

Abstract
Mice lacking the p27Kip1 Cdk inhibitor, like mice lacking Rb, develop pituitary tumors involving pars intermedia melanotrophs, yet p27Kip1 tumors are genetically distinct from Rb derived tumors as they exhibit haploid insufficiency. We compared tumors from mice with p27Kip1 constitutive and tissue specific null mutations to tumors arising in tissue specific Rb knockout mice with the aim of determining whether they are distinguished by quantitative or qualitative differences. The rate of p27Kip1 knockout tumor development was strongly influenced by strain background due to polygenic strain modifiers in the C57BL/6J versus 129S4 strains but, unlike a prior report of Rb mutants, this impacted tumor incidence but not the tumor spectrum. p27Kip1 tumors were oligoclonal or polyclonal based on studies of X-chromosomal inactivaton of Dock11. In contrast, Rb null tumors which developed into monoclonal neoplasms even in the absence of a requirement for Rb mutant clonal selection. Rb null tumors exhibited a higher proliferation rate and developed ischemic necrosis associated with an aberrant vasculature. p27Kip1 null tumors maintained normal vascular density, through a tumor cell dependent mechanism, but were more often hemorrhagic. Gene expression profiles distinguished p27Kip1 from Rb null tumors including significant differences in expression of Rb and E2F signature genes. Rb null tumors expressed higher levels of VEGF which, in other systems, is associated with dilated vessels, ineffective perfusion and tissue hypoxia. Mouse models lacking p27Kip1 and Rb may help us better understand the pathophysiology of MEN syndromes, retinoblastoma and other cancers that disrupt these important cell cycle inhibitors
Keywords: p27Kip1, Rb, Cdk inhibitor, Strain modifier, Clonality, Gene expression, Pituitary tumor, Knockout mouse, Cell cycle, Angiogenesis
Introduction
Inactivation of the retinoblastoma tumor suppressor, Rb, is a common attribute of cancer cells. It may occur through inactivating mutations, as in the case of human retinoblastoma, where loss of Rb protein unleashes the E2F family of transcription factors. However, loss of Rb protein is not necessary for its inactivation, as the cyclin dependent kinases, such as cyclin D/Cdk4, normally inactivate Rb through phosphorylation. Thus activating mutations of cyclin D, or inactivating mutations of CDK inhibitors may be oncogenic as a consequence of loss of Rb function.1 Homozygous disruption of the Rb gene in mouse models is lethal during embryonic development but Rb heterozygous mice and genetic mosaics containing Rb null and wildtype cells are viable and develop spontaneous tumors of the pituitary pars intermedia (intermediate lobe).2, 3
Interestingly loss of the p27Kip1 Cdk inhibitor leads to spontaneous tumors in the pituitary pars intermedia in addition to increasing overall animal growth and causing hyperplasia of the thymus and spleen.4–6 Because p27 knockout mice develop tumors in the same tissue as Rb null animals is has been presumed that Rb mediates the p27−/− tumor phenoytpe. However, in addition to cyclin D complexes, p27Kip1 inhibits both Cdk1 and Cdk2, both of which are capable of phosphorylating substrates other than Rb so the mechanism of tumor suppressor activity of p27Kip1 may involve other pathways. Mice harboring deletions of both Cdk2 and p27Kip1 develop pituitary tumors so it would appear that Cdk2 is dispensable for the pituitary tumor phenotype.7 Loss of p27Kip1 leads to increased overall animal growth, hyperplasia of the thymus and spleen, and spontaneous tumors in the pituitary pars intermedia.4–6 Likewise, mice lacking p18Ink4c, an inhibitor specific to D-type cyclins, develop spontaneous tumors of the pars intermedia melanotrophs, the predominant cell type in this tissue.7 The p18Ink4c knockout mouse has other phenotypes similar to p27 null mice including increased animal size and organ hyperplasia which reinforces the supposition that the Rb pathway mediates both the growth and pituitary tumor phenotypes.
Although the p27Kip1 gene is seldom mutated in human cancers the high degree of genomic instability in solid tumors leads to haploid loss of the gene relatively frequently.8 Unlike Rb, haploid loss of p27Kip1 and p18Ink4c has been shown to be sufficient to induce tumor phenotypes without the presence of mutational inactivation of the remaining allele.6, 9 In human cancers low expression of p27Kip1 portends a poor clinical outcome for a remarkably wide variety of malignancies, including cancer of the breast, lung, and prostate. 10–12 Recently the MENX mutation in rats, a model of multiple endocrine neoplasia, was mapped to a frameshift in the p27Kip1 gene. In addition, a familial case of MEN-1 was ascribed to a p27Kip1 non-sense mutation.13 Thus the link between CDK inhibitor mutations and endocrine neoplasias in mouse and rat models may be prescient of a larger role of these molecules in human endocrine neoplasias.
Despite the fact that deletions of p27Kip1, p18Ink4c, or Rb cause pars intermedia tumors in murine models they may have different mechanisms of tumor suppression. The combination of p27Kip1 and p18Ink4c disruptions, for example, increased the rate of pituitary tumor development, compared to deletion of either gene alone.7 In addition, combined deletions of p27Kip1 and p18Ink4c, or p27Kip1 and Rb, led to the development of multiple endocrine neoplasms with tumors of the anterior pituitary, thyroid c-cells, and adrenal gland.14, 15 The interpretation of these findings may be complicated by strain effects as Rb knockout mice showed a higher frequency of endocrine neoplasms outside the pars intermedia on the C57BL/6 background compared to 129.16
The expression of pro-opiomelanocortin (POMC), the pro-hormone of α-MSH, is relatively specific to the pars intermedia. It has been exploited in the development of both POMC-Cre and POMC-Flp transgenic mice, which are capable of driving tissue specific deletion of genes flanked by LoxP and Flp, respectively.17, 18 We used the POMC-Cre transgene to drive Rb mutation in the pars intermedia and compared the impact of homozygous deletion of Rb to deletion of p27Kip1 in terms of tumor kinetics, strain modifiers, pathophysiology, and global gene expression.
