

Abstract
Poly(ADP-ribose)glycohydrolase (PARG) is the major enzyme capable of rapidly hydrolyzing poly(ADP-ribose)(PAR) formed by the diverse members of the PARP enzyme family. This study presents an alternative splice mechanism by which two novel PARG protein isoforms of 60 kDa and 55 kDa are expressed from the human PARG gene, termed hPARG60 and hPARG55, respectively. Homologous forms were found in the mouse (mPARG63 and mPARG58) supporting the hypothesis that expression of small PARG isoforms is conserved among mammals. A PARG protein of ∼60kDa has been described for decades but with its genetic basis unknown, it was hypothesized to be a product of posttranslational cleavage of larger PARG isoforms. While this is not excluded entirely, isolation and expression of cDNA clones from different sources of RNA indicate that alternative splicing leads to expression of a catalytically active hPARG60 in multiple cell compartments. A second enzyme, hPARG55 that can be expressed through alternative translation initiation from hPARG60 transcripts is strictly targeted to the mitochondria. Functional studies of a mitochondrial targeting signal (MTS) in PARG exon IV suggest that hPARG60 may be capable of shuttling between nucleus and mitochondria, which would be in line with a proposed function of PAR in genotoxic stress-dependent, nuclear-mitochondrial crosstalk.
Keywords: poly(ADP-ribose) glycohydrolase, poly(ADP-ribose), poly(ADP-ribose) polymerase, alternative splicing, mitochondrial targeting, PARG60, PARG55
Introduction
Poly(ADP-ribosyl)ation is a transient, dynamic and reversible posttranslational modification of proteins involved in a host of biological functions [1, 2] . Poly(ADP-ribose) (PAR) is formed by all PARP enzymes by cleavage of NAD+ into nicotinamide and ADP-ribose which becomes polymerized and transferred to specific target proteins. Genome-wide sequence analyses in human suggests the existence of up to 17 different genes encoding enzymes capable of synthesizing poly(ADP-ribose) (PAR) [3-5], and, so far, ten of them have been shown to be catalytically active. In contrast, only three human poly(ADP-ribose) glycohydrolase (PARG) proteins, PARG111, PARG102 and PARG99 have been described, which are all expressed from one single PARG gene by alternative splicing [6-8] . PARG proteins have been the only enzymes known to efficiently catalyze hydrolysis of poly(ADP-ribose) (PAR) produced by enzymes of the poly(ADP-ribose) polymerase (PARP) family [9] until recently a novel gene encoding a 39 kDa enzyme with low poly(ADP-ribose) glycohydrolase activity has been discovered [10-12] . However, the extent to which this enzyme, (ADP-ribose) hydrolase (ARH3), that is structurally unrelated to PARG, participates in the rapid, DNA damage dependent PAR turnover within the cell has not been elucidated yet.
PARG rapidly cleaves PAR to form monomeric ADP-ribose by both exoglycosidic and endoglycosidic activity. Following DNA damage, PAR metabolism is mediated by PARP-1 and PARP-2 in concert with PARG, and PAR turnover can be extremely rapid with a half-life of the biopolymer of only ∼ 1 minute [13]. Cellular accumulation of PAR beyond a short period of time has been shown to be deleterious [14, 15] and is involved in caspase-independent cell death regulation [16], reviewed in [17]. Alterations of PAR metabolism are causally involved in the pathogenesis of inflammatory [18] and autoimmune disease [19] , ischemia reperfusion injury in brain [20] , heart and intestine [21-23] , neuronal degeneration and neurotoxicity [24] , genetic and genomic instability (reviewed in [25]) and cancer (reviewed in [26, 27] ). Targeting of the poly(ADP-ribose) metabolic pathway has therefore become an important focus in research on cancer therapy (recent reviews in [28-30]), cardiovascular disease intervention [31-34] and a number of other pathophysiological conditions [35, 36] . Because of the potential of PAR metabolism as a drug target, identification of the key players appears to be crucial. While several PARP enzymes have been studied extensively, comparatively little is known about regulation of the catabolic arm of the pathway. However, the function of PARG in the PAR pathway is essential as deletion of the PARG gene results in a chronic, lethally toxic accumulation of cellular PAR [14, 37].
In contrast, a hypomorphic PARGΔ2-3/Δ2-3 mutant mouse that is devoid of exons II and III [18] was shown to be viable and expressing a residual PARG of 63 kDa by alternative splicing of the disrupted gene. This protein, designated mPARG63, is catalytically active and has unique properties [18]. In the present study we tested the hypothesis that the residual PARG proteins in the hypomorphic mouse may in fact be proteins that are naturally also expressed in wildtype mice as well as human. We show that mPARG63 is a naturally occuring protein that is expressed not only in the hypomorphic mutant but also in wildtype mouse tissues. More significantly, we demonstrate the existence of a human homolog, hPARG60, that is expressed in all tissues tested (skin, liver, testis, HeLa). At mRNA level, human PARG60 (hPARG60) is characterized as missing exon V, making it slightly smaller in size than the mouse homolog mPARG63. The data presented support the hypothesis that alternative translation initiation allows for expression of a protein with a consensus mitochondrial targeting sequence of the presequence type with strictly mitochondrial localization, termed hPARG55 (mPARG58 in mouse). The identification of hPARG60 (mPARG63) and hPARG55 (mPARG58) as novel PARG isoforms that associate with the mitochondria is of particular interest with regard to the emerging view that ADP-ribose metabolism may be involved in mitochondria-mediated, caspase independent cell death pathways of biomedical interest [15, 38] .
Materials and Methods
Cloning of human PARG cDNA by 5'RACE and construction of expression vectors
Human total RNA preparations from skin, liver and testis were purchased from Stratagene (La Jolla, CA). Mouse total RNA from liver and testis was also purchased or isolated from PARGΔ2-3/Δ2-3 mouse embryonic fibroblast culture, reversely transcribed and used in subsequent polymerase chain reaction (PCR) amplification reactions as described earlier [6, 7] . A mouse or human PARG gene specific primer which binds in the distal portion of the PARG 5'-UTR (human: hex0for: 5'-CGGAATTCGGGAAAGTGAACGAATCCCGAATCAAAGCGGCGC-3', mouse: mex0for: 5'-CCGATCTCGAGCCGAGTGGAAGCGGC-3') in combination with a specific reverse primer that binds in exon 6 (human: hex6rev: 5'-GTGCAGTCTGAATGAGCTCCCACCGGCTC-3' and mouse: mex6rev: 5'-GCAGTCTGAATGAGCTCCCACCTACTCCCTGC-3') was used to amplify the 5'end of the cDNA including exons I-VI. Full-length cDNA clones were generated by using the forward primers in combination with a primer which specifically binds to the end of the coding region in exon 18 (human: hPARG-exon18rev: 5'-GGACCGGTCCTCAGGTCCCTGTCCTTTGCCCTGAATG-3'), including the translation stop codon, or the corresponding mouse specific primer (mouse: mPARG-exon18rev: 5'-GGACCGGTCTCAGGTGCCTGCCTTCTGTGCC-3'). Purified PCR fragments were cloned into vector pcDNA3.1-TOPO/V5-His (Invitrogen, Carlsbad, CA). Plasmid pcDNA-hPARG60 was obtained by inserting a ∼1600 bp PCR product (Figure 1 A, band 3) into this recipient vector. After sequencing, all hPARG cDNA clones were subcloned into pEGFP-C1 (Invitrogen) to express a N-terminal in-frame enhanced green fluorescent protein (EGFP) fusions of the protein (e.g.: pEhPARG60). In order to facilitate detection of the C-terminal portions of PARGs, in-frame fusions with V5 tags were created resulting in pEhP60V5, pEhPARG102V5 and pEhPARG111V5. Plasmids encoding hypothetical proteins hPARG59 and hPARG53 (pEhPARG59V5 and pEhPARG53V5, respectively) were constructed by editing pEGFP-hPARG111V5 utilizing conventional restriction enzyme-based subcloning methods. Plasmids encoding V5-tagged hPARG protein variants without EGFP portion were phPARG111V5, phPARG102V5, phPARG60V5, phPARG59V5, phPARG55V5 and phPARG53V5. A catalytically inactive control hPARG56d cDNA was generated by site directed mutagenesis, that carries a glutamate (E) to asparagine (N) amino acid exchange of in the position corresponding to position 756 of the human PARG111 amino acid sequence, an amino acid considered critical for glycohydrolase function [39]. Luciferase reporter gene constructs were based on plasmid pGFL [40] encoding an EGFP-luciferase gene fusion and comprised pCMV-Ex1-GFL [7] and pCMV-MTS-GFL1. The latter was constructed by fusion of PARG cDNA exons IV through XVIII, to the translation initiation site of a β-galactosidase gene encoding its first 6 amino acids resulting in a fusion protein: “MSFLTE”/hPARG exons IVVI/EGFPluciferase. By removing the β-galactosidase portion of the cDNA, pCMV-MTS-GFL2 was obtained which has a predicted translation start at the beginning of the MTS in exon IV (“MRRMPRC…”).
Fig. 1. PCR amplification of PARG splice variants obtained from reversely transcribed human (A) and mouse (B) total RNA of diverse tissues.

