

Abstract
We have determined the specificity profile of the homing endonuclease I-AniI and compared it to the conservation of its host gene. Homing endonucleases are encoded within intervening sequences such as group I introns. They initiate the transfer of such elements by cleaving cognate alleles lacking the intron, leading to their transfer via homologous recombination. Each structural homing endonuclease family has arrived at an appropriate balance of specificity and fidelity that avoids toxicity while maximizing target recognition and invasiveness. I-AniI recognizes a strongly conserved target sequence in a host gene encoding apocytochrome b, and has fine-tuned its specificity to correlate with wobble vs. non-wobble positions across that sequence, and to the amount of degeneracy inherent within individual codons. The physiological target site in the host gene is not the optimal substrate for recognition and cleavage: at least one target variant identified during the screen is bound more tightly and cleaved more rapidly. This is a result of the periodic cycle of intron homing, which at any time can present nonoptimal combinations of endonuclease specificity and insertion site sequences in a biological host.
Homing is the transfer of an intervening genetic sequence (either an intron or intein) to a homologous allele that lacks that same sequence 1–3, leading to gene conversion and transmission of the mobile element. Homing endonucleases promote the mobility of these intervening sequences, which include their own reading frames, by generating double strand breaks in homologous alleles that lack the intron or intein. Break repair leads to transfer of the element via homologous recombination, using the allele that contains the homing endonuclease gene as a template. In rare cases, homing endonucleases can also be encoded by free-standing genes 4. In either case, homing endonuclease genes (HEGs) are selfish DNA sequences that are inherited in a dominant, non-Mendelian manner5–7.
Homing endonucleases and their mobile introns are found in virtually all microbial genomes, including phage, bacteria, archaea, protista, and organellar genomes in fungi and algae 1; 2; 8–10. A variety of analyses have indicated that mobile introns and homing endonucleases display a periodic life cycle in these host genomes 5–7: invasion of a DNA target is followed by subsequent vertical transmission, eventual loss of endonuclease activity in individual hosts, loss of the endonuclease reading frame and eventually the intron, and subsequent reinvasion by an active homologue. In the case of phage and prokaryotes, this cycle causes the persistence of introns in genomes that are otherwise subject to strong purifying selection and streamlining 11.
Homing endonucleases recognize long DNA sequences (typically 14 to 40 base pairs), which reduces their toxicity in the host organism. Homing endonucleases tolerate sequence variation at individual base pairs within their targets; this attenuated fidelity enables them to adapt to sequence drift in their target and increases their potential for ectopic transfer. There are at least five discrete homing endonuclease families, each of which has arrived at the optimal balance of specificity vs. fidelity that is most suitable for evolutionary success in their host genomes. These families are classified and named according to their most conserved sequence and structural motifs, and generally are localized and constrained to distinct biological and genomic niches 12.
Members of the largest family, termed ‘LAGLIDADG’ homing endonucleases (LHEs), are most often encoded in archaea and in the mitochondrial and chloroplast genomes of single-cell eukaryotes 13. LHEs are segregated into two subfamilies, based on the presence of either one or two copies of the conserved LAGLIDADG motif per protein chain. Enzymes that contain a single copy of this motif act as homodimers, and recognize consensus DNA targets sites that are constrained to palindromic or near-palindromic symmetry. In contrast, LHEs that have two copies of the motif act as monomers, possess two structurally similar nuclease domains, and are free to evolve towards recognition of asymmetric DNA targets. The protein domains are connected by flexible linker peptides with lengths ranging from 3 residues to over 100 residues 14. Free-standing LAGLIDADG endonucleases (those that are not associated with additional protein domains) recognize DNA sites that range from 18 to 22 base pairs 15–18. They cleave both DNA strands, generating mutually cohesive four base 3′ overhangs 15; 1; 2. Like most nucleases, LHEs require divalent cations for activity.
The structures of several LHEs bound to their DNA targets have been determined. These include three homodimers (I-CreI 21–24, I-MsoI 24 and I-CeuI 25); two pseudosymmetric monomers (I-AniI 26 and I-SceI 27), one artificially engineered chimeric protein (H-DreI 28), and one intein-associated endonuclease (PI-SceI 29). Structures have also been determined of several additional LHEs in the absence of DNA. Although the primary sequences, DNA target specificities and oligomeric architectures of LHEs have diverged significantly, their overall topologies, tertiary folds, and DNA-binding mechanisms are still strongly conserved.
Many LAGLIDADG homing endonucleases are encoded within introns that interrupt highly conserved rDNA genes, each of which can can simultaneously harbor large numbers of separate group I introns with associated homing endonuclease reading frames (for example, the 23S rDNA in Chlorosarcina brevisisinosa) 30; 31. The specificity of two of such endonucleases (I-CreI and I-CeuI, homodimers encoded in introns within the 23S rDNA of Chlamydomonas reinhardtii and Chlamydomonas eugametos, respectively) has been previously measured and found to be strongly correlated with the number and type of protein-DNA contacts found in the DNA-bound complex 24; 25. Mobile introns can also inhabit protein-coding genes in many microbial species, including phage (for example, the DNA polymerase gene in SPO1 and Bastile 32), in bacteria (for example, the nrdE gene in Bacillus anthracis 33) and and in organellar genomes (such as the I-AniI endonuclease described in this work). However, these mobile introns are typically found at fewer sites within individual host genes, and their specificities are generally less well characterized.
The I-AniI protein is a monomeric 250 residue LAGLIDADG endonuclease, encoded within a group I intron that interrupts the mitochondrial apocytochrome b oxidase (cob) gene of Aspergillus nidulans (A.n.) 34. I-AniI recognizes and cleaves a 19 base pair asymmetric DNA target site. The enzyme binds its DNA substrate with approximately nanomolar affinity, and cleaves the noncoding upper strand more rapidly than the coding lower strand 35; 36. Homologous introns and homing endonucleases are found in the same genetic location in related host organisms, including Saccharomyces cerevisiae 37 and Venturia inaequalis 38. These two proteins display 49 and 65% sequence identity, respectively, to I-AniI. Expanded analyses of recently deposited cob sequences from microbial genomics projects indicates that at least one additional position in the gene has been invaded by a mobile intron 39; the specificity of one of these (I-CsmI, from Chlamydomonas smithii) has also been studied in detail, with many of those results agreeing with the analysis described here 40.
I-AniI and its host gene offer an excellent system for a detailed analysis of the coevolution of a homing endonuclease and a corresponding protein-encoding host gene and target site. In this study we describe the specificity profile and overall fidelity of the I-AniI endonuclease at each base pair of its target site using a high-throughput cleavage screen of a randomized substrate library. We identify and characterize one target variant that is a significantly superior substrate as compared to the physiological DNA target sequence in Aspergillus. The specificity profile of the endonuclease is significantly correlated to the reading frame of the host gene, down to the position of individual basepairs at wobble vs. non-wobble positions in individual codons. This analysis illustrates the degree to which homing endonuclease specificity is ‘fine-tuned’ to its natural target site, and also demonstrates the extent to which a viable intron invasion site can be diverged from an optimal DNA target sequence for a homing endonuclease.
RESULTS
Recovery of cleavable DNA substrates
The site specificity of the I-AniI homing endonuclease was assayed across its entire 19 base pair DNA recognition target, by selection of cleavable DNA site variants from a randomized site library under optimal cleavage conditions as described above. The information content (specificity) was calculated at each base pair using the published algorithm of Schneider et al. 41
75 unique cleavable target sequences were identified in the screen (Figures 2 and S2). Enriched from an initial library containing an average of 6.7 mutations per target site, two-thirds of the cleavable substrates (50 of 75) contain a single base pair substitution, while the majority of the remainder contain two (15 of 75) or three (5 of 75) mismatches.
Figure 2. Target sites recovered in a specificity measurement screen of the I-AniI endonuclease.