Material and Methods
Mouse strains
We previously described the generation of mice with constitutive deletion (p27−/−) and floxed alleles (p27L+/L+) of p27Kip1 coisogenic to the 129S4 substrain.17, 19 p27+/− mice were backcrossed 12 generations to C57BL/6J (Jackson Labs) and then bred to the 129S4 mutants. Thusly a cohort of p27−/− on the C57 inbred, 129S4 inbred, F1 hybrid, and F2 hybrid backgrounds (total number 94) were generated. Craniotomies were performed on animals at 9 months of age or sooner if the animals developed morbidities. Pituitaries were photographed in situ, removed, weighed, and either frozen or fixed overnight in PBS + 4% paraformaldehyde. Mice harboring floxed alleles of p27Kip1 (p27L+/L+, 129S4 N10) or Rb (RbF19/F19, 129S4 N3) with and without the POMC-Cre transgene were bred to induce pituitary pars intermedia specific gene deletions.17, 20 Animals of each genotype (n=10) were sacrificed at 6 weeks of age, 15 weeks of age, in addition a survival cohort (n=25 per genotype) which was followed for signs of morbidity up to 9 months.
Histology
Paraffin sections (0.4 μm) of pituitary tumors and normal pituitaries were stained with hematoxylin and eosin. Proliferation was assessed by immunoperoxidase staining with rabbit anti-Ki67 with a hemotoxylin counter-stain. Florescent TUNEL staining was performed on paraffin-sectioned tumors using FragEL kit (Calbiochem). Immunostaining to vascular endothelium was performed with (1:1000) digoxigenin labeled rat monoclonal MECCA32 antibody (courtesy of Andrew Farr, University of Washington) with an anti-digoxigenin alkaline phosphatase and BCIP/NBT color development (Roche).
X-chromosome inactivation
The Mouse Phenome Database was queried for SNPs in mRNA/UTRs which are polymorphic between C57BL/6J and any 129 substrain. Of 6 genes that were PCR amplified and sequenced only Dock11 was found to be polymorphic in the 129S4 substrain vs C67BL/6J (SNP ID: rs13483724). To test for X-chromosome inactivation of Dock11 expression, purified RNA was reverse transcribed with a random primer and then PCR amplified with D5 plus D6 primers (see Supplemental Table 1) x20 cycles followed by nested primers D7 plus D12 for an additional 30 cycles with PCR conditions previously described for p27.19 Dye-deoxy sequencing was conducted with the D11 and D12 primers. An intron prevents amplification of genomic DNA with this approach so genomic DNA was amplified separately with D1 plus D4 followed by nested primers D3 and D12.
Microarray assessment
RNA from large (> 20 mg) pars intermedia pituitary tumors was purified (Trizol, Invitrogen) according to manufacturers recommendations. Similarly RNA was purified from normal wildtype 129S4 mouse pituitary pars intermedias (n=10) which was microdissected away from the pars distalis tissue along the hypophyseal cleft. Samples were treated with DNAse-I at room temperature for 10 minutes and then column purified (RNeasy, Qiagen). 1 hg total RNA was amplified through in vitro transcription (MessageAmp-II, Ambion) and quantified (on an Agilent BioAnalyzer). RNA from p27−/− tumors (n=2) RbF19/F19;POMC-Cre+ (n=2), and pooled wildtype pars intermedia RNA were labeled with Cy5 using an amino-allyl coupling procedure and admixed with Cy3 labeled pooled wildtype pars intermedia RNA, as previously described.21 All samples were run in duplicate on FHCRC murine 22K cDNA microarrays (GEO accession #GPL2931).
Data Analysis
cDNA arrays were imaged and quantified with a GenePix scanner and software. Local background subtraction was not performed to avoid introducing low intensity signal variation. Data processing was performed with R/BioConductor software.22 To summarize, log ratios (log2(Cy5/Cy3)) were sequentially subtracted by loess fitted values, first as a function of signal intensity (log2(Cy5*Cy3)) and then according to 2-dimensional spot location (using Bioconductor’s marray package).23 Inter-array amplitude scaling was performed using median absolute deviation (MAD) of each array. Missing values were imputed using K-nearest neighbor (KNN) and differential gene expression, most strongly correlating with Rb vs p27 null genotype, was identified using a false positive discovery approach (using the pamr Bioconductor package).24 Gene ontologies of differentially expressed genes were surveyed by uploading Entrez Gene IDs to the DAVID 2006 database and performing functional annotation clustering for level 4–5 GO biological process and molecular function terms.25 Weighted Rb null signature gene expression was calculated averaging an individual genes expression for a given genotype (Rb null or p27 null relative to wildtype) multiplied by the statistical weight previously reported by Black EP et al. These weighted values were averaged for all of the all of Rb null signature genes which are present on our array (n=70). Likewise, signature genes characteristic of MEFs lacking the Rb family members p130Rb2 and p107Rb3 (n=85), genes characteristic E2F1 deletion (n=83), and genes which distinguishes E2F3 from E2F1 null cells (n=87) were present on our array and were used to compare Rb and p27Kip1 null pituituary tumors.26
Quantitative PCR
Tumors from p27 null (n=5) and Rb null (n=6) and were compared to 30 week old wildtype pars intermedia from 30 week old mice. Preneoplastic 6 week old p27 null (n=7), and Rb null (n=4) wild type (n=10, pooled) were also obtained and run in parallel. First strand synthesis was performed using ABI RT kit (Applied Biosystems). 1/300 of the cDNA product was assayed with 250 nM primers (see Supplemental Table 2) and SYBR green PCR master mix (Applied Biosystems) and run at 95°C for 15 sec, 60°C for 1 min, x40 cycles in an ABI 7900 machine. All samples were run in triplicate wells with the S16 gene assayed in parallel as a loading control and all assays were duplicated. Results were adjusted first to an S16 loading control and then expressed in terms of the change in cycle number (∆Ct) relative to the 6 week old wildtype sample.
Results
Effect of strain background in p27Kip1 knockout pars intermedia
It has been reported that an Rb null mutation bred into the C57 background also developed decreased incidence of pars intermedia tumors but increased spontaneous tumors of the anterior lobe (pars distalis).16 We therefore assayed the effect of genetic background on the rate of pituitary tumor development in p27−/− mice. By 9 months of age 17/21 (81%) of p27−/− mice coisogenic to the 129S4 substrain developed tumors in excess of 20 mg. In comparison only 1/17 (5.9%) of p27Kip1 knockouts on the C57BL/6J background, 1/14 (7.1%) on the F1 hybrid background, and 3/40 (7.5%) on the F2 hybrid backgrounds (see Figure 1A) developed large tumors in this time frame. The number of large tumors in F2 hybrid mice is less than would be expected if a single genetic modifier suppressed tumor development in the C57BL/6J background with a Mendelian inheritance pattern (p=0.03, Pearson’s Chi-squared test). In addition, the survival of p27−/− 129S4 mice was significantly shorter than C57BL/6J p27−/− animals (Figure 1B) (p = 4.5×10−, chi-squared test). Tumors in C57BL/6J mice arose exclusively in the pars intermedia and other than their smaller size, were histologically indistinguishable from those of their 129S4 counterparts. These data indicate the presence of polygenic strain modifiers that significantly alter pars intermedia tumor development in response to p27Kip1 loss. In contrast to previous reports in Rb+/− the effect is principally on the kinetics of tumor development as because anterior lobe pituitary tumors and other endocrine neoplasms were not observed.