Using a specific forward primer binding to the 5'UTR close to the transcription initiation site (hex0for for human samples, mex0for for the respective mouse samples) in combination with a reverse recognizing binding to exon 6 (hex6rev, mex6rev) or exon 18 (hex18rev, mex18rev) a number of bands were routinely amplified (1-5) and these PCR fragments were cloned, sequenced and used for expression studies. Band 1: full length hPARG111/mPARG110 cDNA. Band 2: double band containing hPARG102/hPARG99 and mPARG101/mPARG 98. Band 3: novel hPARG60/mPARG63 cDNAs. Band 4: hypomorphic PARGΔ2-3/Δ2-3 mouse, identical to Band 3 in the wt mouse. Band 5: mPARG52 with ORF starting in exon V that may be unique to the gene disrupted mouse. These analyses have been reproduced several times in independent experiments and representative data are shown. The sketch at the bottom indicates the position of the primers used in this experiment relative to the full-length PARG mRNA and the position of the translation stop codon.
Computational sequence analyses
Sequence alignments and searches for expressed sequence tages (EST) were performed using the NCBI software BLAST ([41] (http://www.ncbi.nlm.nih.gov/BLAST). Protein sequence analyses were done using PSORTII (http://psort.nibb.ac.jp) and MitoP2 ([42] . The MTS was further analyzed by wheel plots using a program by Webgenetics (http://webgenetics.com/cgibin/wg?form=wheel&ID=0). Binary protein sequence alignments of human and mouse PARG were obtained using ALION (Craig G. Nevill-Manning, Cecil N. Huang & Douglas L. Brutlag, (1997) “Pairwise protein sequence alignment using Needleman-Wunsch and Smith-Waterman algorithms,” personal communication, http://motif.stanford.edu/alion/).
Culture, Transfection and Treatment of Cells
HeLa cells and human embryonal kidney cells (HEK293) (both from ATCC) as well as 129S1/Sv and PARGΔ2-3/Δ2-3 mouse embryonal fibroblasts (MEF) [18] were cultured under standard cell culture conditions (37 deg C, 5% CO2) in DMEM (Sigma, St. Louis, MOa) supplemented with 10% fetal bovine serum (HyClone, Logan, UT). For overexpression of human PARG protein or reporter protein, HEK293 cells were transfected using FuGene6 transfection reagent (Roche, Indianapolis, IN) according to the manufacturer's protocol or utilizing conventional calcium phosphate transfection methods.
Microscopic preparations and immunofluorescence staining
Cells were fixed and indirect immunofluorescence staining was performed as described earlier [7] using the antibodies described further below. Cells were embedded in Vectashield mounting medium (Vector Laboratories, Burlingame,CA) containing 1 μg/ml DAPI (Sigma) or 8μM TOPRO3 (Invitrogen) as DNA counter stains, for conventional microscopy (Nikon Eclipse TE2000-U with a Coolsnap EZ camera system and Image-Pro Plus 5.1 software) or for confocal laser scan microscopy (Leica SP5 system), respectively.
Western blot analyses
of subcellular cell fractions and total cell lysate were performed using standard SDS-PAGE methods with 7.5% and 10% polyacrylamide gels. After electrotransfer to PVDF membranes (Millipore, Billerica, MA), PARG fusions and other proteins were visualized using antibodies as described below.
Antibodies
For analyses of human PARG, two different sets of 2 rabbits each were immunized (Sigma-Genosys, St. Louis, MO). For generation of an antiserum recognizing the N-terminus of PARG (ab 9882) a custom synthetic peptide (Sigma-Genosys) 310KNSCQDSEADEETSPG325 was used. Similarly, an antiserum directed against the C-terminus (designated ab 9877) was obtained using a synthetic peptide 848AYCGFLRPGVSSENL862 (Sigma-Genosys) which is similar to a peptide published before [43]. The antisera were affinity-purified using a kit (Pierce Biotechnology Inc. Rockford, IL). Western Blot analyses and immunostaining were performed using a mouse monoclonal anti-GFP antibody (Clontech, Mountain View, CA), a mouse monoclonal anti-V5 antibody (Invitrogen, Carlsbad, California), mouse monoclonal anti-histones (pan) (Chemicon, Temecula, CA) and anti-PARP1 (C2-10) antibodies (Biomol, Plymouth Meeting, PA) or a rabbit anti-MnSOD antiserum (Stressgen, Ann Arbor, MI) in combination with either fluorescein-coupled goat anti-rabbit antiserum, Cy3-coupled donkey anti-rabbit or TRITC-coupled donkey anti-rabbit secondary antibodies for immunofluorescence analyses (Jackson ImmunoResearch Laboratories, West Grove, PA). Immunoblotting was performed utilizing goat anti-mouse or goat anti-rabbit secondary antibodies coupled to horseradish peroxidase (Jackson ImmunoResearch Laboratories) for enhanced chemoluminescence (ECL) detection of the antibodies listed above.
Subcellular fractionation
nuclear and mitochondrial fractions were isolated from transfected cells using NucleiPURE Prep Nuclei Isolation and Mitochondria Isolation Kits (both Sigma, St. Louis, MO) in accordance with the manufacturer's instructions. The protein content of each fraction was determined using the Bradford Protein Assay (BIORAD, Hercules, CA).
PARG activity
was measured in lysates obtained from subcellular fractions or whole, transfected HEK293 cells [44]. Briefly, the assay is based on a timed in vitro digest of purified, 32P labeled PAR with a subsequent thin layer chromatography step that separates the undigested polymer from the ADP-ribose monomers liberated by PARG activity in the sample. Quantification is performed by scintillation counting of excised spots corresponding to undigested polymer and monomeric ADP-ribose only. Therefore, using this method, mainly exoglycosidic activity is measured. Specific activity of PARG was calculated after measuring protein contents of the corresponding samples. By splitting the samples, a portion of each sample was separated on 15% polyacrylamide gels to show all products formed by the reaction to obtain an estimate of the endoglycosidic activity that was present in the sample [45].
Results
Isolation of a cDNA encoding a novel hPARG60 isoform
PCR amplification of hPARG cDNA (Figure 1A) from reversely transcribed total RNA yielded numerous bands rather than a single product. In order to amplify specifically the 5'ends of cDNA derived from PARG transcripts, a forward primer in the 5' untranslated region (hex0for) in combination with a reverse primer in exon VI (hex6rev) was used (5'UTR, Fig. 1A, left hand side of the gel). These primers produced three bands corresponding to the cDNAs of hPARG111 (band 1, 1880 bp), hPARG102 (band 2, 1448- 1481 bp), as well as hPARG99 (also band 2, 1385 - 1381 bp, forming a double band with hPARG102) as confirmed by subcloning and sequencing of fragments recovered from the gel. However, an additional strong third band of 433 bp was observed (Fig. 1A, band 3) corresponding to a novel PARG splice variant encoding a PARG protein with a predicted molecular weight of ∼60,000 Da and a unique primary structure, termed hPARG60 (GenBank acc. # EF382674). Amplified cDNA molecules from three human tissues, namely testis, skin and liver, yielded identical cDNA sequences, indicating that hPARG60 expression is probably not restricted to specific tissues. A set of primers for amplification of full-length hPARG60 cDNA products (hex0for + hex18rev, right-hand side, Figure 1A) was also used. For further analysis of the gene 3'RACE PCR was performed but no alternative splicing could be detected at the 3'end (data not shown). An open reading frame (ORF) of 1583 bp was amplified, (band 3), cloned and sequenced (right hand side of Fig. 1A). To confirm specificity of the PCR methods, amplified cDNA encoding hPARG111 (band 1, 3193 bp), hPARG102 (2761 - 2794 bp) and hPARG99 (2694 - 2698 bp; again as a double band) were again isolated, cloned and analyzed.
In mouse, PCR amplification of mPARG cDNA from reversely transcribed total testis and liver RNA (C57/BL6) yielded specific bands that encoded mPARG110 (Fig.1B, band 1), mPARG101 and mPARG98 (again as a doublet in band 2). Comparable to the results in human, a cDNA encoding a small mPARG with a predicted mass of 63 kDa could be isolated (mPARG63, GenBank acc. # EF382673), (band 3 of 540 bp, left hand side Fig 1B and 1630 bp product, right hand side where primers mex0for and mex18rev were used to generate full length cDNA PCR products) from these tissues. Mouse PARG63 is larger than its human homolog hPARG60 due to further processing of the human transcript as explained further below. Total RNA isolated from PARGΔ2-3/Δ2-3 MEF cells was included in the PCR analyses as a negative control for expression of the larger PARG isoforms. Mouse PARG63 cDNA isolated from wt testis and liver was of the same size as the mPARG63 expressed from the PARGΔ2-3/Δ2-3 MEF and sequence analyses revealed that their sequences were identical. Interestingly, analyses of PARGΔ2-3/Δ2-3 transcripts indicated the expression of an additional small isoform that appeared to be unique to the hypomorphic mutant (Fig. 1B, band 5). Sequence analyses of the corresponding cDNA showed that it encodes a ∼52 kDa PARG that is structurally similar to hPARG53, i.e. with the translation start in exon V. Because this type of PARG cDNA could not be isolated from human or wt mouse samples, it was not analyzed further in this study.
Small PARG isoforms are endogenously expressed in HeLa cells
Using antibodies specific for the N-terminus (ab9882) or the C-terminus (ab9877) of PARG, immunoblot analyses of HeLa cells transfected with EGFP-hPARG111 or EGFP-hPARG102 fusion proteins allowed for detection of both, ectopically overexpressed hPARG as well as endogenous hPARG proteins (Fig. 2). Typically, ab9882 recognized the overexpressed hPARG111 and hPARG102 EGFP fusion proteins as well as the endogenous hPARG111, hPARG102 and probably hPARG99 proteins which appear as a double or triple band (Fig. 2A). The C-terminus-specific antibody ab9877 recognized those isoforms as well but also an additional, substantial composite band in the range of 55-60 kDa (Fig. 2). Endogenous expression of mRNAs encoding the small hPARG isoforms in HeLa cells was confirmed by PCR analyses (Fig. 2B), as described above, suggesting the expression of one or more smaller PARG isoforms at both, mRNA and protein level in HeLa cells.