In this experiments, 428 substrate targets were sequenced. The wild-type site was isolated twice in this screen and is indicated; individual mismatches in other sites, relative to the wild-type target, are highlighted in yellow. The center of the target site (dividing the left and right half-sites) is indicated with the vertical line. In addition to individual sequences of cleavable substrates, the number of individual parental clones for each target can be identified, based on unique sequences of randomized, addressible flanking bases. Out of 76 unique sequences, 31 appeared more than once, out of which half were generated from multiple parental site clones. The frequency at which individual sites were recovered did not correlate strongly with their performance in kinetic cleavage assays; however at least one site (‘Lib4’) is a much better substrate than the physiological, wild-type target sequence (see Figure 4).
Seven unique sites account for 294 of 428 recovered DNA targets, corresponding to over 65% of the isolated endonuclease substrates in this experiment (Figure 2). The remainder of the cleavable DNA sequences were recovered at much lower frequencies. The enhanced recovery of the most frequently isolated target sequences was not due either to the initial bias of the initial DNA template synthesis (as demonstrated by sequencing and analysis of clones in the naive library), nor to a significant bottleneck in early cycles of the selection (all but one of these frequently recovered substrates arose from multiple parental clones).
As described below, the more frequently recovered DNA target sequences are usually found to be cleaved slightly more rapidly than are those that were recovered only once. One of the most frequently recovered substrates (labeled ‘Lib4’ in Figure 2), containing two basepair substitutions, was found to be a superior substrate as compared to the wild-type sequence from Aspergillus and to other sequences recovered from the library that were tested in detail.
In addition to the cleavable targets analyzed in this study, one clone containing a site that harbored an unusually high number of mismatches (8) relative to the wild-type I-AniI target was recovered once in the final output of the selection. Subsequent in vitro analyses of this site indicated that I-AniI exhibits no cleavage activity on this site under conditions described above, and likely represents a spurious target site sequence that slipped through the target site screen. The remaining target sites that were retested individually for cleavage were viable substrates for the enzyme.
Site specificity and fidelity
While individual base pair substitutions can apparently be tolerated by the endonuclease at many positions under the reaction conditions used in this experiment, on average the cleavable substrate sequences only deviated by 1.4 mismatches from the cognate physiological target site (relative to the average of 7 mutations per site that were encorporated in the naive library). The specificity of DNA recognition at individual base pairs is highly variable across the target site (Figure 3a). Nine positions (−9, −7, −6, −4, −3, +3, +4 +6 and +7) display nearly invariant base preference, four positions (−5, −2, +1 and +8) display little or no sequence preference, and the remaining six sites (−10, −8, −1, +2, +5 and +9) display attenuated specificity. In addition, two positions (−1 and +1) display mild base covariation, with an A→C substitution at position +1 dependent on the conservation of the wild-type A:T base pair at the neighboring −1 position. These positions correspond to the direct center of the DNA target site, and are subject to significant base unstacking upon binding of the enzyme. Such positions in DNA target sequences often display sequence-specific conformational preferences that contribute to overall protein binding specificity.
Figure 3. Specificity of DNA recognition by I-AniI.

Panel a. Information content at individual positions across the I-AniI target site, calculated using recovered sequences from a screen of site variants. The corresponding translated protein sequence of the host gene is provided below the DNA target sequence (note that in fungal mitochondrial genomes, ‘TGA’ encodes for Trp rather than ‘Stop’). The information content (specificity of recognition) was calculated at each basepair according to the method of Schneider, et al., 1986; “2” = invariant base preference). Arrows indicate wobble positions in host coding frame (average information content = 1.0, compared to 1.7 for non-wobble positions). Panel b. Cleavage of individual single base substitutions across the ‘central four’ positions in the target site. Three substitutions (−1G, +2G and +2A) are strongly disfavored; two (+1 G and A) are equivalent or superior to wild-type; the remainder are slightly reduced but within 2-fold of wild-type cleavage efficiency.
Across the central four basepairs of the target site (which are not in direct contact with protein atoms, and are subject to binding specificity primarily as a result of DNA bending), three individual basepair substitutions (−1 T→G; +2 C→G and +2 C →A) completely inhibit target site cleavage, while two substitutions (+1T→C and +1T→A) yield substrate cleavage efficiencies equal or superior to the wild-type target (Figure 3b). The remaining 7 individual subsitutions across the central four bases yield relatively minor changes in cleavage efficiency. The T→A substitution as position +1 is also observed in the hypercleavable ‘Lib4’ target site (Figure 1) and was isolated as an individual substitution from the randomized site library screen.
Figure 1.

Schematic of methodology for in vitro screen for cleavage target site variants. See main and supplementary text for a full description of experimental details. Panel a: Design of the cyclical screen and enrichment for cleavable target sites. Panel b: progression of screen from naive library to final convergence in round 4.
The overall specificity of I-AniI is extremely high, even under optimal in vitro cleavage conditions as described here. The total number of potential DNA target sequences for this endonuclease is 419 or 2.7 × 1011 sites for the 19 basepair target site. Based solely on the information content at each basepair described above, approximately 16384 of these sequences are predicted to be cleavable (corresponding to nine positions that are invariant or nearly so, four positions that can tolerate any of the possible basepairs, and six positions that can approximately accomodate either of two basepairs: 19 × 26 × 44 = 16384). However, this number represents a significant underestimate of the enzyme’s substrate specificity, as it assumes that substitutions at all ten variable positions listed above can be accomodated by the enzyme simultaneously (taking into account neither covariation between adjacent basepairs, or the observation that cleavable substrates isolated in this screen harbor an average of only 1.4 mutations per site). Thus, the number of theoretically cleavable targets should be reduced by at least 7-fold (the ratio of 1.4 mutations per cleavable site on average to the 10 basepair positions that can potentially be altered in any given substrate). This results in a prediction of approximately 2340 cleavable sites out of over 2 × 1011 potential substrate sequences (or one cleavable site per 108 random sequences). Extrapolated to a mammalian genome (~109 basepairs), one would predict that I-AniI should be capable of cleaving approximately 10 sites per genome under optimal in vitro cleavage conditions. However, under less optimal reaction conditions (for example, reduced enzyme concentration, or in the presence of high concentrations of competing DNA binding proteins and highly ordered chromatin structure as found in vivo), as many ‘marginal’ substrates would likely be poorly bound and cleaved. This observation and interpretation is in agreement with many reported studies on the transfection of mammalian and/or human cells with nuclear-localized homing endonucleases, including I-SceI 42; 43 and I-PpoI 44, that demonstrate levels of double strand break foci and cell toxicity that are not significantly above background measurements for untransfected cells. Similarly, the transformation of human cells with the I-AniI reading frame, and subsequent expression and nuclear localization of high levels of active endonuclease, does not appear to cause measurable cell toxicity or elevated genomic double strand breaks, even though at least four potentially cleavable targets are known to exist in the human genome (M.S.K and B.L.S., unpublished observations).
Correlation of I-AniI specificity with constraints of its host target gene sequence
The mitochondrial apocytochrome B (cob) gene in fungi has been the target of at least two historical invasion events by a LAGLIDADG homing endonuclease, one of which is represented by I-AniI and its close homologues in Saccharomyces cerevesiae and Venturia inequalis 37–39. This protein-encoding host gene shows a typical pattern of conserved and nonconserved residue positions, with nonconserved amino acids observed at approximately 25% of the positions in its reading frame (Figure 4). Both group I introns are located at positions in the cob gene that encode exceptionally well-conserved amino acid sequences, each extending over approximately 6 codons. The translated sequence for the I-AniI target site (using the fungal mitochondrial genetic code, where TGA = ‘Trp’, rather than ‘stop’) is ‘WGGFSV’, a peptide sequence that lies within a surface-exposed, tight turn that connects two helices near the enzyme active site (Figure S3). The two glycine residues in this sequence appear to be essential for the structure of this region of the enzyme subunit, as both exhibit backbone dihedral conformations that are disallowed for all other protein residues. Two of the three large hydrophobic residues (W and V) lie in the core of the protein, while the phenylalanine (F) is completely solvent exposed. Apparently, this particular protein sequence is tightly constrained to maintain the function of the enzyme.
