

Abstract
Arachidonic acid-derived epoxides, epoxyeicosatrienoic acids, are important regulators of vascular homeostasis and inflammation, and therefore manipulation of their levels is a potentially useful pharmacological strategy. Soluble epoxide hydrolase converts epoxyeicosatrienoic acids to their corresponding diols, dihydroxyeicosatrienoic acids, modifying or eliminating the function of these oxylipins. To better understand the phenotypic impact of Ephx2 disruption, two independently derived colonies of soluble epoxide hydrolase-null mice were compared. We examined this genotype evaluating protein expression, biofluid oxylipin profile, tissue oxylipin production capacity, and blood pressure. Ephx2 gene disruption eliminated soluble epoxide hydrolase protein expression and activity in liver, kidney, and heart from each colony. Plasma levels of epoxy fatty acids were increased, and fatty acid diols levels were decreased, while measured levels of lipoxygenase- and cyclooxygenase-dependent oxylipins were unchanged. Liver and kidney homogenates also show elevated epoxide fatty acids. However, in whole kidney homogenate a 4-fold increase in the formation of 20-hydroxyeicosatetraenoic acid was measured along with a 3-fold increase in lipoxygenase-derived hydroxylation and prostanoid production. Unlike previous reports, however, neither Ephx2-null colony showed alterations in basal blood pressure. Finally, the soluble epoxide hydrolase-null mice show a survival advantage following acute systemic inflammation. The data suggest that blood pressure homeostasis may be achieved by increasing production of the vasoconstrictor, 20-hydroxyeicosatetraenoic acid in the kidney of the Ephx2-null mice. This shift in renal metabolism is likely a metabolic compensation for the loss of the soluble epoxide hydrolase gene.
Soluble epoxide hydrolase (sEH)3 is a ubiquitous enzyme found in many tissues such as liver, kidney, heart, and ovary (1). sEH catalyzes the degradation of endogenous epoxy lipids such as epoxyeicosatrienoic acids (EETs) to their less active diols (dihydroxyeicosatrienoic acids, DHETs) and hence plays a critical role in the control of EET levels (2). These epoxy lipids are potent vasodilators, regulating cerebral and renal homodynamic and blood pressure (3–5). In addition, EETs inhibit platelet aggregation (6), promote fibrinolysis (7) and have anti-inflammatory properties (8, 9). Whereas deletion of the sEH gene, Ephx2, has been reported to reduce blood pressure in male mice (10), the inhibition of endogenous EET hydrolysis may provide pharmacological benefit in hypertension and acute inflammation (11).
The human Ephx2 gene encodes sEH and consists of 19 exons encoding 555 amino acids (12). There is 73% homology between the human and mouse sEH protein sequences (13), with 100% conservation in the catalytic residues (14). Each monomer of the homodimeric mouse sEH has two distinct domains (14, 15). The N-terminal domain exhibits phosphatase activity, and the C-terminal domain is responsible for the epoxide hydrolase activities (16, 17). Whereas the endogenous role of the phosphatase domain has yet to be elucidated, lipophilic phosphates appear to be excellent substrates (17–19).
Extensive investigations in recent years have established a role for cytochrome P450 (CYP)-derived eicosanoids in the regulation of blood pressure (11). The CYP-derived eicosanoids are synthesized throughout the body but act in a paracrine and autocrine fashion to regulate cellular function. In particular, the kidney and vascular endothelium produce significant levels of CYP-derived eicosanoids with effects on renal tubular transport function and vascular reactivity. Altered levels of renal EETs and hydroxyeicosatetraenoic acids (HETEs) are associated with changes in blood pressure (20–22). Furthermore, CYP epoxygenase activity is increased in animal models of hypertension, including the spontaneously hypertensive rats (21) and a high salt diet (23). The elevation in EET biosynthesis in these models could be a consequence of elevated blood pressure and might represent a compensatory response of the animal to deleterious increases in blood pressure. Moreover, inhibition of epoxygenases by clotrimazole leads to salt-dependent hypertension (24) and an inability to increase P450 epoxygenase activity with excess dietary salt intake in Dahl salt-sensitive rats might contribute to the hypertensive phenotype of these animals (23). In addition, with the onset of obesity-related hypertension the renal expression of CYP epoxygenases declines, while CYP omega hydroxylase and cyclooxygenase 2 expression increase (25, 26). Therefore, it would appear that the interplay of epoxygenase, ω-hydroxylases, and cyclooxygenase in the kidney are important determinants of blood pressure control.
