

Abstract
The mammalian target of rapamycin (mTOR) is a central regulator of cell growth. mTOR exists in two functional complexes, mTORC1 and mTORC2. mTORC1 is rapamycin-sensitive, and results in phosphorylation of 4E-BP1 and S6K1. mTORC2 is proposed to regulate Akt Ser473 phosphorylation and be rapamycin-insensitive. mTORC2 consists of mTOR, mLST8, sin1, Protor/PRR5, and the rapamycin insensitive companion of mTOR (rictor). Here, we show that rapamycin regulates the phosphorylation of rictor. Rapamycin-mediated rictor dephosphorylation is time and concentration dependent, and occurs at physiologically relevant rapamycin concentrations. siRNA knockdown of mTOR also leads to rictor dephosphorylation, suggesting that rictor phosphorylation is mediated by mTOR or one of its downstream targets. Rictor phosphorylation induced by serum, insulin and insulin-like growth factor is blocked by rapamycin. Rictor dephosphorylation is not associated with dephosphorylation of Akt Ser473. Further work is needed to better characterize the mechanism of rictor regulation and its role in rapamycin-mediated growth inhibition.
Keywords: Rictor, Mammalian target of rapamycin, mTOR complex 2, Rapamycin
Introduction
The mammalian target of rapamycin (mTOR) is a key regulator of cell proliferation and growth. Recently mTOR was found to exist in two distinct functional complexes, mTORC1 and mTORC2 [1,2]. mTORC1 consists of mTOR, mLST8 and raptor. Activation of mTORC1 results in phosphorylation of its effectors S6K1 and 4E-BP1, and this pathway is rapamycin-sensitive. In contrast, mTORC2 is thought to be rapamycin-insensitive [1,2]. mTORC2 regulates Akt Ser473 kinase phosphorylation and consists of mTOR, mLST8, sin1, Protor/PRR5, and the rapamycin-insensitive companion of mTOR (rictor) [1–6].
Rapamycin has variable effects on Akt activation in different models and cell lines. Rapamycin increases Akt activation in some cancer cell lines, and this has been attributed to the loss of mTOR/S6K-dependent feedback inhibition of signaling, with loss of phosphorylation and degradation of insulin response substrate-1 [7,8]. In animal models, rapamycin was found to increase phosphorylated Akt (pAkt) in tumor lysates at doses that inhibit Akt signaling in nonmalignant tissues, including endothelial cells [9,10]. An increase in pAkt levels was also noted in tumors from patients treated with the rapamycin analogue everolimus (RAD001, Novartis, Basel, Switzerland) [7]. In contrast, in some cancer cell lines prolonged (24 hour) rapamycin treatment inhibits the assembly of mTORC2 and Akt Ser473 phosphorylation [11]. Human leukemia cells treated with temsirolimus have dissociation of mTORC2 as well as mTORC1 [12]. Furthermore, samples from patients with hematologic malignancies treated on Phase I/II trials with rapamycin analogues temsirolimus or everolimus demonstrated a decrease in pAkt Ser473 levels. Thus it appears that in some cells or under certain conditions rapamycin may affect mTORC2 assembly through a yet unknown mechanism.
Recently rapamycin analogue temsirolimus (Wyeth, PA) was approved by the Food and Drug Administration for renal cell carcinoma, and rapamycin and its analogues are currently in clinical trials for the treatment of many other tumor types. Better understanding the molecular consequences of mTOR inhibition is critical for rapamycin and its analogues to be most effectively used in the clinic.
Here we report that rictor, previously thought to be rapamycin-insensitive, is in fact regulated at the phosphorylation level by rapamycin. Knockdown of mTOR with siRNA also leads to a decrease in rictor phosphorylation, suggesting that phosphorylation of rictor may be mediated by mTOR or by one of its downstream targets. Rapamycin-mediated dephosphorylation of rictor is observed in multiple cell lines representing a spectrum of aberrations in Akt/mTOR signaling. Thus, the results of our study suggest that rapamycin regulates components of mTORC2 as well as mTORC1.