Figure 4. Conservation of apocytochrome B (‘cob’) gene from sequenced microbial genomes, and position of known inserted group I introns.

For the A.n. cob protein sequence, a set of homologous sequences was collected using an iterative PSI-BLAST search of the non-redundant protein sequence database. A multiple sequence alignment was made from the blast output, allowing the for the calculation of information content (IC) at each position. The IC values were linearly normalized from zero to 1, with zero corresponding to the least conserved position and 1 corresponding to the most conserved positions. The arrows point to conserved regions of the host gene targeted by I-CsmI and I-AniI.
Comparison of the specificity of I-AniI at each basepair in its cognate target sequence with the reading frame of the host gene demonstrates a very strong correlation between specificity at individual DNA bases, and the constraints on each of those bases imposed by the degeneracy of the amino acid code (Figure 3a). The average information content at non-wobble positions in the target sequence is approximately 1.7, whereas the same measurement of specificity at wobble positions indicates a much lower value of 1.0 (i.e. the endonuclease is more tolerant of target site variation at those positions). Furthermore, the reduced specificity at the wobble positions of the host gene is most pronounced for three of the more degenerate codons in the target site, that encode two glycines and one valine, respectively. In contrast, two of the wobble positions that display only slightly reduced, ‘intermediate’ specificities correspond to the host codons for Tryp and Phe (each of which allow only two possible bases at their wobble position). Therefore, the correlation of specificity of the homing endonuclease, relative to sequence constraints on the host gene coding sequence, is extraordinarily ‘fine-tuned’ to the most possible forms of genetic variation that can occur in a highly constrained target position. Once encorporated into a host target site, it seems likely that subsequent vertical transmission of a homing endonuclease gene is maximized by continuing to evolve to be particularly tolerant of those DNA base substitutions that are silent during protein translation, and thus are likely to experience accelerated genetic drift.
The one notable ‘outlier’ in the specificity profile and trend described above is the lack of obvious specificity at position +1. While there is not an obvious explanation for this result, it is worth noting that this position happens to be one of two that is decidedly ‘suboptimal’ for recognition (being one of two basepairs that are mutated in a highly cleavable ‘Lib4’ site, described below), which may be reflected in the low measured information content at that position.
Comparison of wild-type and variant target sites as endonuclease substrates
In order to determine if the relative recovery of various DNA target sequences was strongly correlated with their behavior as endonuclease substrates, a panel of sites from the screen was characterized in more detail using in vitro cleavage assays (Figure 5). Five of the most frequently isolated targets, five of the targets isolated once each, and the wild-type target from Aspergillus, were subjected to individual kinetic cleavage analyses. These experiments demonstrate that the more ‘successful’ targets from the screen are cleaved an average of 50% more rapidly than those that were isolated only once each.
Figure 5. Relative behavior of isolated target sites as cleavage substrates for I-AniI.

Panel a: Kinetics of cleavage under reaction conditions as described in ‘methods’. Extent of cleavage quantitated at 30, 60, and 120 minutes. The numbers above the individual substrate bars correspond to the number of times each sequence was isolated in the screen, as shown in the first column of Figure 1. Note the complete rapid digestion of ‘Lib4’ relative to the wild-type target sequence. Panel b: Relative binding of wild-type and ‘Lib4’ substrates by the I-AniI endonuclease, determined by isothermal titration calorimetry. The Kd values are averages of multiple independent experiments conducted on separate days, which reproducibly indicate that the ‘Lib4’ sequence is bound approximately 10-fold more tightly than the wild-type sequence. While the estimated molar ratios of binding for the experiments shown in these panels are slightly above and below 1:1, respectively, the average of multiple runs in each case indicates a 1:1 binding stoichiometry of protein to DNA duplex. The variation in this value illustrated in this figure is typical of ITC runs with homing endonuclease/DNA systems.
One of those target sites (‘Lib4’) displayed much more efficient cleavage than the wild-type target (Figure 5). Binding analyses of the relative affinity of this substrate compared to the wild-type target indicated that this target site is bound approximately 10-fold more tightly (with a reduction in ΔGbinding of −1.5 kcal/mol) than the wild-type sequence. This improved affinity is largely driven by a significant improvement (−4.5 kcal/mol) in the enthalpy change (ΔH) upon binding, which implies that the new target site provides new contact points (presumably additional hydrogen bonds) to the protein.
This target site contains two basepair substitutions, both in the right DNA half-site, at basepairs +1 and +8. The endonuclease does not display obvious direct contacts to either of these basepairs in the wild-type complex, also implying that these two substitutions might introduce novel interactions in the protein-DNA interface that enhance affinity and improve cleavage efficiency. Subsequent cleavage analyses of two separate sites that each harbor a single individual substitution present in the ‘Lib4’ site demonstrate that both contribute to tighter binding and more rapid cleavage, with the majority of improved recognition derived by the A→T substitution at position +8 (data not shown). Neither the ‘Lib4’ sequence, nor a sequence containing the single mismatch at position +1 is found (both of which would encode a Phe to Leu point mutation in the translated host protein) is found in a search of all currently sequenced cob genes. In contrast, approximately 30 known cob sequences contain the ‘Lib4’ mismatch at basepair +8, which corresponds to a silent mutation at the wobble position within the valine codon.
Given the evolutionary dynamics of mobile introns and homing endonucleases (and most if not all other invasive genetic elements), it would not be surprising if many naturally isolated combinations of endonuclease and homing site sequences represented non-optimal pairing of enzyme specificity and substrate sequence, as described above for I-AniI. A variety of analyses have indicated that mobile introns and homing endonucleases display a periodic life cycle in their host genomes 5–7. Horizontal transfer into a novel DNA target is followed by subsequent vertical transmission and expansion across closely related homologous genes, eventual loss of endonuclease activity in individual hosts, loss of the endonuclease reading frame and eventually the intron, and subsequent reinvasion by an active homologue. Estimates of the periodicity of intron invasion, loss and reinvasion are typically several million years.
The structure of the DNA-bound endonuclease complex: details of recognition and cleavage
The I-AniI/DNA complex structure 26 was extended to 2.4 Å resolution, giving a more detailed view of both the protein/DNA interface and the I-AniI active site when compared to the previously determined structure. The structure consists of a full-length enzyme monomer and a 28-bp DNA duplex containing the 19-bp native homing site from the Aspergillus mitochondrial COB intron. The refined structure additionally includes 47 water molecules, 10 of which mediate contacts in the protein/DNA interface. Three metal ions were modeled into the overlapping active sites and and the DNA was observed to be uncleaved. Details of the active site metal coordination are provided in supplementary information and Figure S4.
The variability of specific recognition and fidelity across the DNA target site is a function of the contacts made between the endonuclease and the individual basepairs of the substrate. Side chains from the β-sheet DNA recognition surface of each individual I-AniI domain contact nucleotide bases in each of the two corresponding DNA target half sites (Figure 6). All contacts to nucleotide bases are made in the major groove. Eighteen of the 38 phosphate groups (47%) are in direct contact with the protein. A total of 18 direct contacts, ten of which are water-mediated, are made between hydrogen-bond partners on the protein and corresponding hydrogen-bond donors and acceptors on DNA bases in the target site. This corresponds to roughly 30% of the potential hydrogen-bond sites across the DNA target’s major groove.
Figure 6.