To expand on the phenotypic characterization of the Ephx2-null mouse, animals were obtained from two independently derived and housed colonies of mice. The two colonies were either derived at the National Institutes of Health (10), or by Lexicon Genetics Inc. (The Woodlands, Texas). The latter, was exclusively for Boehringer Ingelheim Pharmaceuticals Inc. (Ridgefield, CT). Previous studies have shown that attenuation of endogenous EETs hydrolysis via sEH inhibition is a potentially useful pharmacological strategy for blood pressure regulation (4, 9, 27). In this regard, our main goal for this study was to perform a broad profile of the Ephx2-null phenotype and subsequently to evaluate blood pressure regulation under resting state as well as stressed conditions. We hypothesized that elevated endogenous EETs would have an advantageous role in blood pressure homeostasis and protect the overall health of the animal under abnormal conditions in which blood pressure is compromised.
EXPERIMENTAL PROCEDURES
Animals and Genotypic Analysis
Mice on a 129X1/SvJ × C57BL/6 background with targeted disruption in the Ephx2 gene were obtained from Chris Sinal, Dalhousie University, Halifax, NS, Canada under a National Cancer Institute Material Transfer Agreement 1-16268-04 (10). These mice backcrossed onto a C57BL6 genetic background three generations prior to use in this study. This colony is referred as the NIH colony, and was used to rederive a new colony by embryo transfer. Routine genotyping of sEH-null mice was performed as published (10) using the following primers F1, 5′-TGAGATTGGCACGAC-CCTA-3′; R2, 5′-TTGAAACCTGGGCTGGACG-3′; and Neo R3, 5′-AGTTCATTCAGGGCACCG-3′. Primer F1/R2 predicts a 290-base pair product for the wild type allele (Fig. 1A). For the null allele, primers F1/R3 predict a 560-base pair product of a neomycin resistance sequence as described previously (Fig. 1A and Ref. 10).
FIGURE 1. Ephx2-null mice of the NIH and BI colonies lack a complete sEH gene and express no detectable sEH protein.
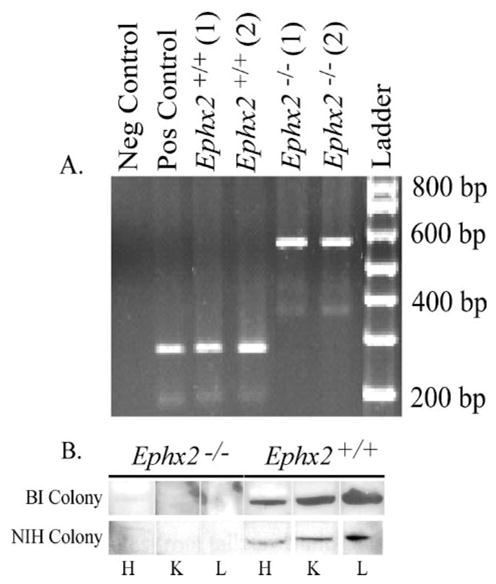
A, representative genotype analysis of wild type and Ephx2-null mice from the NIH colony. Negative and positive controls show no band product or a 290-bp band, respectively. Genomic DNA of sEH amplified from mouse tail shows a 290-bp gene product of sEH from NIH colony wild type. The Ephx2-null mice show a higher gene product of 560 bp from the neomycin resistant gene. Genotypic analysis was performed in all mice in each breeding colony. B, tissue distribution of sEH protein from wild type and Ephx2-null mice from both colonies. Immunoblots demonstrate expression of the 62.5 kDa protein in heart (H), kidney (K), and liver (L) of wild type (+/+) and lack of sEH protein expression in Ephx2-null mice (−/−) of both colonies.
A second colony of sEH-null mice was independently derived for Boehringer Ingelheim Pharmaceuticals, Inc. The heterozygous Ephx2 founder mice were created at Lexicon Genetics Inc. by gene trapping using random insertional mutagenesis with retroviral vector VICTR48 as described by Zambrowicz et al. (28). The integration site of the targeting cassette is in intron two. The heterozygous Ephx2 mice were then mated to establish a colony in the C57BL/6 albino × 129Sv/Ev mixed genetic background. Once obtained, Ephx2-null homozygotes were paired for subsequent breeding. Mice were maintained on sterile normal rodent diet (PicoLab rodent 20 from LabDiet, Richmond, IN) and bottled water ad libitum. Mice at age of 12–20 weeks were used and matched wild type mice were purchased from Taconic Farms. These animals were housed and bred in the Department of Cardiovascular Disease at Boehringer Ingelheim Pharmaceuticals, Inc. and are referred to as the BI colony.