Fig. 2. Endogenous and ectopic hPARG expression in HeLa cells.
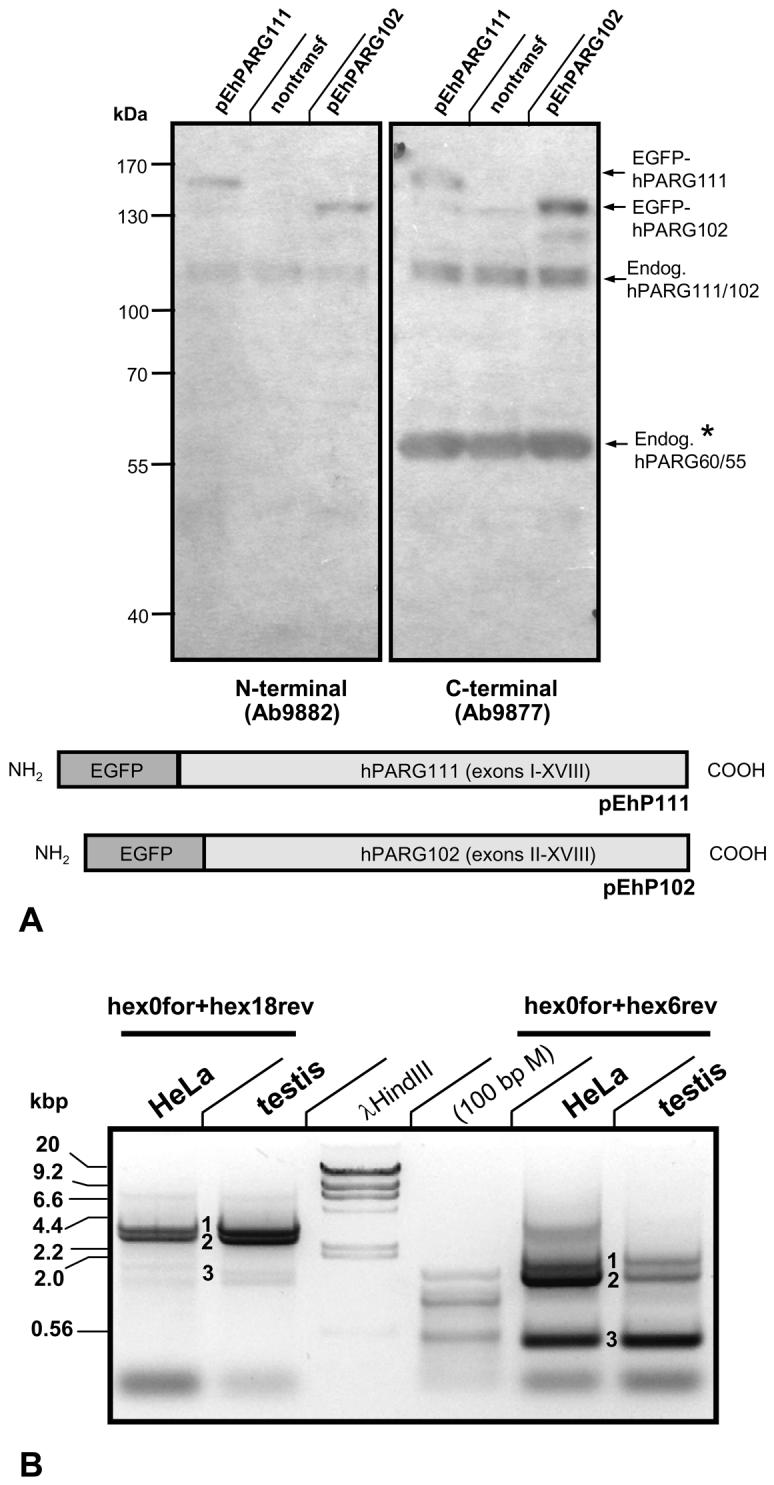
(A) Immunoblot analyses of pEhP111/pEhP102 transfected HeLa cells using two anti-peptide antibodies generated in rabbits that are either N-terminus-specific (ab9882) or C-terminus-specific (ab9877). Both antibodies recognize the overexpressed PARG protein but, as expected, only ab9877 and not ab9882 specifically detects endogenous PARG protein in the range of 55-60 kDa. (B) PCR amplification of PARG splice variants obtained from reversely transcribed HeLa cell total RNA. Using the same primer sets as in Fig. 1, the bands representing hPARG111 (1), hPARG102 /hPARG99 (2), and the novel hPARG60 (3) were reproducibly amplified.
Small PARG isoforms are expressed by activation of an alternative splice donor site in the 5' region of PARG transcripts
Analyses of both human and mouse PARG PCR products showed that their 5' ends are very similar and indicated the presence of a previously undescribed, highly conserved alternative splice donor site (SDalt) upstream of the ORF encoded by exon I (Fig. 3) in both species which is consistent with a consensus splice donor sequence of the type (G/C)AG|G(T/C)NNGT. In both species SDalt is located downstream of the previously reported facultative splice donor sites SD1a, SD1b and SD1c which are utilized for expression of the 102 kDa and 99 kDa PARG isoforms [7]. Alternative splicing using SDalt in combination with splice acceptor site SA3 leads to skipping of exons I, II and III, bringing facultative exon Ia in frame with exon IV (Figure 4A, 4B).
Fig. 3. Schematic view of the human and murine PARG gene 5' regions.

The putative transcription initiation start [6] is indicated by a circled +1, along with the position of the forward primers (underlined italics) used to generate the cDNA molecules presented in Figure 1. Shaded boxed regions represent open reading frames with light grey boxes indicating the hPARG111/mPARG110 ORF. In hPARG60 and mPARG63, usage of a facultative exon (exon Ia, dark gray box) leads to alternative N-terminal protein sequences of 16 amino acids not found in the three long forms of the enzyme in both species. While facultative splice donor sites SD1a-c have been reported to be involved in generation of hPARG102, hPARG99 and the respective mouse proteins, they are not employed in the expression of the 60 kDa splice variant in either species. Instead, a splice donor site, termed SDalt here, facilitates elimination of exons 1, 2 and 3, bringing exon 1a in frame with exon 4.
Fig. 4. Schematic view of alternative PARG gene 5' splicing reactions leading to expression of hPARG60/mPARG63.

(A). Usage of alternative splice donor site SD1alt in conjunction with splice acceptor site SA3 results in elimination of exons I, II and III and formation of a new protein N-terminus encoded by facultative exon Ia (A) and (B). Alignment (C) of resulting human PARG60 (upper) and murine PARG63 (lower) sequences indicates a high degree of evolutionary conservation of the two proteins except for the skipping of exon V and the N-terminal regions encoded by facultative exon Ia. The amino acids sequence within the brackets is not expressed in hPARG55 and mPARG58. The MTS sequence is highlighted by an open box. (D) Splicing that retains exon V in the hPARG mRNA, as found in the mRNA molecules encoding hPARG111, 102 and 99 is shown, including the splice donor site SD4a (open box) facilitating this process. (E) Alternative in-frame splicing of exon IV to exon VI is only possible because of the presence of SD4b (open box) overlapping SD4a, leading to skipping of exon V. (F) Skipping of exon V (E) in human may be caused by a sequence variation in SD5 ( g instead of t, in bold) that weakens the splice donor consensus sequence GAG | GTAAGA (mouse) to GAG | GGAAGA (human), (marked by * in (A)).
For both, human and mouse, two most likely translation possibilities can be predicted:
(i) Translation can start in exon Ia with the first available methionine encoded in frame, giving rise to a putative16 amino acid (aa) N-terminus present in the proteins of both, human and mouse (Fig. 3). This sequence is unique in that it is not present in any of the other PARG proteins and that it is not very conserved from mouse (MVRGAPRRRRSPSLSG) to human (MVQAGAEKDAQSISLR), although the two share 75% homology on DNA level (Figure 3). Translation initiation is predicted to be efficient in both, given a good resemblance of their start sites, …GGAAUGG…, with the consensus sequence A/GNNAUGG [46] , where essential nucleotides are underlined.
(ii) Translation can start at two potential downstream start codons present in exon IV, encoding M53 and M56 in hPARG60 (Figure 4C), corresponding to M461 and M464 in the hPARG111 protein. These translation initiation sites match the consensus sequence (see above) well enough to be potentially functional: GAGAUGA (M461) and AGAAUGC (M464). For comparison, the hPARG111 translation initiation is encoded by AGCAUGA (M1) and alternative translation initiation from large PARG transcripts has been described before [6, 7] , supporting the view that most likely both events, (i) and (ii) occur in vivo.
Skipping of exon V in human gives rise to a PARG transcript that is absent in mice
All isolated hPARG60 but none of the mPARG cDNA molecules were lacking the sequence information of aa 486-526 of hPARG111, corresponding to the 40 amino acids encoded by exon V (Figure 3, 4A, D, E) plus the last amino acid, V486, of exon IV. The skipping of exon V was unexpected as this type of splicing would be expected to lead to a frame shift mutation in exon VI and termination of translation within the first three codons of exon VI (Figure 4D, E, F). Genomic sequence analyses using NCBI BLAST, however, revealed the presence of two overlapping splice donor site motifs at the 3'-end of exon IV (SD4a, SD4b) that alternatively allow for correct splicing of exon IV to either exon V or exon VI. In this context, a nucleotide variation between the mouse and human sequence in SD5 was noted, leading to an alteration of the splice donor sequence GAG|GTAAGA (mouse) to GAG|GGAAGA, which is predicted to negatively affect splicing efficiency of exon V to exon VI in human (Figure 4D), thus favoring splicing of exon IV to exon VI.