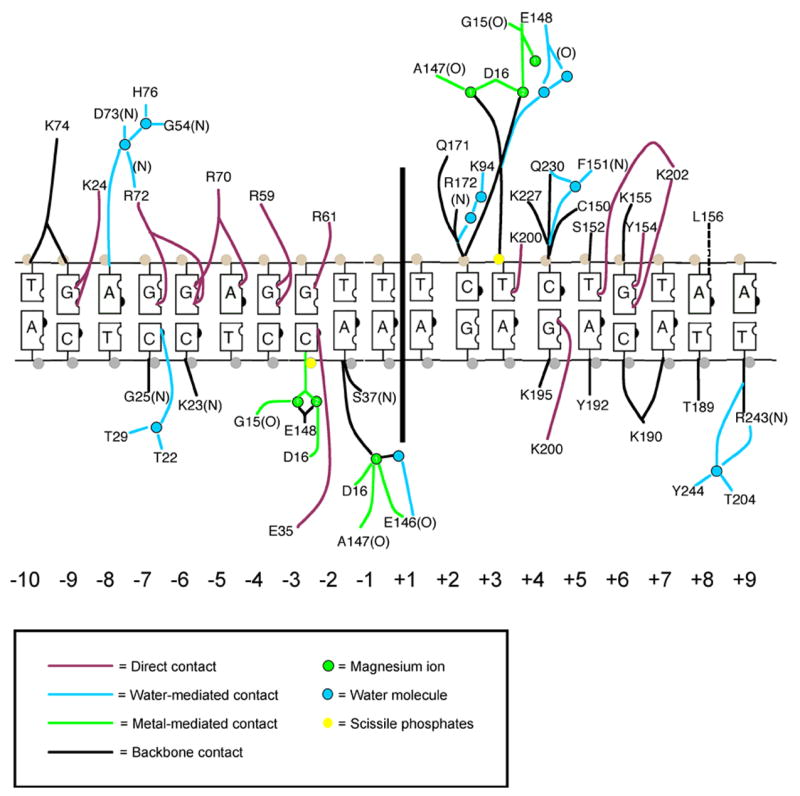
I-AniI/DNA interface and contacts, modeled upon refinement of structure to 2.4 Å resolution. The top strand corresponds to the noncoding strand of the cob host gene. The acidic residues from the LAGLIDADG motif that are involved in metal binding are D16 and E148.
I-AniI interacts with its DNA target through a strikingly asymmetric distribution of contacts, with 12 contacts made to bases in the left half site, and five to bases in the right half site. In addition, the majority of direct nucleotide base contacts (15 of 18; 83%) are made in the top, noncoding strand of the target DNA. Arginine contacts to a run of purine bases in the top strand of the left half site account for eight of the direct contacts. These interactions correspond to the DNA target sequence 5′-GGAGG-3′ from base pairs −7 to −3, and protein residues Arg 72, Arg 70, Arg 59, and Arg 61. Two of these residues, Arg 59 and Arg 72, contact two neighboring purine residues simultaneously.
Unlike the previously studied I-CreI LAGLIDADG homing endonuclease 24, I-AniI exhibits a relatively loose correlation between measured information content at individual base pairs and the number of direct contacts at each position within the target site. Positions +7 and +9, for example, exhibit high information content (1.9 and 1.6, respectively), yet I-AniI makes no direct contacts to nucleotides at these positions. One partial explanation for this difference in behavior might be that whereas I-CreI appears to be highly optimized for recognition of its cognate target site in Chlamydomonas (displaying a cognate Kd value of ~ 1 nM), I-AniI is clearly less well optimized towards its target site in Aspergillus, as described above: at least two positions in the target site (not visualized in the ‘wild-type’ complex) yield tighter binding when mutated, and may provide additional contacts that improve the correlation between structure and binding specificity.
In addition, the structure of I-CreI was determined at 1.9 Å resolution, allowing us to model a total of 28 water molecules that mediate contacts in the protein/DNA interface, accounting for about half of the protein-DNA contacts. In contrast, only ten water molecules can be confidently modeled into the I-AniI protein/DNA interface in the 2.4 Å resolution structure. Therefore, the structure may be missing several important contact-mediating water molecules from the protein/DNA interface that can only be visualized at higher resolution.
The coordinates of the I-AniI/WT DNA complex have been deposited in the RCSB database; accession code 2QOJ.
DISCUSSION
The success or failure of a homing endonuclease lineage is dictated by the balance of specificity and fidelity across its target site, relative to host constraints of the same sequence
Consideration of the I-AniI homing endonuclease and its most well-studied cousins from the LAGLIDADG family, particularly similar studies on I-CsmI from the same host gene 40, illustrates that the most successful homing endonucleases display several evolutionary mechanisms for optimizing their recognition of and persistance in genomic host sites:
(1) Such elements have arrived, by random sampling, at substrate specificities that correspond to highly conserved, and therefore static, sequences in potential host genes. This observation has been made previously for mobile introns specific both for structural RNA genes (for example, 30; 45) and for protein-encoding genes (for example, 46). In this latter study, the homing endonuclease I-TevI (a member of the much less specific GIY-YIG endonuclease family, generally found in phage genomes) was shown to bind a DNA target site in its phage thymidylate synthase host gene that corresponds to functionally critical active site residues in the host enzyme, including one critical base common to all known TS gene sequences.
If a host gene is essential or valuable to the host organism, then purifying selection pressure across the gene is likely to be higher and therefore more likely to produce ‘static’ sequence targets for invasion. Furthermore, once an endonuclease and its element (intron or intein) have become established, only a precise deletion of the element from the host gene would re-establish an intron or intein-less allele for reinvasion. Any ‘sloppy’ deletion event would likely create a mutation within a critical region, rendering the gene product non-functional. Thus, endonucleases select against deletion by targeting essential genes, especially if the target site corresponds to an active site or other functionally critical region of the protein.
(2) Homing endonucleases optimize an appropriate balance of relative specificities at individual basepairs of the target site that corresponds to the underlying constraints imposed by the host gene sequence (for additional experimentally validated examples, including a similar study of the I-CsmI homing endonuclease from the mitochondrial gene in Chlamydomonas smithii 40, see 16; 18; 47–49). In the case of I-CsmI, the endonuclease as shown to display a “slight but significant tendency…to cleave substrates containing a silent or tolerated amino acid change more efficienty than nonsilent or nontolerated ones”.40
In the case of host genes encoding structural tRNA and rRNA molecules, constraints imposed by the gene product itself would include conservation of basepairs and longer range tertiary interactions required for function in the folded RNA molecules. These constraints can be quite strong, as only four unique bases are encoded within these nucleic acids. Therefore, many highly structured RNA molecules tend to be extremely well conserved across biological kingdoms and therefore somewhat static targets for invasion by homing endonuclease-intron systems. Additionally, the preponderance of base-paired structural elements in RNA structures increases the amount of palindromic symmetry in many potential target sites, encouraging the success and persistance of homodimeric homing endonucleases. These host genes can thus serve as a reservoir for such proteins, which can then undergo gene duplication and fusion into monomeric endonucleases, which greatly expands their available target site repertoire.
In contrast, host genes that encode proteins appear to be more challenging targets for invasion, for at least three reasons: (i) the combination and interactions of the twenty possible amino acids within unique protein fold families are more readily and rapidly diverged during evolution than are the interactions of nucleic acids; (ii) the genetic code for amino acid sequences is partially degenerate, allowing synonymous (silent) mutations that can inhibit homing endonuclease binding while maintaining the structure and function of the host protein; and (iii) because protein coding sequences are inherently less symmetric than are those that code for rRNA and tRNA.