Materials and methods
Cell lines, cultures and reagents
MDA-MB-468, BT-549, BT-474 and MCF7, cell lines were obtained from the American Type Tissue Culture Collection (Manassas, VA). All cell lines were cultured in DMEM/F12 supplemented with 10% fetal bovine serum at 37°C and humidified in 5% CO2.
Dimethyl sulphoxide (DMSO), insulin like growth factor-I and insulin were purchased from Sigma Chemical Company (St. Louis, MO).
Western blot analysis
Cultured cells were washed with cold PBS and lysed in lysis buffer as described elsewhere [13]. 25–50 μg of lysate was separated by SDS-PAGE on a 4% or 7.5% gel, and transferred to 0.2 μm polyvidine difluoride membrane (Bio-Rad Laboratories, Hercules, CA). Membranes were blocked with 0.1% casein or 5% bovine serum albumin in tris-buffered saline and probed with antibodies to rictor (1:2000) (#A300–459A, Bethyl Labs, Montgomery, TX) or (1:1000) (#2140), mTOR (1:250) (#2972), raptor (1:125) (#4978), Akt (1:1000) (#9272), phospho-Akt (Ser473) (1:1000) (#9271) (all from Cell Signaling Technology, Beverly, MA) and β-actin (1:10000) (Sigma Chemical Co.). The immunoblots were visualized by Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE).
Immunoprecipitation
MDA-MB-468 cells (1 × 107) were lysed with 1 ml lysis buffer and immunoprecipitated following protein A agarose kit manufacturer’s instructions (KPL, Inc., Gaithersburg, MD). One microgram of rictor antibody was added to protein extract.
Calf intestinal phosphatase (CIP) dephosphorylation
Cultured cells were lysed or immunoprecipitated. Samples were suspended in CIP buffer and incubated 60 minutes at 37°C with the enzyme (New England Biolabs, Ipswich, MA).
Small interfering RNA (siRNA)
To silence mTOR, a single transfection of either a single mTOR siRNA sequence or a siRNA pool duplex was performed using DharmaFECT 1 reagent according to manufacturer’s protocol (Dharmacon, Inc., Lafayette, CO). The single mTOR siRNA target sequence used was CCCUGCCUUUGUCAUGCCU [2]. The pool siRNA target sequences were GGCCAUAGCUAGCCUCAUAUU, CAAAGGACUUCGCCCAUAAUU, GCAGAAUUGUCAAGGGAUAUU, CCAAAGCACUACACUACAAUU. The single and pool sequence mTOR and pool negative control siRNAs were purchased from Dharmacon, Inc. The single sequence negative control is purchased from Ambion, Inc (Austin, TX).
Results and discussion
In order to determine the effect of rapamycin on mTORC2 signaling, we initially studied the effect of rapamycin on MDA-MB-468 breast cancer cells, which are rapamycin-sensitive in vitro and in vivo [13,15,16]. On SDS-PAGE analysis and western blotting for rictor, we noted a significant and very reproducible increase in rictor electrophoretic mobility upon rapamycin treatment, suggestive of rictor dephosphorylation. Treatment of cell lysates with calf intestinal phosphatase (CIP) returned the mobility of rictor in the vehicle-treated cells to that of rapamycin-treated cells, suggesting that the mobility shift seen is indeed attributable to rictor phosphorylation (Fig. 1A). Similarly, when rictor was immunoprecipitated with anti-rictor antibodies, a mobility difference was observed between rictor protein immunoprecipitated from vehicle- and rapamycin-treated cells, and phosphatase treatment again equalized rictor mobility, confirming that the mobility shift seen is due to rictor dephosphorylation (Fig. 1B).
Fig. 1.