Interestingly, no human PARG cDNA that lacked exons I, II and III (i.e. hPARG60) and at the same time contained the sequence information of exon V could be isolated, suggesting that skipping of this exon is the rule rather than the exception in human. Recent entries of EST data from a variety of tissues into the NCBI database support this notion, e.g.: AW816686, DA800072, DA121686, including testis (DB073432), cornea (CV574127), trachea (DB220314), liver (DW413098), and uterus (DB277286, DB281918). Notably, no hPARG111, hPARG102 or hPARG99 cDNA have been reported in which exon V was skipped. An alignment of hPARG60 and mPARG63 proteins is given in Figure 4C. According to our findings and the majority of available EST clone data, the C-terminus of mPARG proteins lacks 8 aa that are present in the hPARG cDNA clones. This difference accounts for the slightly different protein sizes and accordingly the different nomenclature of human and mouse PARGs (e.g. hPARG111 vs. mPARG 110) protein variants throughout this study. Figure 5 and Table 1 give an overview over PARG isoforms in human and mouse cells. For human PARGs lacking the amino acids corresponding to exon 5, the two possible translation products are designated hPARG60 and hPARG55. The two short mouse PARG variants are mPARG63 and mPARG58, respectively. Figure 5 also shows two isoforms that were constructed for additional characterizations of PARG expression, but for which there is no evidence of natural occurrence.
Fig. 5. PARG mRNA splice variants and resulting protein structures.

Open reading frames are represented by shaded boxes. Alternative splicing yields messages encoding 102 kDa and 99 kDa PARGs in human (100 kDa and 98 kDa in mouse) but also shorter PARG proteins of less than 65 kDa that were identified in both human and mouse RNA preparations. In hPARG60 and mPARG63 usage of a facultative exon (exon Ia) leads to alternative N-terminal protein sequences of 16 amino acids not found in the three long forms (i.e. 111 kDa, 102 kDa and 99 kDa) of the enzyme in both species. In human, exon V was consistently skipped in all cDNAs encoding hPARG60 isolated in this study but never in the other mRNAs encoding the three large protein variants. Skipping of exon V appears to be specific for human and was absent in murine cDNA preparations. Proteins hPARG55 and mPARG58 (indicated by *1)) are most likely not encoded by specific mRNAs but may be expressed from hPARG60 and mPARG63 messages, respectively, by alternative translation start codon usage as previously observed in the large PARG cDNAs [7]. *2) Human PARG59 and hPARG53 were not encoded by natural cDNAs obtained by RT-PCR but were constructed for the purpose of studying MTS function in this study.
Table 1.
Overview over human and murine PARG isoforms. The molecular weight of the proteins are theoretically calculated sizes according to amino acid composition and protein length.
| PARG splice variant | Species | MW [kDa] | Designation of skipped exon(s) | Splice sites used for (alternative) splicing | Exons translated in protein | Predominant subcellular localization |
|---|---|---|---|---|---|---|
| hPARG111 | H.s. | 111 | Full-length | SD1 to SA1, SD4 to SD5 | I - XVIII | Nuclear |
| hPARG102 | H.s. | 102 | I | SD1a,b to SA1, SD4a to SA4 | II - XVIII | Cytoplasmic |
| hPARG99 | H.s. | 99 | I, II | SD1c to SA2, SD4a to SA4 | III- XVIII | Cytoplasmic |
| hPARG60 | H.s. | 60 | I, II, III, V | Sd1alt to SA3, SD4b to SA5 | Ia, IV, VI – XVIII |
Cytoplasmic & Mitochondrial |
| hPARG55 | H.s. | 55 | I, II, III, V | Sd1alt to SA3, SD4b to SA5 | IV, VI -XVIII |
Mitochondrial |
| mPARG110 | M.m. | 110 | Full-length | SD1 to SA1, SD4 to SA4 | I - XVIII | Nuclear |
| mPARG101 | M.m. | 101 | I | SD1a,b to SA1, SD4 to SA4 | II- XVIII | Cytoplasmic |
| mPARG98 | M.m. | 98 | I, II | SD1a,b to SA2, SD4 to SA4 | III - XVIII | Cytoplasmic |
| mPARG63 | M.m. | 63 | I, II, III | Sd1alt to SA3, SD4 to SA4 | Ia, IV - XVIII |
Cytoplasmic& Mitochondrial |
| mPARG58 | M.m. | 58 | I, II, III | Sd1alt to SA4, SD4 to SA4 | IV - XVIII | Mitochondrial |
Abbreviations: H.s., Homo sapiens; M.m., Mus musculus; SD, splice donor site; SA, splice acceptor site
Human PARG60 is catalytically active in vitro
CMV immediate early promoter-driven expression plasmids of different hPARG proteins as in-frame fusions to EGFP were transfected into HEK293 cells in order to examine to what extent the transfected cells show alteration of PARG activity (Figure 5). With very similar transfection efficiencies, as judged by the percentages of EGFP positive cells, PARG activity was 4 to 4.5 fold higher in the pEhPARG102 transfected cells as compared to cells that had received the control vector pEGFP-C1 (p <<0.001) while transfection of a catalytically dead mutant, that carries a glutamate (E) to asparagine (N) amino acid exchange in the position corresponding to position 756 of the human PARG111 amino acid sequence, had no effect on activity. In contrast, pEhPARG60 transfected cells had elevated PARG activity of approximately 2-fold (p < 0.005) in vitro, while activity in cells expressing a catalytically dead mutant was in the range of the activity observed in control cells. The TLC based PARG assay allows measuring of mainly exoglycosidic activity using protein-free, long-chain PAR as a substrate and does therefore not allow for evaluation of endoglycosidic PARG activity for which no assay is currently available.
Polyacrylamide gel electrophoresis of the PAR cleavage products did not yield any indication of an obvious alteration of endoglycosidic versus exoglycosidic activity mode (Figure 6B). In order to confirm this finding, these assays were also repeated with extracts from wildtype 129S1/Sv and PARGΔ2-3/Δ2-3 mouse embryonic fibroblasts (data not shown). Because the latter cells lack all three large PARG isoforms, an alteration of the enzymatic mode would have been evident. However, while the overall specific PARG activity was 4fold higher in wildtype cells, at equal activities in the extracts normalized to cell numbers, no difference in the PAR cleavage products was detectable in the hypomorphic mutant cells under these experimental conditions.
Fig. 6. Human hPARG60 is enzymatically active.

In vitro poly(ADP-ribose) glycohydrolase assays using 32P labeled PAR were employed to measure relative exoglycosidic PARG activities in extracts of transiently transfected HEK293 cells expressing EGFP-hPARG fusion proteins (A). PARG activity in extracts of HEK293 cells transfected with the parental vector pEGFP-C1 was defined as 100% and the scale reflects the percentage of PARG activity in the other samples relative to this control. Nontransfected cells and cells transfected with pEhP102d, encoding an enzymatically dead mutant (E756N in full-length hPARG111) of PARG102 generated by site-directed mutagenesis, were used as controls. The negative control samples (“neg. con.”) contained no protein (background). Positive controls used 6.75 ng of recombinant bovine PARG. Significance analyses using pooled data from three independent experiments were done using the Student's T-test (p<0.005).
(B) Aliquots of samples measured in PARG assays (A, see above) were subjected to ADPR polymer PAGE autoradiograph analyses to visualize 32P labeled products of poly(ADP-ribose) digests. Data were pooled from three independent experiments each carried out at least in quadruplicates for these analyses and a representative autoradiograph is shown.
