylation of β-catenin to lead its degradation (Yamamoto, H, et al., 1999) and promote cell injury (Chen, S, et al., 2001, Li, F, et al., 2006, Wu, WB, et al., 2003). We demonstrate that phosphorylation of β-catenin was increased during NO exposure, but mGluRI activation with DHPG alone or in conjunction with NO exposure blocks phosphorylation of β-catenin 6 hours following NO exposure (Fig. 5). Furthermore, the ability of mGluR activation to modulation the phosphorylation and activity of β-catenin is tied to the presence of Akt1, since gene silencing of Akt1 expression leads to phosphorylation of β-catenin during NO exposure. Our work also suggests that blockade of β-catenin phosphorylation is associated with its translocation from the cytoplasm to the nucleus to allow it to assist with a known “pro-survival” cellular program. Exposure to NO alone maintains β-catenin in the cytoplasm of neurons, but administration of DHPG (750 μM) with or without NO exposure insures the translocation of β-catenin from the cytoplasm to the nucleus (Fig. 6, 7).
In summary, we illustrate that activation of mGluRI pathways can afford neuroprotection against oxidative stress and free radical injury, but these pathways are reliant upon closely integrated cellular mechanisms. In particular, Akt1 becomes a central regulator for the mGluRI pathway and its activation is critical for neuronal protection, since pharmacological inhibition of the PI 3-K pathway or gene silencing of Akt1 expression abrogate the protective capacity of mGluRI activation. In addition, the post-translational modification of the Akt1 substrate FOXO3a that requires inhibitory phosphorylation and prevention of its eventual degradation through caspase 3 activity appears necessary for mGluRI protection. These pathways of Akt1 and FOXO3a are closely aligned with the prevention of Bim expression and the maintenance of β-catenin integrity to insure its translocation from the cytoplasm to the nucleus to permit the induction of a “pro-survival” cellular program. Further knowledge of the metabotropic cellular pathways required for neuronal protection should continue to promote the fruitful development of these G-proteins for therapeutic management of disorders of the nervous system.
ACKNOWLEDGEMENTS
This research was supported by the following grants (KM): American Diabetes Association, American Heart Association (National), Bugher Foundation Award, Janssen Neuroscience Award, LEARN Foundation Award, MI Life Sciences Challenge Award, and NIH NIEHS (P30 ES06639).
REFERENCES
- Albasanz JL, Dalfo E, Ferrer I, Martin M. Impaired metabotropic glutamate receptor/phospholipase C signaling pathway in the cerebral cortex in Alzheimer's disease and dementia with Lewy bodies correlates with stage of Alzheimer's-disease-related changes. Neurobiol Dis. 2005;20(3):685–93. doi: 10.1016/j.nbd.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Baskys A, Fang L, Bayazitov I. Activation of neuroprotective pathways by metabotropic group I glutamate receptors: a potential target for drug discovery? Ann N Y Acad Sci. 2005;1053:55–73. doi: 10.1196/annals.1344.006. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, Tsichlis P. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17(3):313–25. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- Blandini F, Braunewell KH, Manahan-Vaughan D, Orzi F, Sarti P. Neurodegeneration and energy metabolism: from chemistry to clinics. Cell Death Differ. 2004;11(4):479–84. doi: 10.1038/sj.cdd.4401323. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96(6):857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Campusano JM, Abarca J, Forray MI, Gysling K, Bustos G. Modulation of dendritic release of dopamine by metabotropic glutamate receptors in rat substantia nigra. Biochem Pharmacol. 2002;63(7):1343–52. doi: 10.1016/s0006-2952(02)00870-5. [DOI] [PubMed] [Google Scholar]
- Charvet C, Alberti I, Luciano F, Jacquel A, Bernard A, Auberger P, Deckert M. Proteolytic regulation of Forkhead transcription factor FOXO3a by caspase-3-like proteases. Oncogene. 2003;22(29):4557–68. doi: 10.1038/sj.onc.1206778. [DOI] [PubMed] [Google Scholar]