(3) Many persistent homing endonucleases also acquire secondary activities that provide an advantage to the host, such as assisting in intron splicing, thus generating selective pressure to help maintain endonuclease fold and stability 37; 50; 51. In the case of I-AniI, the homing endonuclease has also evolved to become an essential cofactor for splicing of its cognate group I intron. This ‘maturase’ activity is required for successful expression of the cytochrome b oxidase gene 52; 53. In such a case, one would expect that the loss of endonuclease activity due to random mutations would be reduced, because additional purifying selection pressure will act at many positions on the endonuclease reading frame to preserve its folding and stability, in order to maintain function of the host gene. While a small number of mutations in I-AniI can upcouple its homing endonuclease and maturase activities 26; 35; 54, the number of random mutations in the LHE gene that can abrogate homing activity without reducing the fitness of the host (by decreasing expression of the cob gene) is far fewer than for a comparable homing endonuclease with no additional function. The moonlighting relationship of the two activities can thereby ‘stabilize’ the endonuclease reading frame in the host genome.
The cost to the endonuclease of developing this secondary ‘moonlighting’ activity as a mechanism to help maintain its primary function as a DNA endonuclease is that each activity imposes different, partially overlapping constraints on the identity of individual amino acids in the LHE scaffold. While some of those sequence constraints are primarily structural and thus help to preserve both activities, others are distinct to the two separate functions of the protein and are potentially noncomplementary. Thus, the protein may have difficulty in optimizing either or both activities during evolution, leading to a mobile element that is relatively stable in its particular biological host, but at the cost of reduced efficiency for horizontal transfer.
Concluding remarks: several properties of wild-type homing endonucleases should be optimized for gene targeting
LAGLIDADG homing endonucleases (LHEs) are under intense study by several groups as possible reagents for gene targeting applications 55; 56. The experiments reported here, as well as a variety of published studies from several groups 55, clearly indicate that (i) the optimal DNA substrate of an LHE does not necessarily coincide with its physiological target in its cognate biological host, and (ii) most if not all homing endonucleases, while inherently very specific due to their long target sites, are capable of even greater specificity because they have evolved to only be specific enough to avoid host toxicity.
This study demonstrates that the I-AniI endonuclease, and presumably its cousins throughout the LAGLIDADG family display more than sufficient specificity to be potential reagents for gene-specific applications. However, wild-type homing endonucleases often display a variety of biophysical characteristics that are suboptimal for use in biotech or therapeutic applications, as a result of the lack of host pressure to maintain their homing function. This can include instability and/or insolubility upon expression and nonoptimal DNA affinity for various purposes. As well, the presence of additional biochemical activities on the same protein scaffold (such as protein splicing for intein-associated homing endonucleases, or protein and RNA binding for various maturase-homing endonuclease proteins) can impose constraints on their structure that interferes with optimal DNA cleavage activity. In the case of I-AniI, maturase activity may be responsible for conservation of surface residues on the C-terminal domain that reduce the protein’s solubility and also impart RNA-binding function that may interfere with its DNA endonuclease activity. Even with these caveats, wild-type homing endonucleases have been used in many studies to drive gene conversion events, with little no attendent issues of cell toxicity or elevated nonspecific DNA cleavage 44; 5, and appear promising enough as reagents for targeted genetic applications that it is well worth the effort required to optimize their stability, specificity and biochemical behavior. As a case in point, during the experiments described in this and an earlier study on I-AniI, we were able to easily (i) eliminate RNA intron association as competing binding activity, via a single point mutation 26; (ii) Improve the solubility and solution behavior of the purified protein, via two mutations on its surface distant from the DNA-binding surface; and (iii) identify a substrate variant of the endonuclease target site that is bound and cleaved significantly more efficiently than the wild-type targets.
MATERIALS AND METHODS
Materials
The I-AniI homing endonuclease was expressed and purified from E. coli strain BL21 (DE3)lysS as previously described 26, with the exception that expression was induced at 37°C for 2 to 3 hours after the culture had achieve early log growth phase (OD600 ~ 0.6). Wild-type I-AniI was used for the substrate selection experiments. For binding assays, two point mutations (F80K and L232K) were introduced that improve the solubility of the enzyme and facilitate measurements using isothermal titration calorimetry. Both mutations are far removed from the DNA-binding surface of the enzyme, and are on exposed surface loops (Figure S1).
DNA oligonucleotides for generation of the randomized substrate library, and for subcloning and amplification steps, were purchased from Operon, Inc. (50 nmole scale, salt-free). DNA oligonucleotides for cocrystallization studies and for isothermal titration calorimetry were ordered from IDT, Inc. (1 micromole scale; HPLC and salt-free purifications respectively).
In vitro site specificity screen
A plasmid library containing randomized target site variants for the I-AniI endonuclease was constructed using a straightforward protocol described here and shown schematically in Figure 1a. Additional details of the protocol for library construction are provided in supplementary information. A single ‘template’ oligonucleotide was designed and synthesized that contains, in order, a 5′ EcoRI restriction site for subcloning, the randomized target site (mixed at each position in the target site at a ratio of 60% wild-type base and 13.3% of the three possible substitutions), a 3′ XhoI restriction site for subcloning, and a non-degenerate primer binding site for generation of corresponding randomized double stranded DNA. After annealing a universal primer to the 3′ end of the template, the double-stranded duplex was generated by polymerase extension using Klenow fragment. The site library was digested to yield cohesive ends for subcloning, gel purified and ligated into linearized BlueScript plasmid. To measure the complexity and quality of the library (and subsequently, the progression and convergence of the selection method), randomly selected members of the library were sequenced, and the complexity and average mutational load of the naive library was calculated at each base (Figure 1b). A complexity of 106 unique sites was generated, containing on average 6.7 mutations in each unique target site variant.
The basepair positions that immediately flank the I-AniI target site were also randomized, and thus served as unique sequence ‘labels’. This allowed us to distinguish, for any sites that were recovered more than once, those that arose independently from separate parental clones vs. those derived from single parental clones as a result of a ‘bottleneck’ in the cloning or screening process.
The naïve library was digested for 2 hours at 37°C with purified enzyme under optimal conditions: 10 nM DNA substrates and 10 nM enzyme (a 1:1 ratio is required because LAGLIDADG endonucleases do not readily release free product ends), 50 mM Tris-Cl pH 7.5, 50 mM NaCl, 10 mM MgCl2 and 1 mM DTT. This protocol provides for optimal cleavage of DNA targets by the endonuclease, and therefore yields the most conservative estimate of its specificity. Typically, approximately 1% of the naive library is prone to cleavage in the initial round of digests. Linearized products were purified, religated, and passed on to the next round of selection, and cleavable sites were sequenced at each cycle to monitor progression of the selection. Cycles of site digests and recovery were continued until convergence (which was observed when the number of mutations per site, and the percent of unique sites, both reach a minimum). Progression of the screen and the enrichment of cleavable DNA sequences is illustrated in Figure 1b of supplementary materials.
From the final pool of recovered DNA substrates, 428 cleavable target sites were sequenced, in order to align sufficient unique target sequences to allow accurate calculation of information content and basepair covariation within the site. All sequences were determined in a high-throughput 96-well plate sequencing format using the HLA genotyping facility at the Hutchinson Center.
Using an alignment of all cleavable target sites identified in this screen, the specificity of DNA recognition by I-AniI was calculated at each base pair position of the target site using the method of Schneider et al. 41. This method generates a descriptor of specificity at each DNA basepair (termed ‘information content’, quantitated as ‘bits’ of information) by summing the probability of each possible base being found at each position across the site, relative to the background base content expected at each position based on library design. This form of analysis can be directed against collections of either naturally occurring sites that are recognized by a single protein, or sites that are recovered from a library screen as described in this study. Information content at individual base pairs can range from 0 bits, corresponding to complete degeneracy (25% probability of any base at a position) to 2 bits, corresponding to no degeneracy (100% chance of a unique base at that position).