Figure 1. Pituitary tumor growth and mortality.
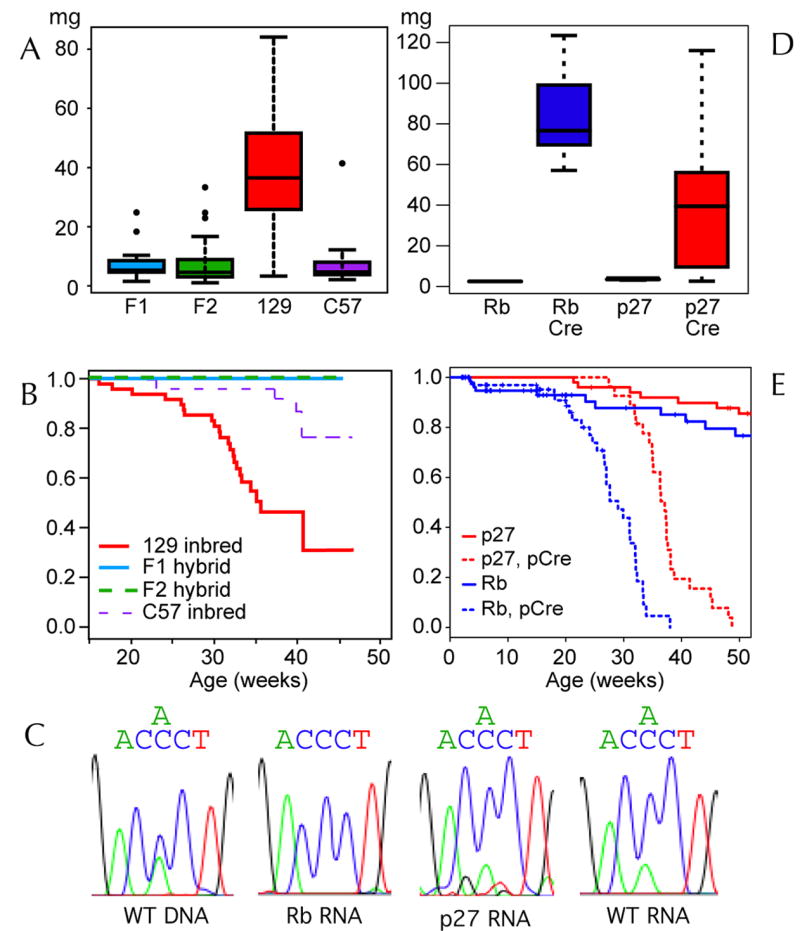
Box and whisker plots demonstrating pituitary tumor mass (A) from p27−/− mice graphed as a function of genetic background. F1 and F2 hybrids are compared to 129S4 and C57BL/6J inbred animals. Mean values (center bar) are shown within the middle quartiles (box), the range (whiskers) and outliers (points). The fraction of surviving animals is plotted (B) as a function of strain background. Sequence plots (C) of a PCR amplified Dock11 SNP are shown using genomic DNA from F1 hybrid tissue (F1DNA), and RNA from normal pars intermedia (WT RNA), an Rb null tumor (Rb RNA) and a p27 null tumor (p27 RNA) as the starting material. Pituitary tumor mass in mice with tissue specific mutations (D) of Rb (Rb/Cre) and p27Kip1 (p27/Cre) is graphed with control animals which lack the Cre transgene. The survival (E) of Rb and p27Kip1 pituitary tissue specific animals is also plotted.
Clonality of p27Kip1 −/− tumors
Unlike the case of Rb+/− mice regions containing early atypical proliferation (or so called EAPs) have not been observed in younger p27−/− mice.27 Thus it is difficult to specify the time of onset of tumor in p27−/− pars intermedia tumors. Regions of focal abnormality may be harder to discern because the cellular morphology of p27Kip1 null tumors is less altered than Rb tumors. Another possibility is that p27−/− mice develop polyclonal tumors such that multiple cells or the entire organ becomes progressively abnormal. An indicator of clonality that may be assessed in hybrid mice is the pattern of an X-chromosomal inactivation strain specific SNPs. We identified a SNP in the Dock11 gene that is polymorphic between C57BL/6J and the 129S4 substrain and showed that Dock11 RNA is expressed in the pars intermedia. We reasoned that, since the bulk of pars intermedia tumors are comprised of melanotroph cells, sequence analysis of RT-PCR amplified tumor RNA would detect only one allele in the case of monoclonal tumors arising in animals heterozygous for the Dock11 SNP. However, 4 out of 4 female p27−/− mice, heterozygous for Dock11, expressed both copies of the allele in their pituitary tumors at levels comparable to normal pars intermedia tissue (Figure 1C). In comparison, 0/2 tumors arising in RbF19/F19; POMC-Cre+ females, also heterozygous for Dock11, expressed both alleles of Dock11. In summary, this data indicates that, in contrast to Rb, p27Kip1 knockout pituitary tumors are typically not monoclonal, and that they develop as a diffuse process at rates determined by polygenic strain modifiers.
Rb versus p27Kip1 null tumors induced by POMC-Cre
In order to compare pars intermedia tumor suppression by Rb and p27Kip1 in a similar genetic context we backcrossed the RbLoxP targeted mutation into the 129S4 strain background and induced pituitary specific homozygous mutations with the POMC-Cre transgene (RbF19/F19; POMC-Cre+). Rb mutants exhibited significantly shorter survival (see Figure 1E) at a median of 29 weeks compared to 37 weeks for mice with POMC-Cre induced p27Kip1 deletion (p27L+/L+; POMC-Cre+) (p = 7×10−8 chi-squared test). In addition by 15 weeks RbF19/F19; POMC-Cre+ mice developed tumors with an average weight of 30 mg, comparable to the average weight of lethal p27 knockout tumors at 30 weeks yet at 15 weeks the Rb mutants showed little signs of morbidity. Subsequently the Rb knockout tumors induced morbidity, mostly through compression of the adjacent brain tissue. However, in 4 Rb null mice the tumor cells penetrated the meninges and tracked along the under surface of the cerebrum, and event never observed in either p27L+/L+;POMC-Cre+ or p27−/− animals. In addition, the histologic appearance of Rb null and p27Kip1 null tumors is remarkably dissimilar.