BI Colony Blood Pressure
All procedures were conducted in accordance with approved IACUC protocols. Mean blood pressure (BP) and heart rate (HR) were monitored continuously in conscious, unrestrained female (20 weeks old) and male (12–20 weeks old) wild type and sEH-null mice using DSI telemetry (St. Paul, MN). Mice were surgically implanted with DSI TA11PA-20 telemetry transmitters and allowed to recover for a minimum of 7 days prior to study. Following recovery, mice were singly housed in plastic boxes placed directly on top of the telemetry receiver. Mean BP and HR data were recorded for 60 s every 20 min for the duration of the monitoring period. Individual mean BP and HR data were averaged over a 12-h cycle.
NIH Colony Blood Pressure and LPS Challenge
All procedures were conducted in accordance with approved IACUC protocols. Mean systolic blood pressure (SBP) and HR were measured in conscious, restrained wild type and Ephx2-null mice, starting at 8 weeks of age. Systolic blood pressure was measured by tail cuff plethysmography using a BP-2000 blood pressure analysis system (Visitech Systems, Apex, NC). Mice (n = 4 per genotype) were trained for 6 days to become acclimated to the Visitech System. For SBP and HR, twenty measurements were recorded daily and the last ten measurements averaged for a given mouse.
Western Blot Analysis
The expression of sEH or CYP4A proteins were analyzed by homogenization of tissues as described earlier (29). Briefly, protein concentrations were determined using the Pierce bicinchoninic acid protein assay. Protein aliquots from each tissue were resolved on 10–12% sodium dodecyl sulfate-polyacrylamide gels, transferred to nitrocellulose membranes, and immunoblotted with polyclonal antibodies against human sEH ((30); 1:1,000) or polyclonal antibodies against rat CYP4A (Chemicon International, Temecula, CA) followed by horseradish peroxidase-conjugated secondary antibody (1:10,000). β-Actin antibodies (1:10,000) were purchased from Sigma, followed by anti-mouse horseradish peroxidase-conjugated secondary antibody (1:10,000). Chemiluminescent detection using ECL reagent from Amersham Biosciences (Piscataway, NJ) was carried out on x-ray film.
sEH Activity Assay
Soluble epoxide hydrolase activity was performed as previously described (29). Briefly, homogenates were diluted with sodium phosphate (100 mM, pH 7.4; 0.1 mg/ml bovine serum albumin) and chilled (on ice) until assayed. Assays were initiated by adding 1 μl of 5 mM [2-3H]-trans-1,3-diphenylpropene oxide (3H-tDPPO; [S]final = 50 μM) in N,N′-dimethyl-formamide to 100 μl of tissue homogenate. Tubes were incubated at 30 °C for 5 min. Reactions were quenched with 60 μl of methanol and 200 μl of isooctane (31). Controls for interfering glutathione-transferase activity were prepared as described previously, but were quenched with 60 μl of methanol and 200 μl of hexanol (31). A 40-μl aliquot of the aqueous phase was analyzed for the presence of the radioactive diol product by liquid scintillation counting. Reactions were performed in triplicate. Results are means ± S.D. of three separate experiments.
Sample Collection and Oxylipin Extraction
Blood was collected from phenobarbital-anesthetized mice by cardiac puncture in EDTA-rinsed syringes. Each sample was immediately spun (10 min, 400 × g), and the plasma was separated and stored at −80 °C until analysis. Urine was collected from male Ephx2-null or wild type mice (n = 3) housed 24 h in metabolic chambers. Food and water were provided ad lib. Urine was collected into dry ice-chilled tubes containing butylated hydroxytoluene and EDTA at 0.2 mg and 10 mg of triphenylphosphine. Samples were stored at −80 °C until analysis. Oxylipins were extracted by solid phase extraction. Detailed protocols are provided as Supplemental Data under “Oxylipin extraction” (32).