Discrete cleavage assays of base pair variants at the ‘central four’ basepair positions
In addition to the screen from a randomized site library described above, all individual single basepair mismatches were generated across the central four positions of the I-AniI target site, and tested individually for relative cleavage activity. This was done because these four positions, which are located in between the two scissile phosphates, are within a region of significant protein-induced DNA bending, and are not in direct contact with the enzyme. Therefore, recognition of these bases is largely driven by indirect readout of sequence-specific DNA conformational preferences, and is prone to particularly strong patterns of high or low specificity and fidelity that is independent of protein contacts.
The target site variants containing individual basepair mismatches across these four positions were generated using the same templated polymerase extension protocol described above, except that four separate mutant oligonucleotide templates were used (each randomized at only one of the four central positions of the target site). These templates were combined in a single reaction mixture, annealed with a common universal primer, converted to individual double-stranded duplexes, and cloned in a single ligation and transformation step into BlueScript. In this case, each product construct contains a single point substitution within the central four bases of the target site. Individual constructs were isolated, prepped and sequenced, and a complete matrix of all twelve point substitutions across the central four bases was generated. All substrate concentrations were normalized and aliquotted for individual digests, and each target site variant was individually tested in parallel cleavage assays.
Sequence alignment and information content of the apocytochrome B (cob) host gene
For the A.n. cob protein sequence, a set of homologous sequences was collected using an iterative PSI-BLAST search of the non-redundant protein sequence database. Using the output, a multiple sequence alignment consisting of the 500 most homologous cob sequences was created, corresponding to protein sequence identities ranging from 55% to 95% and E-values ranging from 10−105 to 10−164. The degree of conservation of each protein residue in the host gene was observed by calculating the information content (IC), as described previously 57. For simplicity, the IC values were linearly normalized from 0 to 1, with 1 corresponding to the most conserved positions.
Cleavage assays
Cleavage assay reactions were carried out using target sites in supercoiled plasmid substrates. The I-AniI cleavage buffer and digest condition were both identical to that used for the site library screen: 2 hours at 37°C with 10 nM DNA substrates and 100 nM enzyme in 50 mM Tris-Cl pH 7.5, 50 mM NaCl, 10 mM MgCl2 and 1 mM DTT. Reactions were terminated by the addition of Stop Buffer (2 % SDS, 100 mM EDTA, 20 % glycerol, and 0.2 % Bromophenol Blue). Samples were run on an 0.8% agarose gel, stained with ethidium bromide and imaged with an Eagle Eye II and EagleSight version 3.22 (both Stratagene, La Jolla, CA). Quantification was performed with program ImageJ 1.32j.
Isothermal titration calorimetry
Two sets of DNA oligonucleotides containing either the wild-type I-Ani target site (5′CCTTCCCTGAGGAGGTTTCTCTGTAAACCCTTCC 3′ and its complement; target site is bold) or the ‘hypercleavable’ LIB4 target identified during target site screening experiments (5′CCTTCCCTGAGGAGGTTACTCTGTTAACCCTTCC 3′ and its complement; base pair substitutions relative to wild-type are underlined) were purchased from Operon Biotechnologies. After suspending to 1 mM in STE buffer, complementary strands were combined in a 1:1 ratio and annealed by incubating at 95°C for 10 minutes and slowly cooling to room temperature to form DNA target sites for Isothermal Titration Calorimetry (ITC) binding analysis. G-C anchors at each oligonucleotide end promote stable heterodimer formation and prevent hairpin formation of the pseudo-palindromic site.
Protein and DNA samples were dialyzed overnight into Ani ITC buffer (50 mM Tris, pH 7.5, 50–100 mM NaCl, 10 mM CaCl2) at 4°C. Dialyzed samples were quantified by UV spectroscopy, using extinction coefficients to calculate concentrations. Protein concentrations were further confirmed by Bradford assay as well as by estimation on SDS page gels against commercial protein standards. Sample concentrations ranged from 4–14 μM I-AniI and 22–50 μM dsDNA target.
A VP-ITC MicroCalorimeter was used for all experiments, with the protein sample in the cell and the DNA target in the auto-pipette. Individual runs consisted of 30–40 injections of 5–10 μL each, depending on sample concentrations, and were conducted at 30°C with a 351 rpm stirring speed. A full experiment consisted of three experimental runs injecting DNA into the protein-containing cell, as well as a control run injecting DNA into buffer. Data were fit using Origin 7 SR2 software; both sets of data were well modeled by the one-site fitting algorithm in Origin, and KD values were calculated from the resulting fit parameters.
X-ray crystallography
The resolution of the I-AniI-DNA cocrystal structure was extended to 2.4 Å after screening crystals for improved diffraction behavior. Complementary DNA strands were annealed by incubating for 5 minutes at 90 °C and then allowed to cool to room temperature. The protein was diluted to a concentration of 5 mg/mL, mixed with a 1.2 molar excess of DNA duplex at room temperature and used directly for cocrystallization. Crystals containing wild-type DNA target site were grown at 22 °C by vapor diffusion. A 500 microliter reservoir containing 100 mM KCl, 20 mM MgCl2, 50 mM sodium citrate pH 5.0 and 16 to 22 % PEG 3350 was equilibrated against a 2 microliter drop containing a 1:1 mixture of the protein/DNA complex and the reservoir solution. Crystals grew as plates within two weeks, and were flash-frozen in mother liquor described above, augmented with 30% PEG 3350 and 20% glycerol v/v. The crystals are space group P21 and diffract to approximately 2.4 Å resolution.
The structure of the I-AniI/DNA complex was solved by molecular replacement using CNS 58 with the lower-resolution I-AniI/DNA complex structure as an initial search model. The structure was modeled in COOT 59, and refined to 2.4 Å using REFMAC with 5% of the data set aside for cross-validation. The final refinement statistics are Rwork/Rfree = 0.216/0.276. Geometric analysis of the structures using PROCHECK 60 indicates over 90% of the residues are in the most favored region. Refinement statistics are provided in Table 1.
Table 1.