Human PARG60/55 but not hPARG102 are associated with the mitochondria
After transfection of plasmids pEhPARG111V5, pEhPARG102V5, and pEhPARG59V5 followed by subcellular fractionation and immunoblot analyses using anti-EGFP and anti-V5 tag antibodies, only hPARG59 but not hPARG111 or hPARG102 fusion protein was detectable in the mitochondrial fraction (Figure 7A). Plasmid pEhPARG59 encodes an experimental, hypothetical and probably not naturally occurring hPARG protein encoded by exons IV-XVIII with a translation start in exon IV and therefore hPARG59 reflects the situation found in mPARG58 where exon V is always present. However, transfection of pEhPARG60 gave similar results in this assay (data not shown). At first, mitochondrial localization of the transfected proteins was puzzling as EGFP as the N-terminal portion of these fusion proteins does not have a mitochondrial targeting sequence required for mitochondrial import. However, computer analyses revealed the presence of a putative mitochondrial targeting sequence (MTS) in the 3' half of exon IV downstream of the first available translation start codon in that exon (Figure 4C). Provided that alternative translation initiation may give rise to a hPARG55 (Figure 5), the identified MTS would be located at the N-terminus and could therefore act as a cleavable presequence for mitochondrial import by receptor translocase complexes TOM and TIM, thereby mediating mitochondrial localization. In a wheel plot projection, an amphitropic α-helical structure of the sequence can be predicted which is a common feature of many N-terminal mitochondrial import presequences (Figure 7B). Although it is not clear whether the secondary structure of the aa stretch encoded by exon Ia is truly α-helical or not, applying the same projection method to the sequence including exon Ia would not yield any clear amphipathic configuration if an α-helix was present (Figure 7C). Typically, presequence type MTS are proteolytically removed from the imported proteins at mitochondrial entry. Western blot analyses of HeLa cells transfected with expression plasmids for V5-tagged hPARG60 (527 aa), hPARG59 (516 aa), hPARG55 (475 aa) and hPARG53 (464 aa) (Figure 7B) showed that the protein band observed in the hPARG55 lane has slightly greater electrophoretic mobility than expected. If the MTS presequence was cleaved off from hPARG55 protein during mitochondrial import, the remaining protein would have a predicted molecular weight of less than 53 kDa (458 aa), i.e. slightly smaller than hPARG53 (464 aa), (Figure 7B, lanes hPARG55V5 and hPARG53V5), further suggesting functionality of the identified MTS. Yet, mitochondrial localization of the transfected PARG proteins could not be readily explained by the presence of a presequence-type MTS in the middle of the EGFP-hPARG fusion proteins. In order to further confirm functionality of the putative MTS as an internal or an N-terminal signal, two different luciferase reporter plasmids, pMTS-GFL1 and pMTS-GFL2, respectively, were constructed. The two plasmids both contain PARG exons IV, V and VI N-terminally fused to an EGFP-luciferase reporter gene but they differ in the presence or absence of an engineered, highly efficient β-galactosidase translation initiation site upstream of exon IV. By using this approach, the start of the potential MTS is positioned 38 amino acids downstream of a new, artificial translation start (encoding MSFTLTE…, followed by the PARG sequence KEQWET…of exon IV) and at the same time the amino acids encoded by facultative exon Ia were removed. Again, using subcellular fractionation and subsequent luciferase activity measurements, a preferential localization of the recombinant protein in the mitochondrial fraction was detected (data not shown). However, using fluorescence microscopy in order to detect the localization of EGFP-hPARG60 fusion in transfected cells, the protein seemed to reside mainly in the cytoplasm and the nucleus and specific mitochondrial localization was only detectable in a subpopulation of the transfected cells (not shown). The same result was obtained using the EGFP-luciferase constructs containing an artificial β-galactosidase N-terminus. In contrast, transfected cells expressing only the EGFP-luciferase reporter protein showed no preferential localization of the reporter (Figure 8, right panel). Plasmid pMTS- GFL2 does not provide an additional translation initiation site upstream of the MTS, but allows for immediate translation initiation within exon IV, and was constructed in order to investigate the ability of the MTS encoded region to act as an N-terminal signal without additional sequences. The MTS-EGFPluciferase reporter protein encoded by this plasmid was strictly targeted to the mitochondria (Figure 8, left panel) in virtually all transfected cells. In order to confirm the presence of PARG activity in association with mitochondria, PARG and PARP activities in pure nuclear and mitochondrial fractions isolated from untreated HEK293 cells were measured (data not shown). Because the sizes and protein contents of the two fractions were very different, the specific activities for PARG and PARP were calculated. Specific PARG activity, i.e. the number of pmol of PAR converted into free ADP-ribose per minute per mg of protein, was higher in mitochondrial fractions than in nuclear extracts although the total activity was consistently higher in the nucleus. In contrast, PARP activity was much higher in the nucleus and comparatively little specific activity was found in the mitochondria. These findings support the view that PARG is able to reside in the mitochondrial fraction. Immunofluorescence analyses using HeLa cells transfected with expression plasmids encoding V5-tagged hPARG102, hPARG60, hPARG59, an enzymatically inactive mutant hPARG56d, and hPARG53 (see Figure 5) confirmed that the MTS targets PARG to the mitochondria (Figure 9). Anti-V5 antibodies detected hPARG102 in the cytoplasm (Figure 9A), as previously reported [7, 43]. Similarly, in hPARG60 transfected cells, signals were seen mainly in the cytoplasm (Figure 9B). as expected from immunoblot experiments which showed that a EGFPhPARG59 fusion, i.e. a protein similar to hPARG60 in that it lacks an N-terminal MTS due to the presence of the EGFP portion, was present in all subcellular fractions tested (Figure 7A). In contrast, hPARG59 alone, an artificial protein designed for this study, was nearly exclusively located in the mitochondria, (Figure 9C) which was not dependent on enzymatic activity, as demonstrated by transfection of an hPARG56d expression plasmid encoding an inactive mutant enzyme (Figure 9D). In hPARG59, the putative MTS is located at the N-terminus functioning as an external MTS of the presequence type. Overexpression of hPARG55, which is likely naturally expressed from alternatively translated hPARG60 transcripts (Figure 5 & Figure 7D), and which has the same predicted translation initiation site as hPARG59 but lacks exon V, shows that mitochondrial localization was dependent on the presence of exon IV and correct translation start codon usage (Figure 9E), while exon V was dispensable. Absence of exon IV abolished mitochondrial targeting as hPARG53 was found in the cytoplasm and the nucleus (Figure 9F). Natural expression of hPARG53 has not been shown and the cDNA encoding this protein was engineered for the purpose of this study. Taken together, these data indicate that the MTS located in exon IV can act as an efficient N-terminal signal in the naturally occurring PARG splice isoform hPARG55, and also as a less efficient, internal signal in hPARG60, as in the latter case only a fraction of the protein colocalized with the mitochondria, which was predominantly observed in the Western Blot analyses of purified mitochondrial cell fractions. The Western Blot analyses also revealed that the MTS does not seem to be active in the longer PARG splice variants, e.g.hPARG111 and hPARG102. It should be noted that because these studies were done with overexpressed proteins, detection of the corresponding endogenous enzymes would be necessary to confirm these data. However, all PARG isoforms are known to be expressed at very low levels and antibodies sensitive enough to detect such low levels are not currently available. In addition, antibodies recognizing the peptide encoded by exon 1a in mouse or man could not be generated due to insufficient immunogenicity of that region so that the available antibodies that detect the novel small PARG proteins will also always recognize the larger PARGs as well.
Fig. 7. Overexpressed human PARG59 but not hPARG102 or hPARG111 is associated with the mitochondria.


(A) Immunoblot analyses of subcellular fractions obtained from HeLa cells transiently transfected with N-terminal EGFP fusions of hPARG59, hPARG102 or hPARG111 using V5-tag or EGFP-specific antibodies show that the PARG MTS can act as an internal protein signal in the presence of an N-terminal peptide sequence that is upstream of the actual MTS. This resembles the situation in hPARG60 and mPARG63 where the MTS is not strictly N-terminal but downstream of a 16 aa peptide sequence encoded by the facultative exon Ia (s. Figure 5). Purity of the mitochondrial fractions was assessed by detection of MnSOD as a mitochondrial marker and histones, as well as PARP1 as nuclear marker proteins. No contamination of the mitochondrial fractions by nuclei was detected (shown for transfection with hPARG59, data not shown for hPARG102 and hPARG111). (B) Immunoblot analyses of HeLa cells transiently transfected with plasmids for overexpression of hPARG isoforms with C-terminal V5 tags, using anti-V5 antibodies. The * indicates an apoptotic caspase cleavage product of the transfected hPARG111. (C) Wheel projection of hPARG and mPARG N-termini indicating the presence of putative amphipathic α-helices in the actual MTS peptide sequences but not the amino acid sequences encoded by the N-terminally located facultative exon Ia in human or mouse. This computer-based prediction- and analysis tool of mitochondrial targeting signals is based on the classic assumption that these signals are typically alpha-helical in their secondary structure where basic, mostly lysine (K) and arginine (R), and hydrophobic amino acid residues should be located on opposite sides of that α-helix to achieve amphipathic properties.
Fig. 8. An N-terminal PARG MTS directs an EGFP reporter protein to the mitochondria.
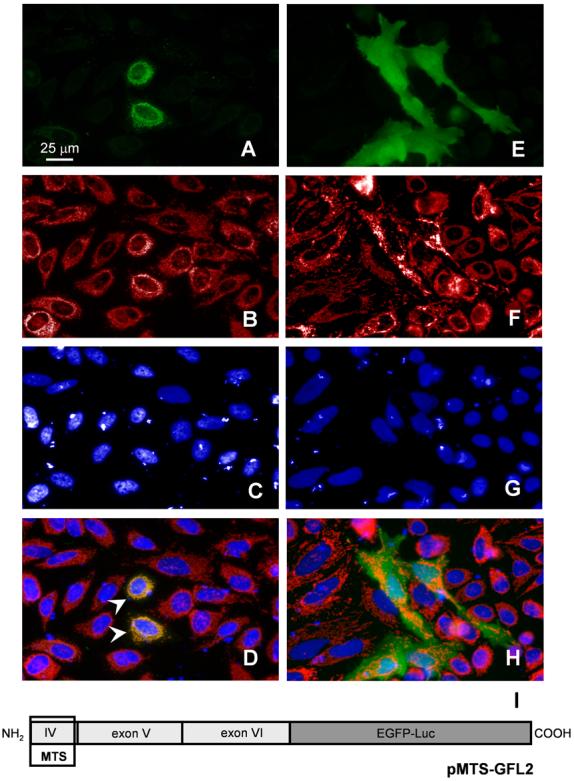
AntiMnSOD immune fluorescence stain of HeLa cells transiently transfected with pMTS-GFL, encoding a C-terminal in-frame fusion of an EGFP-firefly luciferase reporter gene with the putative MTS located in exon 4. The N-terminal portion of the encoded protein is identical to the one proposed for hPARG55 and comprises aa 461-526 of human PARG. (A) GFP fluorescence of the fusion protein (pMTS-GFL) (B) MnSOD staining with a polyclonal rabbit antibody and anti-rabbit Cy3 utilized in order to label mitochondria, (C) DAPI stain of the nuclei and (D) merge of A, B, and C, where arrowheads indicate areas of colocalization. Panel E-F show control cells transfected with a vector expressing the GFL reporter without the PARG MTS (pRSV-GFL). Conventional fluorescence microphotographs, Nikon T 3000, 40x objective, taken with a Nikon Coolpix camera in combination with ImagePro software.