Oxylipin Production Assay
Polyunsaturated fatty acid oxidase activities were determined in S9 fractions using modifications of previously reported procedures (29). Fatty acid epoxygenase, ω-hydroxylase, mid-chain hydroxylase, and prostaglandin synthase activities were obtained simultaneously. Briefly, aliquots (2 mg of protein) were suspended in 100 μl of 0.1 M sodium phosphate buffer (pH 7.4) and incubated (30 °C, 30 min) with or without arachidonic acid (100 μM) in the presence of an NADPH regenerating system. Reactions were halted with methanol containing analytical surrogates, and samples were centrifuged to remove precipitated protein. Arachidonate oxidation products were quantified in the isolated supernatant by HPLC/MS/MS. In the absence of arachidonic acid, no oxidation products were detected. Each reaction was performed in duplicate and mean specific activities were expressed in fmol/min/mg protein.
Oxylipin Analysis
Epoxides, diols, alcohols, and ketones of linoleate and arachidonate were quantified in all samples. Thromboxanes and prostaglandins were also measured in plasma and tissue production assays, but not urine because of matrix-dependent ion suppression. These oxylipins were quantified by HPLC/MS/MS using internal standard methods (32). Detailed protocols including HPLC (supplemental Table S1) and mass spectroscopy (supplemental Table S2) parameters are provided as Supplemental Data. Plasma and urine epoxide and diol surrogate recoveries were acceptable in all samples (see supplemental Table S3). Recoveries of tetradeuterated 6-keto-prostaglandin F1α (d4–6-keto-PGF1 α) were acceptable in plasma (see supplemental Table S3) and used to quantify 6-keto-PGF1α. Epoxide surrogate hydrolysis was less than 3% for all assays. Reagent blanks showed background levels below detection limit, and analytical replicates showed precision to be within 15% for greater than 80% of the analytes present at levels greater than ten times the method detection limits.
Statistics
Values are expressed as mean ± S.D. or S.E., as indicated. Data were analyzed by Student’s t test or ANOVA, as indicated under the figure legends. Values were considered significantly different if p < 0.05.
RESULTS
sEH Gene, Protein Expression, and Activity
The sEH gene was not detected in either colony of Ephx2-null mice (Fig. 1A). Data are shown for the NIH colony. Identical results were obtained with the BI colony (data not shown). Evaluation of sEH protein expression was performed on tissues from representative animals from each colony. Polyclonal antibodies highly selective to sEH recognized a protein band of 62.5 kDa in liver, kidney, and heart of wild type sEH mice from the BI and NIH colonies (Fig. 1B), but not in Ephx2-null mice (Fig. 1B). sEH was also observed in the lungs, spleen, adipose, and testis (data not shown) as previously reported (1). The distribution of the sEH protein was in agreement with published data as previously summarized (33). The results from the sEH tDPPO hydrolase activity assays are shown in Table 1. As expected based on the immunoblots, hydrolase activity was observed in wild type, but not in Ephx2-null tissues. Notably, the sEH activity measured in heart was significantly different between the Ephx2+/+ mice from the two colonies (Table 1). sEH activity was ten times higher in heart extract from the BI colony. However, the rank order activity in both colonies was the same.
TABLE 1. sEH specific activities in liver, kidney, and heart tissue homogenates from the wild-type and Ephx2-null mice.
Tissues were extracted as described under “Experimental Procedures” and examined for sEH activity in S9 fractions using tDPPO as a substrate. Ephx2-null mice show no epoxide hydrolase activity toward tDPPO in any of examined tissues in two different Ephx2-null mouse colonies originated from NIH or BI. All results are means ± S.D. of three separate experiments as described under “Experimental Procedures.”