- Chen J, Heinke B, Sandkuhler J. Activation of group I metabotropic glutamate receptors induces long- term depression at sensory synapses in superficial spinal dorsal horn. Neuropharmacology. 2000;39(12):2231–43. doi: 10.1016/s0028-3908(00)00084-8. [DOI] [PubMed] [Google Scholar]
- Chen S, Guttridge DC, You Z, Zhang Z, Fribley A, Mayo MW, Kitajewski J, Wang CY. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J Cell Biol. 2001;152(1):87–96. doi: 10.1083/jcb.152.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang J, Li F, Maiese K. mGluRI Targets Micro-glial Activation and Selectively Prevents Neuronal Cell Engulfment Through Akt and Caspase Dependent Pathways. Curr Neurovasc Res. 2005a;2(3):197–211. doi: 10.2174/1567202054368317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Akt1 drives endothelial cell membrane asymmetry and microglial activation through Bcl-x(L) and caspase 1, 3, and 9. Exp Cell Res. 2004a;296(2):196–207. doi: 10.1016/j.yexcr.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br J Pharmacol. 2003;138(6):1107–1118. doi: 10.1038/sj.bjp.0705161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation. 2002;106(23):2973–9. doi: 10.1161/01.cir.0000039103.58920.1f. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Activating Akt and the brain's resources to drive cellular survival and prevent inflammatory injury. Histol Histopathol. 2005b;20(1):299–315. doi: 10.14670/hh-20.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Attempted Cell Cycle Induction in Post-Mitotic Neurons Occurs in Early and Late Apoptotic Programs Through Rb, E2F1, and Caspase 3. Curr Neurovasc Res. 2006;3(1):25–39. doi: 10.2174/156720206775541741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Erythropoietin requires NFkappaB and its nuclear translocation to prevent early and late apoptotic neuronal injury during beta-amyloid toxicity. Curr Neurovasc Res. 2005c;2(5):387–99. doi: 10.2174/156720205774962683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: Novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005d;75(3):207–46. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Stress in the brain: novel cellular mechanisms of injury linked to Alzheimer's disease. Brain Res Brain Res Rev. 2005e;49(1):1–21. doi: 10.1016/j.brainresrev.2004.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li FQ, Maiese K. Employing new cellular therapeutic targets for Alzheimer's disease: A change for the better? Curr Neurovasc Res. 2005f;2(1):55–72. doi: 10.2174/1567202052773508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Maiese K. The NAD+ precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3a and mitochondrial membrane potential. J Cereb Blood Flow Metab. 2004b;24(7):728–43. doi: 10.1097/01.WCB.0000122746.72175.0E. [DOI] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS. Glycogen synthase kinase-3 beta activity is critical for neuronal death caused by inhibiting phosphatidylinositol 3-kinase or Akt but not for death caused by nerve growth factor withdrawal. J Biol Chem. 2000;275(44):34266–71. doi: 10.1074/jbc.M006160200. [DOI] [PubMed] [Google Scholar]
- Dewar D, Chalmers DT, Graham DI, McCulloch J. Glutamate metabotropic and AMPA binding sites are reduced in Alzheimer's disease: an autoradiographic study of the hippocampus. Brain Res. 1991;553(1):58–64. doi: 10.1016/0006-8993(91)90230-s. [DOI] [PubMed] [Google Scholar]
- Dietrich D, Beck H, Kral T, Clusmann H, Elger CE, Schramm J. Metabotropic glutamate receptors modulate synaptic transmission in the perforant path: pharmacology and localization of two distinct receptors. Brain Res. 1997;767(2):220–7. doi: 10.1016/s0006-8993(97)00579-9. [DOI] [PubMed] [Google Scholar]
- Doonan F, Cotter TG. Apoptosis: A potential therapeutic target for retinal degenerations. Curr Neurovasc Res. 2004;1(1):41–53. doi: 10.2174/1567202043480215. [DOI] [PubMed] [Google Scholar]
- Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol. 2003;162(4):613–22. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh SH, Kim SH, Kwon H, Park Y, Kim KS, Song CW, Kim J, Kim MH, Yu HJ, Henkel JS, Jung HK. Epigallocatechin gallate protects nerve growth factor differentiated PC12 cells from oxidative-radical-stress-induced apoptosis through its effect on phosphoinositide 3-kinase/Akt and glycogen synthase kinase-3. Brain Res Mol Brain Res. 2003;118(12):72–81. doi: 10.1016/j.molbrainres.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Fork-head transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419(6904):316–21. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- Lee RK, Wurtman RJ, Cox AJ, Nitsch RM. Amyloid precursor protein processing is stimulated by metabotropic glutamate receptors. Proc Natl Acad Sci USA. 1995;92(17):8083–7. doi: 10.1073/pnas.92.17.8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Winding through the Wnt pathway during cellular development and demise. Histol Histopathol. 2006;21(103124) doi: 10.14670/hh-21.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Lin SH, Maiese K. The metabotropic glutamate receptor system protects against ischemic free radical programmed cell death in rat brain endothelial cells. J Cereb Blood Flow Metab. 2001;21(3):262–75. doi: 10.1097/00004647-200103000-00010. [DOI] [PubMed] [Google Scholar]
- Liu F, Gong X, Zhang G, Marquis K, Reinhart P, Andree TH. The inhibition of glycogen synthase kinase 3beta by a metabotropic glutamate receptor 5 mediated pathway confers neuroprotection to Abeta peptides. J Neurochem. 2005;95(5):1363–72. doi: 10.1111/j.1471-4159.2005.03474.x. [DOI] [PubMed] [Google Scholar]
- Maiese K, Ahmad I, TenBroeke M, Gallant J. Metabotropic glutamate receptor subtypes independently modulate neuronal intracellular calcium. J Neurosci Res. 1999;55:472–485. doi: 10.1002/(SICI)1097-4547(19990215)55:4<472::AID-JNR7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ, Li F. Driving cellular plasticity and survival through the signal transduction pathways of metabotropic glutamate receptors. Curr Neurovasc Res. 2005a;2(5):425–46. doi: 10.2174/156720205774962692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Li F, Chong ZZ. New avenues of exploration for erythropoietin. JAMA. 2005b;293(1):90–5. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Swiriduk M, TenBroeke M. Cellular mechanisms of protection by metabotropic glutamate receptors during anoxia and nitric oxide toxicity. J Neurochem. 1996;66(6):2419–28. doi: 10.1046/j.1471-4159.1996.66062419.x. [DOI] [PubMed] [Google Scholar]
- Maiese K, Vincent A, Lin SH, Shaw T. Group I and Group III metabotropic glutamate receptor subtypes provide enhanced neuroprotection. J Neurosci Res. 2000;62(2):257–272. doi: 10.1002/1097-4547(20001015)62:2<257::AID-JNR10>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Maiese K, Vincent AM. Membrane asymmetry and DNA degradation: functionally distinct determinants of neuronal programmed cell death. J Neurosci Res. 2000;59(4):568–80. doi: 10.1002/(SICI)1097-4547(20000215)59:4<568::AID-JNR13>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Manahan-Vaughan D. Group 1 and 2 metabotropic glutamate receptors play differential roles in hippocampal long-term depression and long-term potentiation in freely moving rats. J Neurosci. 1997;17(9):3303–11. doi: 10.1523/JNEUROSCI.17-09-03303.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchal M, Rholam M, Brakch N. Abnormalities of peptide metabolism in Alzheimer disease. Curr Neurovasc Res. 2004;1(4):317–323. doi: 10.2174/1567202043362117. [DOI] [PubMed] [Google Scholar]
- Paschen W. Endoplasmic reticulum dysfunction in brain pathology: Critical role of protein synthesis. Curr Neurovasc Res. 2004;1(2):173–181. doi: 10.2174/1567202043480125. [DOI] [PubMed] [Google Scholar]
- Quiroz JA, Singh J, Gould TD, Denicoff KD, Zarate CA, Manji HK. Emerging experimental therapeutics for bipolar disorder: clues from the molecular pathophysiology. Mol Psychiatry. 2004;9(8):756–76. doi: 10.1038/sj.mp.4001521. [DOI] [PubMed] [Google Scholar]