Dephosphorylation of rictor with rapamycin treatment. (A) MDA-MB-468 cells were treated with vehicle (DMSO) or 100 nM rapamycin for 24 h. Cell lysate was incubated with or without CIP. 4% SDS-PAGE and western blotting with anti-rictor antibody demonstrates a migration shift with dephosphorylation. (B) Following immunoprecipitation (IP), IP product was incubated with or without CIP, and probed with anti-rictor antibody. Cell lysate was used as a control. (C) MCF7 cells were grown for 24 h in serum-free media. Cells were untreated or pre-treated with 100 nM rapamycin for 30 minutes and were cultured for another 24 h under the following conditions: serum-free medium, medium containing 10% serum, 10 μg/ml insulin, or 100 ng/ml IGF-1. (D) MDA-MB-468 cells were transfected with a pool duplex siRNA sequence for mTOR or with a pool negative control siRNA. Cell lysates were prepared at 72 and 96 hours after transfection and western blotting was performed for rictor, mTOR and raptor. (E) MDA-MB-468 cells were transfected with single sequence mTOR siRNA or negative control siRNA. Western blotting was performed for rictor, mTOR and raptor.
We hypothesized that rictor phosphorylation is stimulated through the mTOR signaling pathway. To test this hypothesis we exposed MCF7 cells to overnight serum starvation followed by stimulation with one of three mitogenic stimuli known to activate mTOR signaling: serum, insulin and insulin-like growth factor. Pretreatment of cells with rapamycin blocked rictor phosphorylation in the presence of all three mitogens (Fig. 1C). Knockdown of mTOR by either a pool siRNA duplex for mTOR, or a single mTOR siRNA sequence, also led to a decline in rictor phosphorylation, suggesting that rictor phosphorylation is mediated by mTOR or one of its downstream targets (Fig. 1D and Fig. 1E).
Rapamycin-induced rictor dephosphorylation in a time and dose-dependent fashion. Dephosphorylation of rictor in MDA-MB-468 cells was notable by 2 hours, and was significant by 4 hours and persisted for at least 24 hours (Fig. 2A). Rictor dephosphorylation occurred in physiologically relevant doses (1–100nM) (Fig. 2B).
Fig. 2.
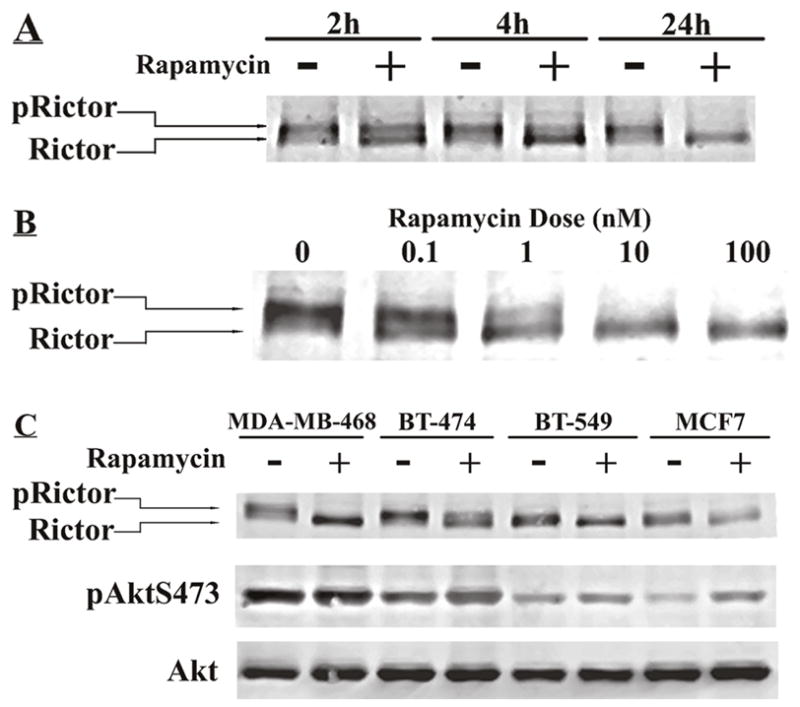
Effect of rapamycin concentration and duration of treatment on rictor dephosphorylation. (A) MDA-MB-468 cells were treated with DMSO or 100 nM rapamycin for 2, 4 or 24 h. Western blotting was performed for rictor. (B) Cells were treated with rapamycin at varying concentrations for 24 h. Western blotting was performed for rictor. (C) Four rapamycin sensitive breast cancer cell lines were treated with vehicle or 100 nM rapamycin for 24 h. Western blotting was performed for rictor, pAkt Ser473 and total Akt.