Tumors arising in p27Kip1 deficient animals mice display a cellular morphology more closely resembling normal pars intermedia melanotrophs and invariably display some degree of hemorrhage or blood filled lakes. RbF19/F19; POMC-Cre+ tumor cells, in contrast, are smaller, more spindle shaped, with pale cytoplasm. Rb tumors contain a spongy meshwork of necrosis interspersed with cords of viable tumor. This was apparent in 15 week old Rb null tumors and became more dramatic at older ages. Centrally located within each region of viable Rb null tumor was a small vessel which was identified by MECCA32 staining, a marker of endothelial cells (see Figure 2D), and the presence of luminal RBCs. Despite being hemorrhagic, large p27Kip1 deficient tumors maintained a microvascular density similar to wildtype pars intermedia tissue without areas of cellular necrosis. TUNEL staining of RbF19/F19; POMC-Cre+ and p27L+/L+;POMC-Cre+ tumors show a low level of TUNEL positivity within the tumor tissue (Figure 2E). However, RbF19/F19; POMC-Cre+ tumor cords are often bounded by a rim of TUNEL positive cells. Taken together these data indicate that Rb null tumors are incapable of maintaining adequate angiogenesis, outgrow their limited blood supply, and undergo ischemic necrosis when they reach a critical distance from tissue capillaries. In contrast the p27Kip1 deficient tumors are capable of maintaining normal vessel density during their growth but are prone to hemorrhage.
Figure 2. Histologic comparison of p27Kip1 vs Rb null pars intermedia tumors.
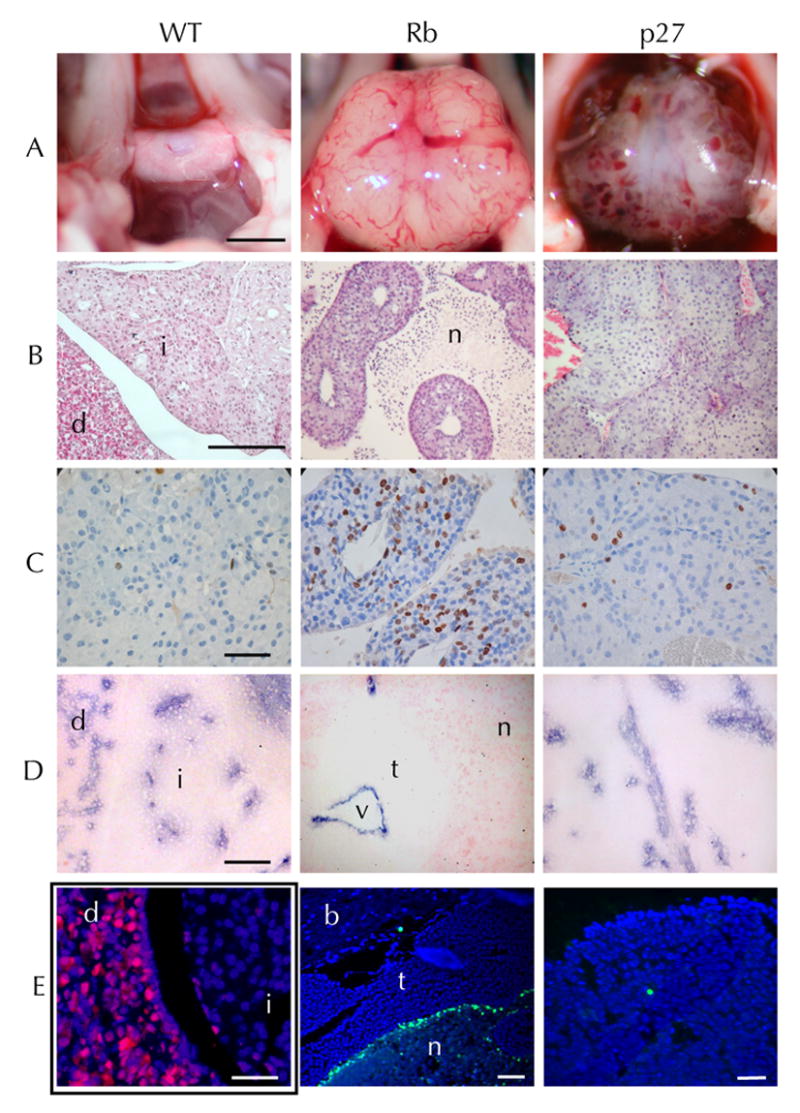
Appearance of the pituitary in situ (A) on the floor of the cranium in a wildtype mouse (WT) comparison to a large RbF19/F19;POMC-Cre+ tumor (Rb) p27L+/L+;POMC-Cre+ pituitary tumor (p27) with extensive hemorrhage. Hematoxylin and eosin (B) stained normal pituitary showing the pars distalis (d), pars intermedia (i). An Rb null tumor with cords of viable tumor and ischemic necrosis (n), and a hemorrhagic p27 null pars intermedia tumor. Ki67 immunoperoxidase staining (C) indicates proliferating cells. TUNEL staining of Rb null tissue with normal brain (b) adjacent to tumor (t) and lined by a region of bright TUNEL positivity on the edge of an area of ischemic necrosis (n). MECCA32 immunostaining (D) highlights the endothelial cells and reveals the capillary density of normal pituitary compared Rb null tumor with a central vessel (v) surrounded circumferentially by viable tumor (t) and peripheral necrosis (n). Anti-p27 immunofluorescence (E left) of a p27L+/L+;POMC-Cre+ pituitary at 6 weeks demonstrates nearly complete loss of p27 expression (red) from the pars intermedia (i) compared to the adjacent pars distalis (d). TUNEL staining (E mid, right) is shown for a p27 null and Rb null tumor (green) showing brain (b), tumor (t) and necrotic (n) areas (blue, DAPI). Scale bars (A 2mm, B 0.5mm, C-E 0.1mm)
Differential gene expression of Rb and p27Kip1 deficient pituitary tumors
To explore the basis of phenotypic differences between pituitary tumors arising in RbF19/F19; POMC-Cre+ versus those arising p27L+/L+; POMC-Cre+ mice we assessed global patterns of RNA transcript expression. Fluorescently labeled cDNA was hybridized to a 22K mouse cDNA spotted array while RNA pooled from wildtype pars intermedia tissue was used as a common reference and was run in parallel as a negative control. Hierarchical clustering of array data indicates that pituitary tumors as a group cluster separately from wildtype pars intermedia tissue. In addition RbF19/F19;POMC-Cre+ tumors clustered independently from p27Kip1 null tumors.