Crystallographic data and refinement Statistics for wild-type I-AniI/DNA
| Data Collection | |
| Space Group | P21 |
| Resolution (highest shell) | 50-2.4 (2.47 - 2.4) |
| Unit Cell dimensions (Å) | a = 60.4 b = 72.7 c = 61.1 β = 103.9 |
| Wavelength | 1.000 |
| Total Reflections | 48322 |
| Unique Reflections | 14643 |
| Completeness (%) | 92.1 (76.75) |
| Redundancy | 3.3 (2.1) |
| Rmerge | 0.035 (0.140) |
| Average I/σI | 18.5 (5.7) |
| Refinement | |
| Rwork | 0.219 (0.229) |
| Rfree | 0.276 (0.320) |
| r.m.s.d. bond length (Å) | 0.001 |
| r.m.s.d. bond angles (°) | 1.25° |
| Protein residues | 275 |
| Nucleotides | 60 |
| Metal ions | 3 |
| Water molecules | 47 |
| Ramachandran Distribution (% core, allowed, generous, disallowed) | 92.3, 5.4, 1.6 0.7 |
| Avg. B-factors (Å2) | 29.9 |
Supplementary Material
Acknowledgments
The gene encoding I-AniI was originally provided as a kind gift of Drs. Richard Waring and Mark Caprara. We thank Ray Monnat, Jr. and Andy Scharenberg for many critical contributions and suggestions to experimental design and interpretation; Jill Bolduc for collection and processing of crystallographic data, Dan Geraghty and lab (Clinical Research, FHCRC) for access to high-throughput sequencing instrumentation and associated assistance and training, and Betty Shen for assistance and instruction in model building and refinement. We also thank an anonymous reviewer for allowing us to directly quote their comments in the discussion. This work was supported through NIH grant R01 GM49857 (BLS), NIH training grant T32 GM09657 (MSK) and an NSF predoctoral fellowship (AMS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Belfort M, Perlman PS. Mechanisms of intron mobility. J Biol Chem. 1995;270:30237–40. doi: 10.1074/jbc.270.51.30237. [DOI] [PubMed] [Google Scholar]
- 2.Dujon B. Group I introns as mobile genetic elements: facts and mechanistic speculations--a review. Gene. 1989;82:91–114. doi: 10.1016/0378-1119(89)90034-6. [DOI] [PubMed] [Google Scholar]
- 3.Dujon B, Belfort M, Butow RA, Jacq C, Lemieux C, Perlman PS, Vogt VM. Mobile introns: definition of terms and recommended nomenclature. Gene. 1989;82:115–8. doi: 10.1016/0378-1119(89)90035-8. [DOI] [PubMed] [Google Scholar]
- 4.Belle A, Landthaler M, Shub DA. Intronless homing: site-specific endonuclease SegF of bacteriophage T4 mediates localized marker exclusion analogous to homing endonucleases of group I introns. Genes and Dev. 2002;16:351–362. doi: 10.1101/gad.960302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho Y, Qiu YL, Kuhlman P, Palmer JD. Explosive invasion of plant mitochondria by a group I intron. PNAS USA. 1998;95:14244–14249. doi: 10.1073/pnas.95.24.14244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. Journal of Molecular Biology. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 7.Turmel M, Cote V, Otis C, Mercier JP, Gray MW, Lonergan KM, Lemieux C. Evolutionary transfer of ORF-containing group I introns between different subcellular compartments (chloroplast and mitochondrion) Molecular Biology & Evolution. 1995;12:533–45. doi: 10.1093/oxfordjournals.molbev.a040234. [DOI] [PubMed] [Google Scholar]
- 8.Curcio MJ, Belfort M. Retrohoming: cDNA-mediated mobility of group II introns requires a catalytic RNA. Cell. 1996;84:9–12. doi: 10.1016/s0092-8674(00)80987-3. [DOI] [PubMed] [Google Scholar]
- 9.Lykke-Andersen J, Aagaard C, Semionenkov M, Garrett RA. Archaeal introns: splicing, intercellular mobility and evolution. Trends in Biochemical Sciences. 1997;22:326–31. doi: 10.1016/s0968-0004(97)01113-4. [DOI] [PubMed] [Google Scholar]
- 10.Chevalier BS, Stoddard BL. Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Research. 2001;29:3757–74. doi: 10.1093/nar/29.18.3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edgell DR, Belfort M, Shub DA. Barriers to intron promiscuity in bacteria. J Bacteriology. 2000;182:5281–5289. doi: 10.1128/jb.182.19.5281-5289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stoddard BL. Homing endonuclease structure and function. Quarterly Reviews of Biophysics. 2005;38:49–95. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- 13.Chevalier B, Monnat RJ, Stoddard BL. The LAGLIDADG homing endonuclease family. In: MBe, editor. Homing endonucleases and inteins. Vol. 16. Springer Verlag; Berlin: 2005. pp. 34–47. [Google Scholar]
- 14.Dalgaard JZ, Klar AJ, Moser MJ, Holley WR, Chatterjee A, Mian IS. Statistical modeling and analysis of the LAGLIDADG family of site-specific endonucleases and identification of an intein that encodes a site-specific endonuclease of the HNH family. Nucleic Acids Res. 1997;25:4626–38. doi: 10.1093/nar/25.22.4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Durrenberger F, Rochaix J-D. Characterization of the cleavage site and the recognition sequence of the I-CreI DNA endonuclease encoded by the chloroplast ribosomal intron of Chlamydomonas reinhardtii. Mol Gen Genet. 1993;236:409–414. doi: 10.1007/BF00277141. [DOI] [PubMed] [Google Scholar]
- 16.Perrin A, Buckle M, Dujon B. Asymmetrical recognition and activity of the I-SceI endonuclease on its site and on intron-exon junctions. EMBO Journal. 1993;12:2939–47. doi: 10.1002/j.1460-2075.1993.tb05956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dalgaard JZ, Garrett RA, Belfort M. Purification and characterization of two forms of I-DmoI, a thermophilic site-specific endonuclease encoded by an archaeal intron. J Biol Chem. 1994;269:28885–28892. [PubMed] [Google Scholar]
- 18.Agaard C, Awayez MJ, Garrett RA. Profile of the DNA recognition site of the archaeal homing endonuclease I-DmoI. Nucleic Acids Res. 1997;25:1523–30. doi: 10.1093/nar/25.8.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colleaux L, D’Auriol L, Galibert F, Dujon B. Recognition and cleavage site of the intron-encoded omega transposase. Proc Natl Acad Sci USA. 1988;85:6022–6. doi: 10.1073/pnas.85.16.6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson AJ, Yuan X, Kudlicki W, Herrin DL. Cleavage and recognition pattern of a double-strand-specific endonuclease (I-CreI) encoded by the chloroplast 23S rRNA intron of Chlamydomonas reinhardtii. Gene. 1992;119:247–251. doi: 10.1016/0378-1119(92)90278-w. [DOI] [PubMed] [Google Scholar]
- 21.Chevalier BS, Monnat RJ, Jr, Stoddard BL. The homing endonuclease I-CreI uses three metals, one of which is shared between the two active sites. Nature Structural Biology. 2001;8:312–6. doi: 10.1038/86181. [DOI] [PubMed] [Google Scholar]
- 22.Heath PJ, Stephens KM, Monnat RJ, Stoddard BL. The structure of I-CreI, a group I intron-encoded homing endonuclease. Nature Struct Biol. 1997;4:468–476. doi: 10.1038/nsb0697-468. [DOI] [PubMed] [Google Scholar]
- 23.Jurica MS, Monnat RJ, Jr, Stoddard BL. DNA recognition and cleavage by the LAGLIDADG homing endonuclease I- CreI. Mol Cell. 1998;2:469–76. doi: 10.1016/s1097-2765(00)80146-x. [DOI] [PubMed] [Google Scholar]
- 24.Chevalier B, Turmel M, Lemieux C, Monnat RJ, Stoddard BL. Flexible DNA target site recognition by divergent homing endonuclease isoschizomers I-CreI and I-MsoI. J Mol Biol. 2003;329:253–269. doi: 10.1016/s0022-2836(03)00447-9. [DOI] [PubMed] [Google Scholar]
- 25.Spiegel PC, Chevalier B, Sussman D, Turmel M, Lemieux C, Stoddard BL. The structure of I-CeuI homing endonuclease: Evolving asymmetric DNA recognition from a symmetric protein scaffold. Structure. 2006;14:869–80. doi: 10.1016/j.str.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 26.Bolduc JM, Spiegel PC, Chatterjee P, Brady KL, Downing ME, Caprara MG, Waring RB, Stoddard BL. Structural and biochemical analyses of DNA and RNA binding by a bifunctional homing endonuclease and group I intron splicing factor. Genes Dev. 2003;17:2875–88. doi: 10.1101/gad.1109003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moure CM, Gimble FS, Quiocho FA. The crystal structure of the gene targeting homing endonuclease I-SceI reveals the origins of its target site specificity. J Mol Biol. 2003;334:685–696. doi: 10.1016/j.jmb.2003.09.068. [DOI] [PubMed] [Google Scholar]