| Genotype | BI colony
|
NIH colony
|
||
|---|---|---|---|---|
| Ephx2+/+ | Ephx2−/− | Ephx2+/+ | Ephx2−/− | |
| nmol diol produced × min−1 × mg protein−1 | nmol diol produced × min−1 × mg protein−1 | |||
| Liver | 32 ± 2 | <0.1 | 38 ± 5 | <0.01 |
| Kidney | 18 ± 3 | <0.1 | 16 ± 3 | <0.01 |
| Heart | 14 ± 2 | <0.1 | 1.4 ± 0.2 | <0.04 |
Blood Pressure
The initial description of the Ephx2-null mouse indicated a male-specific hypotensive phenotype (10). To assess the maintenance of this phenotype in the two new colonies, blood pressure and heart rate measurements were performed in NIH colony by tail cuff and in the BI colony by telemetry. In the BI colony, no physiologically relevant differences in mean blood pressure were noted between genotypes (Fig. 2A). There was a significant difference in mean blood pressure noted between female and male Ephx2-null mice but only on the first day of the 6-day monitoring period. Female wild type mice exhibited a trend toward higher overall heart rate levels compared with male wild type and Ephx2-null mice (Fig. 2A). These results are inconsistent with data published previously (10). To further investigate a potential strain difference, blood pressure was also assessed for 10 days in male Ephx2-null mice from the NIH-derived UC Davis colony compared with wild type. The NIH colony also failed to reveal a consistent difference in systolic blood pressure measurements between Ephx2-null and wild type mice (Fig. 2B). Several transient differences were measured on day 4 and 6, but were variable and not sustained. Therefore, no appreciable differences in resting blood pressure were detected between wild type and Ephx2-null genotypes from either of the two independently derived colonies.
FIGURE 2. Elimination of sEH protein expression fails to cause detectable alteration in blood pressure.
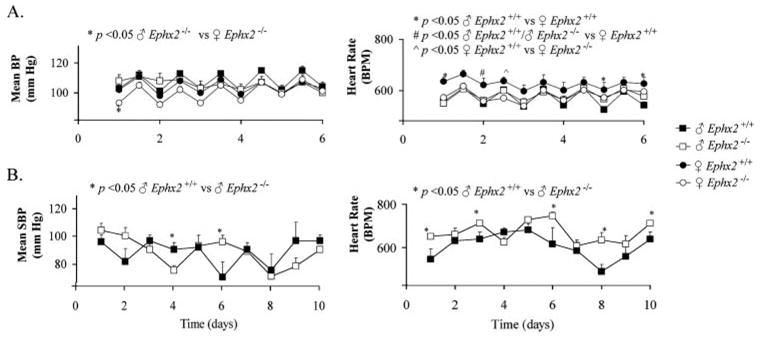
A, mean blood pressure (BP) and heart rate (HR) were recorded via telemetry (12-h averages) in wild type and Ephx2-null mice from the BI colony. B, mean systolic blood pressure (SBP) and HR were recorded by tail cuff measurements in the NIH colony. Results are presented as mean ± S.E. for: ♂Ephx2+/+ (n = 9); ♂ Ephx2−/− (n = 11); ♀Ephx2+/+.(n = 4) and ♀Ephx2−/− (n = 5) in the BI colony and mean ± S.E. for: ♂ Ephx2+/+ (n = 4) and ♂ Ephx2−/− (n = 4) in the NIH colony. Significant differences are indicated by ANOVA with Tukey-Kramer for telemetry and single factor ANOVA for tail cuff plethysmography (p < 0.05).
Plasma Oxylipins
Plasma CYP- and sEH-dependent oxylipin profiles from wild type and Ephx2-null animals from each colony are shown in Table 2 and described graphically in supplemental Fig. S1A. Compared with their wild type counterparts, both the BI and NIH colony Ephx2-null mice showed elevated plasma levels of linoleate epoxides (EpOMEs) and 14(15)-EET. Levels of the linoleate diol 12,13-dihydroxyoctadecaenoic acid (12,13-DHOME) were reduced in these mice. Significant reductions in the 14,15-DHET were also observed in the BI colony Ephx2-null mice. Fig. 3 displays enzymatic substrate to product ratios of plasma oxylipins for wild type and Ephx2-null mice from the BI colony (see Ref. 34 for NIH-derived colony results). Whereas the CYP-epoxy-genase/sEH-dependent epoxidediol ratios were altered by Ephx2 disruption, the sEH-independent, 9-HODE to 9-oxo-ODE ratio was unaffected in the Ephx2-null mice. In addition, the magnitude of the difference between the sEH-dependent substrate to product ratios was correlated with the distance of the oxidized center from the carboxyl carbon (n = 6, r > 0.81, p < 0.05; Fig. 3, inset). This trend mimics the substrate selectivity of the soluble epoxide hydrolase, which preferentially hydrolyzes epoxy fatty acids with epoxides positioned far from the carboxyl carbon as summarized in Newman et al. (33). Changes observed in plasma levels of the measured lipoxygenase- and cyclooxygenase-dependent metabolites were less than 2-fold. The CYP4A metabolite 20-HETE was not detected in plasma extracts (limit of detection, 0.1 nM).