Functional gene ontology clustering, using the top 10% of genes ranked by absolute change relative to wildtype controls, demonstrates that, in general, the functional classification of genes with altered expression is similar in p27Kip1 and Rb null tumors (Figure 3A). Figuring prominently in both tumor types are genes involved in metabolism, kinase regulation and apoptosis. When comparing p27Kip1 null to Rb null tumors, more specific patterns of gene expression emerge. p27Kip1 null tumors had higher levels of expression of genes regulating lipid/carbohydrate metabolism, neurogenesis, transcription, and apoptosis, whereas Rb null tumors had higher levels of expression of genes involved with chromosome organization, mitosis and nucleic acid metabolism (Figure 3B).
Figure 3. Gene ontologies of p27Kip1 and Rb null pituitary tumors.

Pie charts summarizing functional annotation clustering results (based on GO biological process or molecular function level 5 annotations) of the top 10% of all genes ranked by differential expression of (A) p27−/− tumors vs normal wildtype pars intermedia, or (B) Rb null tumor vs. normal. The size of each segment represents the numbers of genes in each group and functional groups are ordered clockwise (from the top) by their statistical significance score. In contrast the genes (C) which are significantly increased in Rb null vs p27−/− tumors (n=82) are given in comparison to (D) the GO classification (BP or MF level 4–5) of genes increased in p27−/− tumors relative to Rb null tumors (n=168). For brevity only one or two descripitive terms for each functional cluster is listed.
Rb and E2F signature gene expression
Of 100 genes previously reported to show altered expression characteristic of Rb null vs. wildtype fibroblast cells, 70 were present on our cDNA array platform. 28 As shown in Figure 4C, RbF19/F19; POMC-Cre+ pituitary tumors showed increased expression of Rb null signature genes, consistent with previous results. Using a similar approach we determined that Rb null tumors also showed increased expression of E2F1 null signature genes, but not genes characteristic of p130Rb2/p107Rb3 nor E2F3 null cells. In contrast, p27Kip1 null tumors showed reduced levels of expression of Rb and E2F1 signature genes compared to wildtype tissues. In summary, this data indicates that Rb and p27Kip1 null pars intermedia tumors are phenotypically similar compared to normal wildtype tissues. Nonetheless, p27Kip1 null tumors are distinguished by a relative lack of Rb and E2F1 signature gene expression.
Figure 4. Rb and p27Kip1 null pars intermedia tumor expression profiles.

Average microarray expression levels of the subset of genes (A) previously described as either Rb, p130/p107, E2F1, or E2F3 signature genes are given for Rb null and p27Kip1 null tumors. Change in expression compared to wildtype for each gene were multiplied by the statistical weights (provided by Black EP et al. 2003, 2005) so the results are positive if they correlate with the reported signature regardless of whether the gene’s expression is increased or decreased. A heat map (B) showing differential expression of 100 genes with the most significant differences in expression in p27−/− vs. Rb null tumor tissue. Average values of replicate arrays are shown from 2 Rb and 2 p27Kip1 null tumors in comparison to arrays with negative control wildtype (WT) tissue. Locations of 41 selected genes are given after heirarchial clustering. QPCR results (C) are shown for 8 genes showing differential expression in Rb null (open triangle), p27Kip1 null (red square) and wildtype (green circle) pars intermedia tissue at 6 weeks of age and in tumors or 30 week old wildtype tissue. Expression values are given in terms of -∆Ct (change in PCR cycle number) relative to wildtype tissue at 6 weeks of age.
Individual genes altered in Rb vs p27Kip1 null tumors
Compared to wildtype pars intermedia, p27Kip1 null pituitary tumors demonstrated altered expression of 330 genes (at an estimated median false positive discovery rate of 15%). Similarly tumors arising in RbF19/F19;POMC-Cre+ mice demonstrated altered expression in 800 genes compared to wildtype pars intermedia tissues. However, tumors were more similar to each other than to normal tissues. Comparing Rb to p27Kip1 null tumors 200 genes showed differential expression (median FDR 20%, supplemental Table 1). The top 100 genes are displayed in a heat map (Figure 4B) along with selected gene names. In many cases p27Kip1 null tumors had higher levels of expression of specific genes compared to their Rb null counterparts. We confirmed the altered expression levels of selected genes with quantitative PCR of cDNA derived from pituitary tumors, as well as pars intermedia tissue from wildtype, and 6 week old Rb or p27Kip1 null animals. Cyclin D2 levels were significantly decreased in both p27 and Rb null tumors as well as in pars intermedia tissue from young p27 null mice compared to wildtype tissues. Similarly pituitary tumors showed increased level of p18Ink4c, p130Rb2 compared to wildtype pars intermedia. Cyclin E2 levels were dimished in older compared to younger wildtype pars intermedia tissue but levels did not drop in either Rb null or p27 null tumors. Rb null tumor showed increased expression of VEGF compared to p27Kip1 null tumors. This argues against a VEGF deficiency as a cause of Rb null tumor hypovascularity. On the other hand, p27Kip1 null tumors displayed increased expression the tissue inhibitor of metalloproteinase 2 (Timp2) which has been implicated in angiogenesis. 29
Discussion
Mice lacking the p27Kip1 Cdk inhibitor develop spontaneous tumors of the pars intermedia, as do mice lacking p18Ink4c and tissue specific deletions of Rb, making them useful for study the pathogenesis of cell cycle inhibitor disruption in cancer. In the case of p27Kip1 null mutation, we observed a strong effect of genetic background on the incidence but not the tissue distribution of pituitary tumors. The strain modifier is polygenic since F2 generation hybrids did not show rates of tumor development predicted by a single gene Mendelian trait. Deletion of p18Ink4c in addition to p27Kip1 has been reported to increase rates of pars intermedia tumors and, additionally, to induce other types of endocrine tumors. Likewise, Rb knockouts were reported to have cause endocrine tumors outside the pars intermedia on the C57BL/6J background compared to 129. In summary, genetic modifiers may suppress tumors of the pars intermedia resulting from p27Kip1 null mutations, but increase the incidence of tumors in other tissues in the case of p18Ink4c or Rb knockouts.