- Sagara Y, Schubert D. The activation of metabotropic gluta-mate receptors protects nerve cells from oxidative stress. J Neurosci. 1998;18(17):6662–71. doi: 10.1523/JNEUROSCI.18-17-06662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Fernandez de Mattos S, van der Horst A, Klompmaker R, Kops GJ, Lam EW, Burgering BM, Medema RH. Cell cycle inhibition by FoxO forkhead transcription factors involves down-regulation of cyclin D. Mol Cell Biol. 2002;22(22):7842–52. doi: 10.1128/MCB.22.22.7842-7852.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazoe T, Doi Y, Arai I, Yoshimatsu A, Fukumoto T, Watanabe S. Both metabotropic glutamate I and II receptors mediate augmentation of dopamine release from the striatum in methamphetaminesensitized rats. Jpn J Pharmacol. 2002;89(1):85–8. doi: 10.1254/jjp.89.85. [DOI] [PubMed] [Google Scholar]
- Sunters A, Fernandez de Mattos S, Stahl M, Brosens JJ, Zoumpoulidou G, Saunders CA, Coffer PJ, Medema RH, Coombes RC, Lam EW. FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J Biol Chem. 2003;278(50):49795–805. doi: 10.1074/jbc.M309523200. [DOI] [PubMed] [Google Scholar]
- Taub J, Lau JF, Ma C, Hahn JH, Hoque R, Rothblatt J, Chalfie M. A cytosolic catalase is needed to extend adult lifespan in C. elegans daf-C and clk-1 mutants. Nature. 1999;399(6732):162–6. doi: 10.1038/20208. [DOI] [PubMed] [Google Scholar]
- Ulus IH, Wurtman RJ. Metabotropic glutamate receptor agonists increase release of soluble amyloid precursor protein derivatives from rat brain cortical and hippocampal slices. J Pharmacol Exp Ther. 1997;281(1):149–54. [PubMed] [Google Scholar]
- Valerio A, Ferrario M, Paterlini M, Liberini P, Moretto G, Cairns NJ, Pizzi M, Spano P. Spinal cord mGlu1a receptors: possible target for amyotrophic lateral sclerosis therapy. Pharmacol Biochem Behav. 2002;73(2):447–54. doi: 10.1016/s0091-3057(02)00835-3. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Maiese K. The metabotropic glutamate system promotes neuronal survival through distinct pathways of programmed cell death. Exp Neurol. 2000;166(1):65–82. doi: 10.1006/exnr.2000.7487. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Mohammad Y, Ahmad I, Greenberg R, Maiese K. Metabotropic glutamate receptors prevent nitric oxide-induced programmed cell death. J Neurosci Res. 1997;50(4):549–64. doi: 10.1002/(SICI)1097-4547(19971115)50:4<549::AID-JNR6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Vincent AM, TenBroeke M, Maiese K. Metabotropic gluta-mate receptors prevent programmed cell death through the modulation of neuronal endonuclease activity and intracellular pH. Exp Neurol. 1999;155(1):79–94. doi: 10.1006/exnr.1998.6966. [DOI] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269(7):5241–8. [PubMed] [Google Scholar]
- Weber JT. Calcium homeostasis following traumatic neuronal injury. Curr Neurovasc Res. 2004;1(2):151–171. doi: 10.2174/1567202043480134. [DOI] [PubMed] [Google Scholar]
- Wong RK, Bianchi R, Chuang SC, Merlin LR. Group I mGluR-induced Epileptogenesis: Distinct and Overlapping Roles of mGluR1 and mGluR5 and Implications for Antiepileptic Drug Design. Epilepsy Curr. 2005;5(2):63–8. doi: 10.1111/j.1535-7597.2005.05207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WB, Peng HC, Huang TF. Disintegrin causes proteolysis of beta-catenin and apoptosis of endothelial cells. Involvement of cell-cell and cell-ECM interactions in regulating cell viability. Exp Cell Res. 2003;286(1):115–27. doi: 10.1016/s0014-4827(03)00105-8. [DOI] [PubMed] [Google Scholar]
- Wymann MP, Bulgarelli-Leva G, Zvelebil MJ, Pirola L, Vanhaesebroeck B, Waterfield MD, Panayotou G. Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol Cell Biol. 1996;16(4):1722–33. doi: 10.1128/mcb.16.4.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Kishida S, Kishida M, Ikeda S, Takada S, Kikuchi A. Phosphorylation of axin, a Wnt signal negative regulator, by glycogen synthase kinase-3beta regulates its stability. J Biol Chem. 1999;274(16):10681–4. doi: 10.1074/jbc.274.16.10681. [DOI] [PubMed] [Google Scholar]