TABLE 2. Plasma CYP- and sEH-dependent oxylipin concentration (n ) determined in wild type (Ephx2+/+) and Ephx2-null (Ephx2−/−) mice from the BI and NIH colonies.
Values shown are mean ± S.E. (n = 3 – 5 per group). Results for 20-HETE were below the 0.1 nM limit of detection and are not shown.
| Oxylipin | BI colony
|
NIH colony
|
||
|---|---|---|---|---|
| Ephx2+/+ | Ephx2−/− | Ephx2+/+ | Ephx2−/− | |
| Epoxygenase-dependent metabolism | ||||
| 12(13)-EpOME | 18.1 ± 1.6 | 124.0 ± 11a | 12.6 ± 0.3 | 77.9 ± 11.7a |
| 9(10)-EpOME | 11.2 ± 2.4 | 47.9 ± 4.2a | 6.3 ± 0.4 | 23.8 ± 0.5a |
| 14(15)-EET | 1.7 ± 0.3 | 3.2 ± 0.2a | 0.4 ± 0.2 | 2.6 ± 0.2a |
| 11(12)-EET | 1.4 ± 0.3 | 1.7 ± 0.1 | 0.4 ± 0.1 | 2.5 ± 0.5 |
| 8(9)-EET | 4.1 ± 0.6 | 5.0 ± 0.9 | 0.4 ± 0.1 | 1.5 ± 0.7 |
| 5(6)-EET | 4.3 ± 1.1 | 3.2 ± 1.0 | 13.3 ± 2.1 | 34.7 ± 10.5 |
|
| ||||
| Soluble epoxide hydrolase-dependent metabolism | ||||
| 12,13-DHOME | 48.9 ± 5.1 | 11.3 ± 1.6a | 16.16 ± 0.02 | 7.2 ± 0.7a |
| 9,10-DHOME | 24.5 ± 2.5 | 24.5 ± 4.5 | 7.4 ± 0.6 | 8.1 ± 0.8 |
| 14,15-DHET | 1.4 ± 0.1 | 0.2 ± 0.1a | 0.6 ± 0.2 | 0.4 ± 0.1 |
| 11,12-DHET | 1.0 ± 0.2 | 0.5 ± 0.1 | 0.36 ± 0.04 | 0.5 ± 0.1 |
| 8,9-DHET | 1.4 ± 0.5 | 1.46 ± 0.57 | 4.9 ± 2.4 | 7.1 ± 1.2 |
| 5,6-DHET | 2.2 ± 0.3 | 1.65 ± 0.24 | 0.3 ± 0.2 | 0.4 ± 0.1 |
p < 0.05 versus wild type (2-tailed t test).
FIGURE 3. Plasma oxylipin substrate:product ratios in the wild type and Ephx2-null mice.
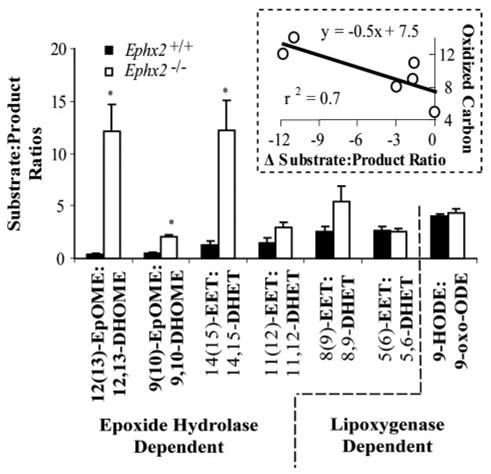
Plasma epoxide:diol ratios were elevated in Ephx2-null mice over the wild type in both the linoleate (EpOME:DHOME) and arachidonate series (EET:DHET). Conversely the sEH-independent 9-HODE:9-oxo-ODE ratio was unaffected in the Ephx2-null mice. The magnitude of the difference between sEH-dependent substrate: product ratios showed a strong correlation (p < 0.05, r > 0.81; Pearsons correlation) with the distance of the oxidized center from the carboxyl carbon (inset box). Significant differences are indicated with an asterisk (*, p < 0.05; 2-tailed t test).