Fig. 9. Subcellular localization of hPARG isoforms.
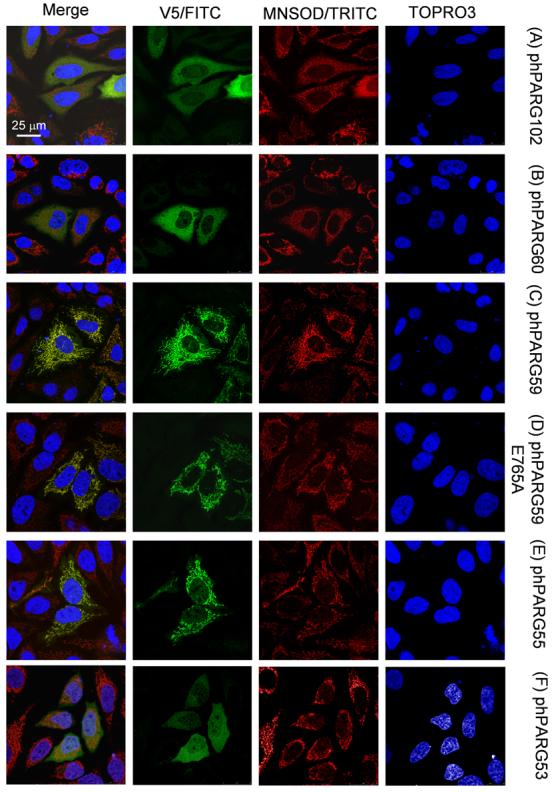
HeLa cells transiently transfected with expression plasmids encoding hPARG isoforms with engineered C-terminal V5-tags were analyzed by indirect immunofluorescence staining using anti-V5 (green) and anti-MnSOD antibodies (red). DNA was counterstained with Topro3 and pseudocolored in blue (confocal laser scan microphotographs, Leica SP5).
Discussion
The results of the study presented here suggest the presence of an additional level of complexity of cellular PAR metabolism due to the catalytic activity of novel small PARG isoforms associated with the mitochondria. This view is supported by several conclusions that can be drawn from this study: (i) Two novel protein isoforms, hPARG60 and hPARG55, were discovered which are capable of PAR hydrolysis. They are conserved from mouse to human and are ubiquitously expressed by alternative splicing in all tissues tested. (ii) The new PARG isoforms lack the putative regulatory A-domain, suggesting that their enzymatic activity may be controlled in a different way than in the large PARG isoforms hPARG111, hPARG102 and hPARG99. (iii) Both, hPARG60 and hPARG55 are associated with the mitochondria, making them potentially important factors in the nuclear-mitochondrial crosstalk involving PAR metabolism. While PARG111 is a nuclear enzyme carrying a nuclear localization signal (NLS) in exon I near the N-terminus [8], PARG 102 and PARG 99 are mainly extranuclear proteins [7, 47] carrying putative nuclear export signals (NES), [43, 48] in the N-terminal A-domain. The presence of the bulk of PARG activity in the cytoplasm remains puzzling as the vast majority of PAR synthesis following genotoxic stress is catalyzed by the predominantly nuclear enzymes PARP-1 and PARP-2 and PAR is therefore detectable mainly in the nucleus after DNA damage. It has therefore been proposed that PARG102 may be able to translocate between the nucleus and cytoplasm under conditions of acute genotoxic stress [5, 9, 43]. In mice, complete PARG gene disruption has been shown to be inconsistent with cellular survival [14]. However, disruption of the mPARG gene by deleting exons II and III did not completely abrogate PARG activity in the hypomorphic PARG Δ2-3/Δ2-3 mouse model devoid of the three large PARG proteins due to expression of 63 kDa and 58 kDa PARG isoforms, presumably from a common mRNA splice variant [18]. These findings indicate that the complete deletion of PARG and the resulting loss of PAR hydrolyzing activity can not be compensated for by ARH3, the only other known PAR catabolizing enzyme, but expression of mPARG63 and mPARG58 is required for cellular viability in this mouse model. In fact, the two proteins appear to be able to fulfill most of the PARG functions in the cell.
Based on the present study, mPARG63 and mPARG58 proteins are homologous to hPARG60 and hPARG55 and the isolation of mPARG63 cDNA from wildtype mouse tissues (Figure 1) supports that expression of a ∼60 kDa PARG is conserved among mammals.
The presence of 50- 60 kDa PARG proteins in different cells or tissues has been reported repeatedly over the last twenty years [43, 49-54]. However, these studies did not lead to cloning of a cDNA encoding the elusive protein and it has been discussed that hPARG60 may be a proteolytic fragment of a larger PARG, e.g. PARG111 (reviewed in [55]). Indeed, larger isolated PARG proteins have been shown repeatedly to be unstable, forming degradation products of 50-65 kDa [5, 54, 56]. Isolation and purification of endogenous bovine PARG from thymus yielded such short proteins and MALDI-TOF analyses of the obtained fraction indicated the presence of several different PARG proteins in the range of 50-65 kDa (Meyer, R. G. and Jacobson, M. K., unpublished results). Interestingly, the splice variants reported here encode PARG proteins that are devoid of the putative regulatory A-domain. Functions of this domain are not yet well understood, but its absence in the PARGΔ2-3/Δ2-3 mouse is responsible for a complex phenotype of hypersensitivity against genotoxic stress [18] and the loss of the three large PARG proteins results in cytotoxic cerebral [20] and splanchnic [21] accumulation of cellular PAR after ischemiareperfusion injury, illustrating its importance in regulating PARG activity, and thus PAR turnover. Recently, it has been reported that PARG physically interacts with PARP-1, regulating its activity and that both form a complex with XRCC1 after MNNG induced DNA damage [57]. The observation that PARP-1 automodification is impaired in the PARGΔ2-3/Δ2-3 mouse and that this alters overall PAR metabolism [58] indicates a role of the A-domain in this interaction and suggests that hPARG60 may interact with PARP-1 differently than hPARG111.
The observed mitochondrial targeting of the fusion proteins containing the MTS in the form of an N-terminal presequence (Figure 7) is also in line with a recent report where a putative mitochondrial targeting sequence encoded in exon IV of the human gene was identified that was postulated to be N-terminal [25]. The results of the western blot analyses, e.g. for EGFP-hPARG59, (Figure 7A) and luciferase activity measurements after subcellular fractionation of transfected cells (data not shown), indicated mitochondrial association also of proteins where the MTS is not N-terminal but preceded by more than 50 amino acid residues, although this was less pronounced. These findings suggest an association of hPARG60 protein with the mitochondria, facilitated by the MTS in exon IV which may be part of an internal targeting signal [59]. In these intrinsic MTS sequences, which are not well characterized, the targeting signal is hidden in the mature protein and can be spread along the length of the protein in a non-canonical way with examples including extrinsic mitochondrial outer membrane proteins. In this scenario, the cryptic hPARG60 MTS may be activated by binding a second factor such as PAR. We hypothesize that hPARG60 may be capable of shuttling between cellular compartments, e. g. in response to DNA damage-induced, PARP-1 and PARP-2 dependent formation of PAR. The fact that PARG may have a second, weak NLS near the C-terminus [43, 48] which would facilitate this type of nucleo-mitochondrial shuttling supports this hypothesis. However, results obtained from overexpression of PARG proteins in the present study may require further confirmatory experiments involving the endogenously expressed proteins to verify the discussed hypotheses of small PARG isoform subcellular localization. In addition, future in-depth investigations will be necessary to determine submitochondrial localization of PARG enzymes.
Interestingly, mitochondrial poly(ADP-ribosyl)ation has been reported repeatedly but it has been controversial since the responsible enzymes could not be clearly identified (reviewed in [60]). The discovery of a PARG protein capable of regulating PAR accumulation in close proximity to, or possibly within mitochondria may be of relevance for the model that overactivation of PARP-1 leads to the breakdown of the mitochondrial membrane potential and subsequent release of apoptosis inducing factor (AIF). In this model, AIF released from the mitochondria rapidly translocates to the nucleus and activates an endonuclease facilitating chromatinolysis and cell death in a caspase-independent manner [61-63]. While it is possible that NAD+ depletion as a result of PAR formation may affect the mitochondrial membrane potential ([64, 65] ), there also is accumulating evidence that PAR itself may be acting as a cell death signaling molecule triggering the formation of a mitochondrial pore transition (MPT) in a yet unknown way [66]. Moreover, it has been shown that monomeric ADP-ribose (ADPR) produced by activation of PARP/PARG enzymes contributes to oxidative stress induced cell death by specifically activating the cell membrane Ca2+ channel TRPM2 by binding to its nudix motif [67, 68] and, hypothetically, the novel PARG isoforms could contribute to the release of ADPR from the mitochondria. Clearly, the function of hPARG60/mPARG63 and hPARG55/mPARG58 and their association with the mitochondria requires to be investigated in more detail in future studies. Although very low PARP activities were measured in extracts of mitochondrial cell fractions (data not shown), the existence of a mitochondrial PARP as a source of PAR that may be a substrate for hPARG55 could neither be conclusively confirmed nor excluded in the present study. Nevertheless, the present study provides a basis facilitating further research concerning the regulation of PAR metabolism and strategies to target this biochemical pathway in various pathologies including cancer and reperfusion injury. Moreover, it may provide a step towards elucidating the existence of an entirely mitochondrial PAR metabolism which is still a controversially discussed possibility.