Our study of X-chromosome inactivation of Dock11 in female pars intermedia tumors indicates that RbF19/F19; POMC-Cre+ tumors are monoclonal as one might expect since tumors arising in Rb+/− mice invariably display loss of heterozygosity.3 However, our RbF19/F19;POMC-Cre+ mice lack both copies of Rb in the entire pars intermedia. Clonal selection in this context implies that an additional mutation was required or that a single clone outgrows the others. In contrast, both copies of X-chromosomal Dock11 were expressed in tumors from p27Kip1 null females, indicating that p27Kip1 null pars intermedia tumors are typically oligoclonal or polyclonal. Thus it is possible that homozygous loss of p27Kip1 is sufficient to induce pars intermedia tumors without additional genetic changes. Hemizygous loss of p27Kip1 was reported to cause pars intermedia tumors only if mice are exposed to the mutagens.6 In the latter case, mutation and loss of expression from the remaining p27Kip1 allele did not occur which implies that tumor development in p27Kip1 hemizygotes requires additional genetic changes even if homozygous loss of p27Kip1 is sufficient.
On the 129S4 genetic background, tissue specific mutation of Rb or p27Kip1 both induced tumors in 100% of mice. Rb tumors, however, achieved a large size more quickly and exhibited, on average a higher proliferation rate. The fact that p27Kip1 tumors exhibited morbidity with relatively smaller tumors may have been due to the presence of tumor-associated hemorrhage, which increased the compressive effect of the tumor on adjacent brain tissue. Pituitary tumors in p27Kip1 null mice maintained a normal vascular density, which implies that they stimulated angiogenesis at rates proportional to the growth of the tumor mass. These new capillaries may have harbored structural or functional abnormalities which was the basis of the hemorrhage. Despite their larger size, Rb null tumors were less often hemorrhagic, showed a decreased vascular density, and displayed morphologic evidence of ischemic necrosis. Thus Rb null tumors appear to outgrow their blood supply due to a deficiency in angiogenesis relative to the rate of tumor growth. It was previously reported that mice lacking the combination of p27Kip1 and p130Rb2 are defective in pituitary tumor angiogenesis and that their bone marrow progenitor cells are deficient in their responsiveness to VEGF.30 In contrast, the pathogenesis of the deficient angiogenesis in our RbF19/F19;POMC-Cre+ mice is tumor cell autonomous since we used the tissue specific POMC-Cre transgene to drive Rb deletion. It is therefore plausible that the gene expression profile of Rb null tumors includes altered expression of extracellular regulators of angiogenesis.
Not surprisingly, pars intermedia tumors arising from p27Kip1 and Rb null tissues have overall gene expression profiles that are relatively similar to one another compared to normal pars intermedia tissue. However, gene expression in p27Kip1 vs Rb null tumors are also distinct from one another. For example, Rb tumors expressed increased levels of genes that were previously reported to be distinguish Rb null or E2F1 null fibroblasts from wildtype controls.26, 28 p27Kip1 null tumors, in contrast, expressed decreased levels of these Rb and E2F1 signature genes. One could postulate that the effect of p27Kip1 loss is mediated partly through another Rb-family member, p130Rb2. Our array data does not support this notion, however, because the expression signature expression characteristic of p130Rb2 and 107Rb3 was decreased to a similar extent in both Rb null and p27Kip1 null tumors. On the other hand, gene expression in p27Kip1 null pituitary tumors correlated more strongly with the E2F3 null gene expression profile than did Rb null tumors.
We used QPCR to confirm the identities of genes differentially expressed in pars intermedia tumors compared to normal tissue. Cyclin D2 RNA was markedly decreased in both Rb null and p27Kip1 null pituitary tumors. However young Rb null animals had normal cyclin D2 levels prior to the development of pituitary tumors. This is somewhat reminiscent of earlier reports that transformed cell lines lacking functional Rb lack cyclin D1 expression whereas Rb null primary MEFs express normal levels of cyclin D1 and cyclin D2.31, 32 Interestingly both Rb and p27Kip1 null tumors showed increased expression of p18Ink4c and p130Rb2. This is consistent with a negative feedback loop that increases the expression of these cell cycle inhibitors which compensates for loss of either Rb or p27Kip1. Loss of both p27Kip1 and p18Ink4c has been shown to cause a synergistic increase in pars intermedia tumors as well as other endocrine tumors, a situation analogous to human MEN syndromes.7, 14 p130Rb2, on the other hand, is thought to compensate for p27Kip1 loss in p27Kip1 knockout MEFs as it has been shown to complex with cyclin E/Cdk2 in conjunction with p21Cip1. 33 The increased level of p130Rb2 in the pituitary tumors is consistent with our observation of a generalized decrease in expression of p130/p107 null signature genes.
We also confirmed the increase in Bnip3 expression from both p27Kip1 and Rb null pituitary tumors. Bnip3 is a Bcl2 family member that may be induced by erk5 or by hypoxic stress.34 In the Rb null tumors VEGF was also substantially increased. Thus the lack of adequate vascularization of the Rb null tumors does not appear to be due to a failure to induce VEGF in response to hypoxia. To the contrary, Rb null tumors were characterized by large tortuous vessels surrounded by tumor necrosis. Chronic elevation of VEGF itself can lead to dilated, disorganized vasculature which is leaky and causes hypoxia (reviewed by Jain).35 Thus the elevated VEGF may be an intrinsic feature of Rb null pituitary tumors and a major contributing factor to the Rb tumor phenotype. This notion is supported by the observation that Rb expression in human non-small cell lung carcinomas negatively correlates with VEGF expression.36
Vooijs, et al. previously noted that POMC-Flp mediated pituitary specific Rb knockouts have a modest increase in apoptosis in the pars intermedia prior to the development of pituitary tumors but no increased apoptosis in tumors themselves. Our TUNEL and endothelial staining of more advanced tumors indicates that the cystic spaces of Rb null tumors are actually areas of ischemic necrosis. The necrotic areas contain a meshwork of dilated vessels, each surrounded by ring of viable tumor cells that are sharply demarcated by a layer of apoptotic cells at their periphery. Studies of Rb−/− mice demonstrate lethality at embryonic day 13.5 with widespread cellular apoptosis. 37, 38 A major contributing factor is a placental defect effecting trophoblast development. Rb−/− embryos show intrinsic neurogenic defects and loss of Rb may sensitize hepatocytes to the effect of placental ischemia.39, 40 Likewise, Rb null melanotroph tumors may be predisposed to ischemic necrosis due to a propensity towards apoptosis compounded by ineffective angiogenesis. Despite the increased expression of E2F1 signature genes in Rb null tumors, E2F1 is not necessarily the cause of aberrant VEGF expression and angiogenesis defects. The inhibitor of differentiation 2 (Id2) is one of many proteins capable of binding Rb and in doing so it inhibits Rb function. Id2−/− deletion in combination with hemizygous Rb loss (Rb+/−) reduced the severity of the pituitary tumor phenotype including a reduction in the number and proliferation rate of early tumor lesions (EAPs), VEGF production and tumor vascularity.41
In the majority of genes where p27Kip1 null tumors showed increased expression relative to Rb null tumors both types of tumors showed reduced expression compared to normal wildtype tissues. One exception was tissue inhibitor of metalloproteinase-2 (Timp2) which displayed increased expression in p27Kip1 null tumors produced compared to either Rb null tumors or normal controls. Timp2 an extracellular metalloproteinase inhibitor that is capable of binding endothelial cell α2β1 integrin and inhibiting their proliferation.29 Rb tumors, on the other hand, were more likely to express E2F1 signature genes, including cyclin E2 and ubiquitin specific protease-1 (Usp1) a protein which deubiquitinates FancD2 and regulates the Fanconia anemia protein pathway.42 Nonetheless it is unclear as to whether there is direct connection between the altered growth and angiogenesis in Rb null versus p27 null tumors and the effects of altered effects of Rb and E2F signature genes themselves.