- 28.Chevalier BS, Kortemme T, Chadsey MS, Baker D, Monnat RJ, Stoddard BL. Design, activity and structure of a highly specific artificial endonuclease. Molec Cell. 2002;10:895–905. doi: 10.1016/s1097-2765(02)00690-1. [DOI] [PubMed] [Google Scholar]
- 29.Moure C, Gimble F, Quiocho F. Crystal structure of the intein homing endonuclease PI-SceI bound to its recognition sequence. Nature Struct Biol. 2002;9:764–770. doi: 10.1038/nsb840. [DOI] [PubMed] [Google Scholar]
- 30.Turmel M, Gutell RR, Mercier J-P, Otis C, Lemieux C. Analysis of the chloroplast large subunit ribosomal RNA gene from 17 Chlamydomonas taxa: three internal transcribed spacers and 12 group I intron insertion sites. J Mol Biol. 1993;232:446–467. doi: 10.1006/jmbi.1993.1402. [DOI] [PubMed] [Google Scholar]
- 31.Lucas P, Otis C, Mercier JP, Turmel M, Lemieux C. Rapid evolution of the DNA-binding site in LAGLIDADG homing endonucleases. Nucleic Acids Res. 2001;29:960–9. doi: 10.1093/nar/29.4.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Landthaler M, Shen BW, Stoddard BL, Shub DA. I-BasI and I-HmuI: two phage intron-encoded endonucleases with homologous DNA recognition sequences but distinct DNA specificities. J Mol Biol. 2006;358:1137–51. doi: 10.1016/j.jmb.2006.02.054. [DOI] [PubMed] [Google Scholar]
- 33.Nord D, Torrents E, Sjoberg BM. A Functional Homing Endonuclease in the Bacillus anthracis nrdE Group I Intron. J Bacteriol. 2007;189:5293–301. doi: 10.1128/JB.00234-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ho Y, Kim SJ, Waring RB. A protein encoded by a group I intron in Aspergillus nidulans directly assists RNA splicing and is a DNA endonuclease. PNAS USA. 1997;94:8994–9. doi: 10.1073/pnas.94.17.8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chatterjee P, Brady KL, Solem A, Ho Y, Caprara MG. Functionally distinct nucleic acid binding sites for a group I intron-encoded RNA maturase/DNA homing endonuclease. J Mol Biol. 2003;329:239–251. doi: 10.1016/s0022-2836(03)00426-1. [DOI] [PubMed] [Google Scholar]
- 36.Geese WJ, Waring RB. A comprehensive characterization of a group IB intron and its encoded maturase reveals that protein-assisted splicing requires an almost intact intron RNA. J Mol Biol. 2001;308:609–622. doi: 10.1006/jmbi.2001.4609. [DOI] [PubMed] [Google Scholar]
- 37.Lazowska J, Claisse M, Gargouri A, Kotylak Z, Spyridakis A, Slonimski PP. Protein encoded by the third intron of cytochrome b gene in Saccharomyces cerevisiae is an mRNA maturase. Analysis of mitochondrial mutants, RNA transcripts proteins and evolutionary relationships. J Mol Biol. 1989;205:275–289. doi: 10.1016/0022-2836(89)90341-0. [DOI] [PubMed] [Google Scholar]
- 38.Zheng D, Koller W. Characterization of the mitochondrial cytochrome b gene from Venturia inaequalis. Curr Genet. 1997;32:361–366. doi: 10.1007/s002940050289. [DOI] [PubMed] [Google Scholar]
- 39.Mouhamadou B, Ferandon C, Barroso G, Labarere J. The mitochondrial apocytochrome b genes of two Agrocybe species suggest lateral transfers of group I homing introns among phylogenetically distant fungi. Fungal Genet Biol. 2006;43:135–45. doi: 10.1016/j.fgb.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 40.Kurokawa S, Bessho Y, Higashijima K, Shirouzu M, Yokoyama S, Watanabe KI, Ohama T. Adaptation of intronic homing endonuclease for successful horizontal transmission. FEBS Journal. 2005;272:2487–2496. doi: 10.1111/j.1742-4658.2005.04669.x. [DOI] [PubMed] [Google Scholar]
- 41.Schneider TD, Stormo GD, Gold L, Ehrenfeucht A. Information content of binding sites on nucleotide sequences. J Mol Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- 42.Alwin S, Gere MB, Guhl E, Effertz K, Barbas CF, 3rd, Segal DJ, Weitzman MD, Cathomen T. Custom zinc-finger nucleases for use in human cells. Mol Ther. 2005;12:610–7. doi: 10.1016/j.ymthe.2005.06.094. [DOI] [PubMed] [Google Scholar]
- 43.Porteus MH. Mammalian gene targeting with designed zinc finger nucleases. Mol Ther. 2006;13:438–46. doi: 10.1016/j.ymthe.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 44.Berkovich E, Monnat RJ, Jr, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9:683–90. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- 45.Turmel M, Boulanger J, Schnare MN, Gray MW, Lemieux C. Six group I introns and three internal transcribed spacers in the chloroplast large subunit ribosomal RNA gene of the green alga Chlamydomonas eugametos. J Mol Biol. 1991;218:293–311. doi: 10.1016/0022-2836(91)90713-g. [DOI] [PubMed] [Google Scholar]
- 46.Edgell DR, Stanger MJ, Belfort B. Coincidence of cleavage sites of intron endonuclease I-TevI and critical sequences of the host thymidylate synthase gene. J Mol Biol. 2004;343:1231–1241. doi: 10.1016/j.jmb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 47.Edgell DR, Shub DA. Related homing endonucleases I-BmoI and I-TevI use different strategies to cleave homologous recognition sites. PNAS USA. 2001;98:7898–903. doi: 10.1073/pnas.141222498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gimble FS, Moure CM, Posey KL. Assessing the plasticity of DNA target site recognition of the PI-SceI homing endonuclease using a bacterial two-hybrid selection system. J Mol Biol. 2003;334:993–1008. doi: 10.1016/j.jmb.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 49.Posey KL, Koufopanou V, Burt A, Gimble FS. Evolution of divergent DNA recognition specificities in VDE homing endonucleases from two yeast species. Nucleic Acids Res. 2004;32:3947–3956. doi: 10.1093/nar/gkh734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weiss-Brummer B, Rodel G, Schweyen RJ, Kaudewitz F. Expression of the split gene cob in yeast: evidence for a precursor of a “maturase” protein translated from intron 4 and preceding exons. Cell. 1982;29:527–536. doi: 10.1016/0092-8674(82)90169-6. [DOI] [PubMed] [Google Scholar]
- 51.Burt A, Koufopanou v. Homing endonuclease genes: teh rise and fall and rise again of a selfish element. Curr Opin Gen Develop. 2004;14:609–615. doi: 10.1016/j.gde.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 52.Solem A, Chatterjee P, Caprara MG. A novel mechanism for protein-assisted group I intron splicing. RNA. 2002;8:412–425. doi: 10.1017/s1355838202029321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lambowitz A, Caprara MG, Zimmerly S, Perlman PS. Group I and group II ribozymes as RNPs: clues to the past and guides to the future. In: Gesteland RF, Atkins JF, Cech TR, editors. The RNA World II. Cold Spring Harbor Laboratory Press; New York: 1999. pp. 451–485. [Google Scholar]
- 54.Downing ME, Brady KL, Caprara MG. A C-terminal fragment of an intron-encoded maturase is sufficient for promoting group I intron splicing. RNA. 2005;11:437–446. doi: 10.1261/rna.7225205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paques F, Duchateau P. Meganucleases and DNA double-strand break-induced recombination: perspectives for gene therapy. Current Gene Therapy. 2007;7:49–66. doi: 10.2174/156652307779940216. [DOI] [PubMed] [Google Scholar]
- 56.Stoddard BL, Monnat RJ, Scharenberg AM. Advances in engineering homing endonucleases for gene targeting: ten years after structures. Progress in Gene Therapy. 2007 in press. [Google Scholar]
- 57.Schueler-Furman O, Baker D. Conserved residue clustering and protein structure prediction. Proteins. 2003;52:225–35. doi: 10.1002/prot.10365. [DOI] [PubMed] [Google Scholar]
- 58.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 59.CCP4: The SERC (UK) collaborative computing project No. 4, a suite of programs for protein crystallography. Daresbury Laboratory; Warrington U. K.: 1979. [Google Scholar]
- 60.Laskowski RJ, Macarthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystall. 1993;26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.