ACKNOWLEDGEMENTS
*We thank Donna Coyle for preparing 32P-labeled PAR and Zhao-Qi Wang, Universitaet Jena, for providing PARGΔ2-3/Δ2-3 3T3 cells. This work was supported by NIH grants CA-43894 (MKJ) and HD048837 (RGM).
Footnotes
This work was supported by NIH grants CA-43894 (MKJ) and HD048837 (RGM).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: Where are we today and where are we going? Microbiol. Mol. Biol. Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D'Amours D, Desnoyers S, D'Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999;342(Pt 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- 3.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 4.Meyer RG, Meyer-Ficca ML, Jacobson EL, Jacobson MK. Enzymes in poly(ADP-ribose) metabolism. In: Burkle A, editor. Poly(ADP-Ribosyl)Ation, Landes Bioscience. 2004. [Google Scholar]
- 5.Gagne JP, Hendzel MJ, Droit A, Poirier GG. The expanding role of poly(ADP-ribose) metabolism: Current challenges and new perspectives. Curr. Opin. Cell Biol. 2006 doi: 10.1016/j.ceb.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Meyer RG, Meyer-Ficca ML, Jacobson EL, Jacobson MK. Human poly(ADP-ribose) glycohydrolase (PARG) gene and the common promoter sequence it shares with inner mitochondrial membrane translocase 23 (TIM23) Gene. 2003;314:181–190. doi: 10.1016/s0378-1119(03)00738-8. [DOI] [PubMed] [Google Scholar]
- 7.Meyer-Ficca ML, Meyer RG, Coyle DL, Jacobson EL, Jacobson MK. Human poly(ADPribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments. Exp. Cell Res. 2004;297:521–532. doi: 10.1016/j.yexcr.2004.03.050. [DOI] [PubMed] [Google Scholar]
- 8.Ohashi S, Kanai M, Hanai S, Uchiumi F, Maruta H, Tanuma S, Miwa M. Subcellular localization of poly(ADP-ribose) glycohydrolase in mammalian cells. Biochem. Biophys. Res. Commun. 2003;307:915–921. doi: 10.1016/s0006-291x(03)01272-5. [DOI] [PubMed] [Google Scholar]
- 9.Davidovic L, Vodenicharov M, Affar EB, Poirier GG. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp. Cell Res. 2001;268:7–13. doi: 10.1006/excr.2001.5263. [DOI] [PubMed] [Google Scholar]
- 10.Oka S, Kato J, Moss J. Identification and characterization of a mammalian 39-kDa poly(ADPribose) glycohydrolase. J. Biol. Chem. 2006;281:705–713. doi: 10.1074/jbc.M510290200. [DOI] [PubMed] [Google Scholar]
- 11.Kernstock S, Koch-Nolte F, Mueller-Dieckmann J, Weiss MS, Mueller-Dieckmann C. Cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of human ARH3, the first eukaryotic protein-ADP-ribosylhydrolase. Acta Crystallograph Sect. F. Struct. Biol. Cryst. Commun. 2006;62:224–227. doi: 10.1107/S1744309106003435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mueller-Dieckmann C, Kernstock S, Lisurek M, von Kries JP, Haag F, Weiss MS, Koch-Nolte F. The structure of human ADP-ribosylhydrolase 3 (ARH3) provides insights into the reversibility of protein ADP-ribosylation. Proc. Natl. Acad. Sci. U. S. A. 2006 doi: 10.1073/pnas.0606762103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Juarez-Salinas H, Sims JL, Jacobson MK. Poly(ADP-ribose) levels in carcinogen-treated cells. Nature. 1979;282:740–741. doi: 10.1038/282740a0. [DOI] [PubMed] [Google Scholar]
- 14.Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC, Stoger T, Poirier GG, Dawson VL, Dawson TM. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17699–17704. doi: 10.1073/pnas.0406182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 16.Yu SW, Wang H, Dawson TM, Dawson VL. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol. Dis. 2003;14:303–317. doi: 10.1016/j.nbd.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 17.Koh DW, Dawson TM, Dawson VL. Mediation of cell death by poly(ADP-ribose) polymerase-1. Pharmacol. Res. 2005;52:5–14. doi: 10.1016/j.phrs.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 18.Cortes U, Tong WM, Coyle DL, Meyer-Ficca ML, Meyer RG, Petrilli V, Herceg Z, Jacobson EL, Jacobson MK, Wang ZQ. Depletion of the 110-kilodalton isoform of poly(ADP-ribose) glycohydrolase increases sensitivity to genotoxic and endotoxic stress in mice. Mol. Cell. Biol. 2004;24:7163–7178. doi: 10.1128/MCB.24.16.7163-7178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masutani M, Nakagama H, Sugimura T. Poly(ADP-ribosyl)ation in relation to cancer and autoimmune disease. Cell Mol. Life Sci. 2005;62:769–783. doi: 10.1007/s00018-004-4509-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cozzi A, Cipriani G, Fossati S, Faraco G, Formentini L, Min W, Cortes U, Wang ZQ, Moroni F, Chiarugi A. Poly(ADP-ribose) accumulation and enhancement of postischemic brain damage in 110-kDa poly(ADP-ribose) glycohydrolase null mice. J. Cereb. Blood Flow Metab. 2005 doi: 10.1038/sj.jcbfm.9600222. [DOI] [PubMed] [Google Scholar]
- 21.Cuzzocrea S, Di Paola R, Mazzon E, Cortes U, Genovese T, Muia C, Li W, Xu W, Li JH, Zhang J, Wang ZQ. PARG activity mediates intestinal injury induced by splanchnic artery occlusion and reperfusion. FASEB J. 2005;19:558–566. doi: 10.1096/fj.04-3117com. [DOI] [PubMed] [Google Scholar]
- 22.Patel NS, Cortes U, Di Poala R, Mazzon E, Mota-Filipe H, Cuzzocrea S, Wang ZQ, Thiemermann C. Mice lacking the 110-kD isoform of poly(ADP-ribose) glycohydrolase are protected against renal ischemia/reperfusion injury. J. Am. Soc. Nephrol. 2005;16:712–719. doi: 10.1681/ASN.2004080677. [DOI] [PubMed] [Google Scholar]
- 23.Shall S, de Murcia G. Poly(ADP-ribose) polymerase-1: What have we learned from the deficient mouse model? Mutat. Res. 2000;460:1–15. doi: 10.1016/s0921-8777(00)00016-1. [DOI] [PubMed] [Google Scholar]
- 24.Ying W, Swanson RA. The poly(ADP-ribose) glycohydrolase inhibitor gallotannin blocks oxidative astrocyte death. Neuroreport. 2000;11:1385–1388. doi: 10.1097/00001756-200005150-00007. [DOI] [PubMed] [Google Scholar]
- 25.Oei SL, Keil C, Ziegler M. Poly(ADP-ribosylation) and genomic stability. Biochem. Cell Biol. 2005;83:263–269. doi: 10.1139/o05-039. [DOI] [PubMed] [Google Scholar]
- 26.Masutani M, Nakagama H, Sugimura T. Poly(ADP-ribose) and carcinogenesis. Genes Chromosomes Cancer. 2003;38:339–348. doi: 10.1002/gcc.10250. [DOI] [PubMed] [Google Scholar]
- 27.Malanga M, Althaus FR. The role of poly(ADP-ribose) in the DNA damage signaling network. Biochem. Cell Biol. 2005;83:354–364. doi: 10.1139/o05-038. [DOI] [PubMed] [Google Scholar]
- 28.Haince JF, Rouleau M, Hendzel MJ, Masson JY, Poirier GG. Targeting poly(ADPribosyl)ation: A promising approach in cancer therapy. Trends Mol. Med. 2005;11:456–463. doi: 10.1016/j.molmed.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 29.Curtin NJ. PARP inhibitors for cancer therapy. Expert Rev. Mol. Med. 2005;7:1–20. doi: 10.1017/S146239940500904X. [DOI] [PubMed] [Google Scholar]
- 30.Bryant HE, Helleday T. Poly(ADP-ribose) polymerase inhibitors as potential chemotherapeutic agents. Biochem. Soc. Trans. 2004;32:959–961. doi: 10.1042/BST0320959. [DOI] [PubMed] [Google Scholar]
- 31.Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 32.Szabo G, Liaudet L, Hagl S, Szabo C. Poly(ADP-ribose) polymerase activation in the reperfused myocardium. Cardiovasc. Res. 2004;61:471–480. doi: 10.1016/j.cardiores.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 33.Chiarugi A. Poly(ADP-ribosyl)ation and stroke. Pharmacol. Res. 2005;52:15–24. doi: 10.1016/j.phrs.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 34.Woon EC, Threadgill MD. Poly(ADP-ribose)polymerase inhibition - where now? Curr. Med. Chem. 2005;12:2373–2392. doi: 10.2174/0929867054864778. [DOI] [PubMed] [Google Scholar]
- 35.Beneke S, Diefenbach J, Burkle A. Poly(ADP-ribosyl)ation inhibitors: Promising drug candidates for a wide variety of pathophysiologic conditions. Int. J. Cancer. 2004;111:813–818. doi: 10.1002/ijc.20342. [DOI] [PubMed] [Google Scholar]