Our results show that p27 null and Rb null tumors are significantly different with respect to both their genetic and phenotypic characteristics. Rb null tumors exhibit a higher proliferation rate and defective angiogenesis. p27 null pars intermedia tumors arise as a polyclonal process which are able to sustain normal vascular density despite their increased growth. These phenotypic differences are accompanied by significant alterations in gene expression profiles including both cell cycle genes and genes known to regulate angiogenesis. The differential effect of p27Kip1 versus Rb deletion may reflect a qualitative difference between the effects of Rb hyperphosphorylation (in the case of p27 deletion) versus outright Rb deletion. On the other hand p27Kip1 may have important activities entirely independent of Rb family members. In addition to its ability to inactivate a variety of Cdks, p27Kip1 deletion has been shown to impair cellular motility by a Cdk independent mechanism.43, 44 The mechanism of increased pathogenesis of p27Kip1 deficient cancer appears not simply to correlate with increased proliferation.45 Our data suggests that p27Kip1 null tumors are capable of maintaining adequate levels of angiogenesis despite their cell cycle activation and that Rb or p27Kip1 protein expression may impact tumor responsiveness to antiangiogenic therapeutics.
Supplementary Material
Acknowledgments
Mice containing a floxed allele of Rb were kindly provided by Anton Berns (Netherlands Cancer Institute). This research was supported by a grant from the National Cancer Institute (CA1000-53, Fero).
Abbreviations
- Cdk
cyclin dependent kinase
- POMC
proopiormelanocortin
- GO
Gene Ontology
- QPCR
Quantitative real time polymerase chain reaction
Contributor Information
Wei-Ming Chien, Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, wchien@fhcrc.org.
Kendra Garrison, Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, kgarriso@fhcrc.org.
Emily Caufield, Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, emilycaufield@hotmail.com.
Jason Orthel, School of Pharmacy, University of Washington, Seattle, WA, jpo8@u.washington.edu.
Matthew L. Fero, Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, mfero@fhcrc.org.
References
- 1.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 2.Williams BO, Schmitt EM, Remington L, Bronson RT, Albert DM, Weinberg RA, Jacks T. Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. Embo J. 1994;13:4251–9. doi: 10.1002/j.1460-2075.1994.tb06745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu N, Gutsmann A, Herbert DC, Bradley A, Lee WH, Lee EY. Heterozygous Rb-1 delta 20/+mice are predisposed to tumors of the pituitary gland with a nearly complete penetrance. Oncogene. 1994;9:1021–7. [PubMed] [Google Scholar]
- 4.Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1) Cell. 1996;85:721–32. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- 5.Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–20. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 6.Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396:177–80. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, Su L, Xiong Y. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. 1998;12:2899–911. doi: 10.1101/gad.12.18.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spirin KS, Simpson JF, Takeuchi S, Kawamata N, Miller CW, Koeffler HP. p27/Kip1 mutation found in breast cancer. Cancer Research. 1996;56:2400–4. [PubMed] [Google Scholar]
- 9.Bai F, Pei XH, Godfrey VL, Xiong Y. Haploinsufficiency of p18(INK4c) sensitizes mice to carcinogen-induced tumorigenesis. Mol Cell Biol. 2003;23:1269–77. doi: 10.1128/MCB.23.4.1269-1277.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singhal S, Vachani A, Antin-Ozerkis D, Kaiser LR, Albelda SM. Prognostic implications of cell cycle, apoptosis, and angiogenesis biomarkers in non-small cell lung cancer: a review. Clin Cancer Res. 2005;11:3974–86. doi: 10.1158/1078-0432.CCR-04-2661. [DOI] [PubMed] [Google Scholar]
- 11.Alkarain A, Slingerland J. Deregulation of p27 by oncogenic signaling and its prognostic significance in breast cancer. Breast Cancer Res. 2004;6:13–21. doi: 10.1186/bcr722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaffer DR, Viale A, Ishiwata R, Leversha M, Olgac S, Manova K, Satagopan J, Scher H, Koff A. Evidence for a p27 tumor suppressive function independent of its role regulating cell proliferation in the prostate. Proc Natl Acad Sci U S A. 2005;102:210–5. doi: 10.1073/pnas.0407362102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Hofler H, Fend F, Graw J, Atkinson MJ. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A. 2006;103:15558–63. doi: 10.1073/pnas.0603877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franklin DS, Godfrey VL, O’Brien DA, Deng C, Xiong Y. Functional collaboration between different cyclin-dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Mol Cell Biol. 2000;20:6147–58. doi: 10.1128/mcb.20.16.6147-6158.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park MS, Rosai J, Nguyen HT, Capodieci P, Cordon-Cardo C, Koff A. p27 and Rb are on overlapping pathways suppressing tumorigenesis in mice. Proc Natl Acad Sci U S A. 1999;96:6382–7. doi: 10.1073/pnas.96.11.6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leung SW, Wloga EH, Castro AF, Nguyen T, Bronson RT, Yamasaki L. A dynamic switch in Rb+/− mediated neuroendocrine tumorigenesis. Oncogene. 2004;23:3296–307. doi: 10.1038/sj.onc.1207457. [DOI] [PubMed] [Google Scholar]
- 17.Chien WM, Rabin S, Macias E, Miliani de Marval PL, Garrison K, Orthel J, Rodriguez-Puebla M, Fero ML. Genetic mosaics reveal both cell-autonomous and cell-nonautonomous function of murine p27Kip1. Proc Natl Acad Sci U S A. 2006;103:4122–7. doi: 10.1073/pnas.0509514103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vooijs M, van der Valk M, te Riele H, Berns A. Flp-mediated tissue-specific inactivation of the retinoblastoma tumor suppressor gene in the mouse. Oncogene. 1998;17:1–12. doi: 10.1038/sj.onc.1202169. [DOI] [PubMed] [Google Scholar]