As it has been proposed that mTORC2 regulates Akt phosphorylation, we next evaluated the effect of rapamycin on the phosphorylation of rictor as well as on pAkt Ser473 in additional cell lines known to have activation of mTOR signaling [15]. Rapamycin-mediated rictor dephosphorylation was observed in these cell lines as well (Fig. 2C). After 24 hours rapamycin treatment, no significant alteration in pAkt Ser473 levels were observed in MDA-MB-468 cells, while the pAkt Ser473 levels increased in the other three cell lines (MCF7, BT549 and BT474), although all four cell lines are sensitive to rapamycin’s growth inhibitory effect [15]. Our finding that rictor undergoes dephosphorylation in the presence of persistent or even increased pAkt Ser473 suggests that either rictor phosphorylation is not critical for mTORC2-mediated pAkt Ser473 phosphorylation, or that feedback loop activation of Akt upon rapamycin treatment may utilize another Akt kinase.
To date rictor has been thought to be the “rapamycin-insensitive companion of mTOR”. Due to the large size of rictor, the rapamycin-induced increase in rictor electrophoretic mobility is only apparent on low percentage gels (5% or less); this may have prevented recognition of its dephosphorylation in previous studies. Further, most studies on rictor have focused on the components of mTORC2 complex, and data from Yang et al suggests that the mTORC2 complex would primarily consist of phosphorylated rictor [4]. It should be mentioned that no increase in rictor mobility with rapamycin treatment was observed in an earlier report [1]. The discrepancy in findings may be attributable to cellular context, study time course, or other differences in experimental design.
Two other studies to date had suggested that rictor phosphorylation may be under regulation [1,4]. In their first report on rictor, Sarbassov et al. noted that in kinase assays with mTOR, a protein of the same apparent molecular weight as rictor gets phosphorylated. Further, in cells metabolically labeled with radioactive phosphate, a reduction in mTOR expression was associated with a decrease in the amount of radioactivity in rictor without decreasing its expression. In cells with reduced mTOR, rictor appeared as a doublet on SDS-PAGE analysis. These data suggested rictor may be a substrate of mTOR. However, they found that treatment of cells with PI3K and mTOR inhibitor LY294002, but not rapamycin, increased rictor mobility. Recently, Yang et al reported that the knockdown of sin1, an essential mTORC2 component required for complex formation and kinase activity, caused an increase in rictor electrophoretic mobility, suggesting that sin1 may modulate rictor phosphorylation [4]. Further they demonstrated that both hypo and hyperphosphorylated rictor was immunoprecipitated by rictor antibody while mTOR preferentially co-precipitated with hyperphosphorylated rictor. These data suggest that dephosphorylated rictor may have a weaker affinity to mTOR or alternately rictor needs to be in a complex with mTOR to be phosphorylated. Further study is needed to determine if modulation of rictor phosphorylation regulates the formation of mTORC2.
In summary, we report that rictor, previously thought to be rapamycin-insensitive, is in fact regulated at the phosphorylation level by rapamycin. Rictor phosphorylation is enhanced by known activators of mTOR signaling and decreased by rapamycin as well as mTOR siRNA. Taken together these data suggest that rictor phosphorylation is indeed regulated by mTOR or one of its downstream targets. Further, our results demonstrate that rapamycin may regulate rictor as well as mTORC1. Further study is needed determine the effect of rictor dephosphorylation on mTORC2 signaling and the role of rictor dephosphorylation in rapamycin-mediated growth inhibition.