- 36.Nguewa PA, Fuertes MA, Valladares B, Alonso C, Perez JM. Poly(ADP-ribose) polymerases: Homology, structural domains and functions. novel therapeutical applications. Prog. Biophys. Mol. Biol. 2005;88:143–172. doi: 10.1016/j.pbiomolbio.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 37.Hanai S, Kanai M, Ohashi S, Okamoto K, Yamada M, Takahashi H, Miwa M. Loss of poly(ADP-ribose) glycohydrolase causes progressive neurodegeneration in drosophila melanogaster. Proc. Natl. Acad. Sci. U. S. A. 2004;101:82–86. doi: 10.1073/pnas.2237114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiarugi A, Moskowitz MA. Cell biology. PARP-1--a perpetrator of apoptotic cell death? Science. 2002;297:200–201. doi: 10.1126/science.1074592. [DOI] [PubMed] [Google Scholar]
- 39.Patel CN, Koh DW, Jacobson MK, Oliveira MA. Identification of three critical acidic residues of poly(ADP-ribose) glycohydrolase involved in catalysis: Determining the PARG catalytic domain. Biochem. J. 2005;388:493–500. doi: 10.1042/BJ20040942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyer RG, Kupper JH, Kandolf R, Rodemann HP. Early growth response-1 gene (egr-1) promoter induction by ionizing radiation in U87 malignant glioma cells in vitro. Eur. J. Biochem. 2002;269:337–346. doi: 10.1046/j.0014-2956.2001.02658.x. [DOI] [PubMed] [Google Scholar]
- 41.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andreoli C, Prokisch H, Hortnagel K, Mueller JC, Munsterkotter M, Scharfe C, Meitinger T. MitoP2, an integrated database on mitochondrial proteins in yeast and man. Nucleic Acids Res. 2004;32:D459–62. doi: 10.1093/nar/gkh137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winstall E, Affar EB, Shah R, Bourassa S, Scovassi IA, Poirier GG. Preferential perinuclear localization of poly(ADP-ribose) glycohydrolase. Exp. Cell Res. 1999;251:372–378. doi: 10.1006/excr.1999.4594. [DOI] [PubMed] [Google Scholar]
- 44.Menard L, Poirier GG. Rapid assay of poly(ADP-ribose) glycohydrolase. Biochem. Cell Biol. 1987;65:668–673. doi: 10.1139/o87-088. [DOI] [PubMed] [Google Scholar]
- 45.Kiehlbauch CC, Aboul-Ela N, Jacobson EL, Ringer DP, Jacobson MK. High resolution fractionation and characterization of ADP-ribose polymers. Anal. Biochem. 1993;208:26–34. doi: 10.1006/abio.1993.1004. [DOI] [PubMed] [Google Scholar]
- 46.Kozak M. Compilation and analysis of sequences upstream from the translational start site in eukaryotic mRNAs. Nucleic Acids Res. 1984;12:857–872. doi: 10.1093/nar/12.2.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossi L, Denegri M, Torti M, Poirier GG, Ivana Scovassi A. Poly(ADP-ribose) degradation by post-nuclear extracts from human cells. Biochimie. 2002;84:1229–1235. doi: 10.1016/s0300-9084(02)00017-2. [DOI] [PubMed] [Google Scholar]
- 48.Shimokawa T, Masutani M, Nagasawa S, Nozaki T, Ikota N, Aoki Y, Nakagama H, Sugimura T. Isolation and cloning of rat poly(ADP-ribose) glycohydrolase: Presence of a potential nuclear export signal conserved in mammalian orthologs. J. Biochem. (Tokyo) 1999;126:748–755. doi: 10.1093/oxfordjournals.jbchem.a022512. [DOI] [PubMed] [Google Scholar]
- 49.Tanuma S, Kawashima K, Endo H. Identification of two activities of (ADP-ribose)n glycohydrolase in HeLa S3 cells. Biochem. Biophys. Res. Commun. 1986;135:979–986. doi: 10.1016/0006-291x(86)91024-7. [DOI] [PubMed] [Google Scholar]
- 50.Tanuma S, Kawashima K, Endo H. Occurrence of (ADP-ribose)N glycohydrolase in human erythrocytes. Biochem. Biophys. Res. Commun. 1986;136:1110–1115. doi: 10.1016/0006-291x(86)90448-1. [DOI] [PubMed] [Google Scholar]
- 51.Tanuma S, Endo H. Purification and characterization of an (ADP-ribose)n glycohydrolase from human erythrocytes. Eur. J. Biochem. 1990;191:57–63. doi: 10.1111/j.1432-1033.1990.tb19093.x. [DOI] [PubMed] [Google Scholar]
- 52.Uchida K, Suzuki H, Maruta H, Abe H, Aoki K, Miwa M, Tanuma S. Preferential degradation of protein-bound (ADP-ribose)n by nuclear poly(ADP-ribose) glycohydrolase from human placenta. J. Biol. Chem. 1993;268:3194–3200. [PubMed] [Google Scholar]
- 53.Maruta H, Inageda K, Aoki T, Nishina H, Tanuma S. Characterization of two forms of poly(ADPribose) glycohydrolase in guinea pig liver. Biochemistry. 1991;30:5907–5912. doi: 10.1021/bi00238a014. [DOI] [PubMed] [Google Scholar]
- 54.Ame JC, Jacobson EL, Jacobson MK. Molecular heterogeneity and regulation of poly(ADPribose) glycohydrolase. Mol. Cell. Biochem. 1999;193:75–81. [PubMed] [Google Scholar]
- 55.Bonicalzi ME, Haince JF, Droit A, Poirier GG. Regulation of poly(ADP-ribose) metabolism by poly(ADP-ribose) glycohydrolase: Where and when? Cell Mol. Life Sci. 2005;62:739–750. doi: 10.1007/s00018-004-4505-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin W, Ame JC, Aboul-Ela N, Jacobson EL, Jacobson MK. Isolation and characterization of the cDNA encoding bovine poly(ADP-ribose) glycohydrolase. J. Biol. Chem. 1997;272:11895–11901. doi: 10.1074/jbc.272.18.11895. [DOI] [PubMed] [Google Scholar]
- 57.Keil C, Grobe T, Oei SL. MNNG-induced cell death is controlled by interactions between PARP-1, poly (ADP-ribose) glycohydrolase and XRCC1. J. Biol. Chem. 2006 doi: 10.1074/jbc.M606470200. [DOI] [PubMed] [Google Scholar]
- 58.Gao H, Coyle DL, Meyer-Ficca ML, Meyer R, Jacobson EL, Wang ZQ, Jacobson MK. Altered poly(ADP-ribose) metabolism impairs cellular responses to genotoxic stress in a hypomorphic mutant of poly(ADP-ribose) glycohydrolase. Exp. Cell Res. 2007 doi: 10.1016/j.yexcr.2006.12.025. in press. [DOI] [PubMed] [Google Scholar]
- 59.Truscott KN, Brandner K, Pfanner N. Mechanisms of protein import into mitochondria. Curr. Biol. 2003;13:R326–37. doi: 10.1016/s0960-9822(03)00239-2. [DOI] [PubMed] [Google Scholar]
- 60.Scovassi AI. Mitochondrial poly(ADP-ribosylation): From old data to new perspectives. FASEB J. 2004;18:1487–1488. doi: 10.1096/fj.04-1841rev. [DOI] [PubMed] [Google Scholar]
- 61.Dawson VL, Dawson TM. Deadly conversations: Nuclear-mitochondrial cross-talk. J. Bioenerg. Biomembr. 2004;36:287–294. doi: 10.1023/B:JOBB.0000041755.22613.8d. [DOI] [PubMed] [Google Scholar]
- 62.Adamczyk A, Czapski GA, Jesko H, Strosznajder RP. Non abeta component of alzheimer's disease amyloid and amyloid beta peptides evoked poly(ADP-ribose) polymerase-dependent release of apoptosis-inducing factor from rat brain mitochondria. J. Physiol. Pharmacol. 2005;56(Suppl 2):5–13. [PubMed] [Google Scholar]
- 63.Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. U. S. A. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ying W, Sevigny MB, Chen Y, Swanson RA. Poly(ADP-ribose) glycohydrolase mediates oxidative and excitotoxic neuronal death. Proc. Natl. Acad. Sci. U. S. A. 2001;98:12227–12232. doi: 10.1073/pnas.211202598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cozzi A, Cipriani G, Fossati S, Faraco G, Formentini L, Min W, Cortes U, Wang ZQ, Moroni F, Chiarugi A. Poly(ADP-ribose) accumulation and enhancement of postischemic brain damage in 110-kDa poly(ADP-ribose) glycohydrolase null mice. J. Cereb. Blood Flow Metab. 2006;26:684–695. doi: 10.1038/sj.jcbfm.9600222. [DOI] [PubMed] [Google Scholar]
- 66.Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. U. S. A. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eisfeld J, Luckhoff A. TRPM2. Handb. Exp. Pharmacol. 2007;179:237–252. doi: 10.1007/978-3-540-34891-7_14. [DOI] [PubMed] [Google Scholar]
- 68.Yang KT, Chang WL, Yang PC, Chien CL, Lai MS, Su MJ, Wu ML. Activation of the transient receptor potential M2 channel and poly(ADP-ribose) polymerase is involved in oxidative stress-induced cardiomyocyte death. Cell Death Differ. 2006;13:1815–1826. doi: 10.1038/sj.cdd.4401813. [DOI] [PubMed] [Google Scholar]