- 19.Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, Kaushansky K, Roberts JM. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996;85:733–44. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- 20.Vooijs M, te Riele H, van der Valk M, Berns A. Tumor formation in mice with somatic inactivation of the retinoblastoma gene in interphotoreceptor retinol binding protein-expressing cells. Oncogene. 2002;21:4635–45. doi: 10.1038/sj.onc.1205575. [DOI] [PubMed] [Google Scholar]
- 21.Pritchard C, Coil D, Hawley S, Hsu L, Nelson PS. The contributions of normal variation and genetic background to mammalian gene expression. Genome Biol. 2006;7:R26. doi: 10.1186/gb-2006-7-3-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo Y, Hastie T, Tibshirani R. Regularized linear discriminant analysis and its application in microarrays. Biostatistics. 2007;8:86–100. doi: 10.1093/biostatistics/kxj035. [DOI] [PubMed] [Google Scholar]
- 25.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 26.Black EP, Hallstrom T, Dressman HK, West M, Nevins JR. Distinctions in the specificity of E2F function revealed by gene expression signatures. Proc Natl Acad Sci U S A. 2005;102:15948–53. doi: 10.1073/pnas.0504300102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nikitin A, Lee WH. Early loss of the retinoblastoma gene is associated with impaired growth inhibitory innervation during melanotroph carcinogenesis in Rb+/− mice. Genes Dev. 1996;10:1870–9. doi: 10.1101/gad.10.15.1870. [DOI] [PubMed] [Google Scholar]
- 28.Black EP, Huang E, Dressman H, Rempel R, Laakso N, Asa SL, Ishida S, West M, Nevins JR. Distinct gene expression phenotypes of cells lacking Rb and Rb family members. Cancer Res. 2003;63:3716–23. [PubMed] [Google Scholar]
- 29.Seo DW, Li H, Guedez L, Wingfield PT, Diaz T, Salloum R, Wei BY, Stetler-Stevenson WG. TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell. 2003;114:171–80. doi: 10.1016/s0092-8674(03)00551-8. [DOI] [PubMed] [Google Scholar]
- 30.Vidal A, Zacharoulis S, Guo W, Shaffer D, Giancotti F, Bramley AH, de la Hoz C, Jensen KK, Kato D, MacDonald DD, Knowles J, Yeh N, Frohman LA, Rafii S, Lyden D, Koff A. p130Rb2 and p27kip1 cooperate to control mobilization of angiogenic progenitors from the bone marrow. Proc Natl Acad Sci U S A. 2005;102:6890–5. doi: 10.1073/pnas.0405823102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lukas J, Bartkova J, Rohde M, Strauss M, Bartek J. Cyclin D1 is dispensable for G1 control in retinoblastoma gene-deficient cells independently of cdk4 activity. Mol Cell Biol. 1995;15:2600–11. doi: 10.1128/mcb.15.5.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bates S, Parry D, Bonetta L, Vousden K, Dickson C, Peters G. Absence of cyclin D/cdk complexes in cells lacking functional retinoblastoma protein. Oncogene. 1994;9:1633–40. [PubMed] [Google Scholar]
- 33.Coats S, Flanagan WM, Nourse J, Roberts JM. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science. 1996;272:877–80. doi: 10.1126/science.272.5263.877. [DOI] [PubMed] [Google Scholar]
- 34.Zhang HM, Cheung P, Yanagawa B, McManus BM, Yang DC. BNips: a group of pro-apoptotic proteins in the Bcl-2 family. Apoptosis. 2003;8:229–36. doi: 10.1023/a:1023616620970. [DOI] [PubMed] [Google Scholar]
- 35.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 36.Ludovini V, Gregorc V, Pistola L, Mihaylova Z, Floriani I, Darwish S, Stracci F, Tofanetti FR, Ferraldeschi M, Di Carlo L, Ragusa M, Daddi G, Tonato M. Vascular endothelial growth factor, p53, Rb, Bcl-2 expression and response to chemotherapy in advanced non-small cell lung cancer. Lung Cancer. 2004;46:77–85. doi: 10.1016/j.lungcan.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 37.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 38.Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–94. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 39.Wu L, de Bruin A, Saavedra HI, Starovic M, Trimboli A, Yang Y, Opavska J, Wilson P, Thompson JC, Ostrowski MC, Rosol TJ, Woollett LA, Weinstein M, Cross JC, Robinson ML, Leone G. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature. 2003;421:942–7. doi: 10.1038/nature01417. [DOI] [PubMed] [Google Scholar]
- 40.Wenzel PL, Wu L, de Bruin A, Chong JL, Chen WY, Dureska G, Sites E, Pan T, Sharma A, Huang K, Ridgway R, Mosaliganti K, Sharp R, Machiraju R, Saltz J, Yamamoto H, Cross JC, Robinson ML, Leone G. Rb is critical in a mammalian tissue stem cell population. Genes Dev. 2007;21:85–97. doi: 10.1101/gad.1485307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lasorella A, Rothschild G, Yokota Y, Russell RG, Iavarone A. Id2 mediates tumor initiation, proliferation, and angiogenesis in Rb mutant mice. Mol Cell Biol. 2005;25:3563–74. doi: 10.1128/MCB.25.9.3563-3574.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D’Andrea AD, Bernards R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 2005;17:331–9. doi: 10.1016/j.molcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 43.Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18:862–76. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McAllister SS, Becker-Hapak M, Pintucci G, Pagano M, Dowdy SF. Novel p27(kip1) C-terminal scatter domain mediates Rac-dependent cell migration independent of cell cycle arrest functions. Mol Cell Biol. 2003;23:216–28. doi: 10.1128/MCB.23.1.216-228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clurman BE, Porter P. New insights into the tumor suppression function of p27(kip1) Proc Natl Acad Sci U S A. 1998;95:15158–60. doi: 10.1073/pnas.95.26.15158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.