Acknowledgments
We thank Toi Clayton Soh for her assistance in manuscript preparation.
This work was supported by NIH1 R01 CA112199 to FMB.
The abbreviations used are
- mTOR
mammalian target of rapamycin
- IGF-1
insulin-like growth factor-I
- pAkt
phosphorylated Akt
- mTORC 1 and 2
mammalian target of rapamycin complex 1 and 2
- Ser473
serine 473
- rictor
rapamycin-insensitive companion of mTOR
- S6K1
ribosomal S6 kinase 1
- PI3K
phosphoinositide-3 kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 2.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 3.Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem. 2005;280:40406–40416. doi: 10.1074/jbc.M508361200. [DOI] [PubMed] [Google Scholar]
- 4.Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20:2820–2832. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearce LR, Huang X, Boudeau J, Pawlowski R, Wullschleger S, Deak M, Ibrahim A, Gourlay R, Magnuson MA, Alessi DR. Identification of Protor as a novel Rictor-binding component of mTOR-complex-2. Biochem J. 2007 doi: 10.1042/BJ20070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woo SY, Kim DH, Jun CB, Kim YM, Vander Haar E, Lee SI, Hegg JW, Bandhakavi S, Griffin TJ, Kim DH. PRR5, a novel component of mTOR complex 2, regulates PDGFRbeta expression and signaling. J Biol Chem. 2007 doi: 10.1074/jbc.M704343200. [DOI] [PubMed] [Google Scholar]
- 7.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol Cancer Ther. 2005;4:1533–1540. doi: 10.1158/1535-7163.MCT-05-0068. [DOI] [PubMed] [Google Scholar]
- 9.Phung TL, Ziv K, Dabydeen D, Eyiah-Mensah G, Riveros M, Perruzzi C, Sun J, Monahan-Earley RA, Shiojima I, Nagy JA, Lin MI, Walsh K, Dvorak AM, Briscoe DM, Neeman M, Sessa WC, Dvorak HF, Benjamin LE. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell. 2006;10:159–170. doi: 10.1016/j.ccr.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stoeltzing O, Meric-Bernstam F, Ellis LM. Intracellular signaling in tumor and endothelial cells: The expected and, yet again, the unexpected. Cancer Cell. 2006;10:89–91. doi: 10.1016/j.ccr.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 11.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 12.Zeng Z, Sarbassov dos D, Samudio IJ, Yee KW, Munsell MF, Ellen Jackson C, Giles FJ, Sabatini DM, Andreeff M, Konopleva M. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood. 2007;109:3509–3512. doi: 10.1182/blood-2006-06-030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mondesire WH, Jian W, Zhang H, Ensor J, Hung MC, Mills GB, Meric-Bernstam F. Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clin Cancer Res. 2004;10:7031–7042. doi: 10.1158/1078-0432.CCR-04-0361. [DOI] [PubMed] [Google Scholar]
- 14.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 15.Noh WC, Mondesire WH, Peng J, Jian W, Zhang H, Dong J, Mills GB, Hung MC, Meric-Bernstam F. Determinants of rapamycin sensitivity in breast cancer cells. Clin Cancer Res. 2004;10:1013–1023. doi: 10.1158/1078-0432.ccr-03-0043. [DOI] [PubMed] [Google Scholar]
- 16.Dong J, Peng J, Zhang H, Mondesire WH, Jian W, Mills GB, Hung MC, Meric-Bernstam F. Role of glycogen synthase kinase 3beta in rapamycin-mediated cell cycle regulation and chemosensitivity. Cancer Res. 2005;65:1961–1972. doi: 10.1158/0008-5472.CAN-04-2501. [DOI] [PubMed] [Google Scholar]