LPS Treatment
Acute inflammation induced by LPS results in hypotension in mice that can be blocked by prophylactic treatment with sEH inhibitors (23). Similarly, the Ephx2-null mice showed a reduced susceptibility to LPS-induced hypotension (Fig. 4). All Ephx2-null mice treated with LPS (10 mg/kg, i.p) survived, while 18% of the wild type mice died within the first 24 h. Systolic blood pressure (Fig. 4) and heart rates were reduced by LPS exposure in both genotypes. Five hours post-LPS administration HR in Ephx2-null mice was 462 bpm, while the wild type mice became so hypotensive that the blood pressure and HR dropped below detectable limits (Fig. 4). Over the next 24–48 h, the HRs in Ephx2-null mice increased to 505 ± 49 and 622 ± 54 beeps per minute (bpm), respectively, whereas HRs in the wild types remained low, 437 ± 27 and 433 ± 21 bpm at 24 and 48 h, respectively. Thus, compared with wild type, survival and recovery from the severe, acute hypotensive response to LPS exposure was enhanced among the Ephx2-null genotype as demonstrated by the attenuated SBP response and mortality rate observed at both 24 and 48 h post-LPS (Fig. 4).
FIGURE 4. Elimination of sEH protein expression reduces hypotensive effects of LPS challenge in mice.
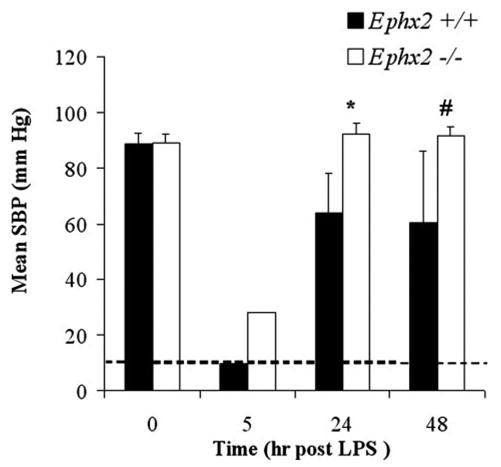
Genotype-specific effects on blood pressure and heart rate in response to LPS challenge. The mean systolic blood pressure (SBP) and heart rate (HR) responses were recorded via tail-cuff in the NIH colony before and periodically after administration of LPS (10 mg/kg, i.p). At 5 h post-LPS administration, the wild type blood pressure and heart rate were below the instrument detection limit (dashed line). Recovery from the acute hypotensive effect of LPS exposure was enhanced among the Ephx2-null mice compared with their wild type counterparts as demonstrated by the attenuated SBP response at both 24 and 48 h post-LPS (single factor ANOVA. *, p < 0.05; #, p < 0.1).
Tissue Oxylipin Production Capacity
Arachidonic acid metabolism was evaluated in the liver and kidney of the wild type and Ephx2-null mice to investigate the potential for compensatory mechanisms in this mutant strain (Table 3). In this assay, the biochemical activity of series of polyunsaturated fatty acid oxidases was determined in S9 fractions using arachidonic acid as a substrate with a NADPH regenerating system. While NADPH can inhibit lipoxygenase activity, it does not alter the relative product profile with fatty acids (35). Metabolite production rates of cyclooxygenase, lipoxygenase, and P450 enzymes are expressed by the concentration of their corresponding metabolites in Table 3 and described graphically in supplemental Fig. S1B.
TABLE 3. Rates of arachidonic acid metabolism determined in BI colony Ephx2 wild type and -null mouse liver and kidney homogenates.
Results are the mean ± S.E. of a minimum of 4 measurements of tissue samples from three mice per group. Differences in means were determined with 2-tailed t tests.
| Liver
|
Kidney
|
|||
|---|---|---|---|---|
| Ephx2+/+ | Ephx2−/− | Ephx2+/+ | Ephx2−/− | |
| fmol × min−1 × mg protein−1 | fmol × min−1 × mg protein−1 | |||
| Epoxygenase metabolism | ||||
| 14(15)-EET | 20.6 ± 5.3 | 170 ± 27a | <0.1 | 22.4 ± 12 |
| 11(12)-EET | <0.1 | <0.1 | <0.1 | 68.7 ± 25a |
| 8(9)-EET | 10.9 ± 0.94 | 93 ± 3.9a | <0.1 | 8.77 ± 1.2 |
| 5(6)-EET | 22.8 ± 6.9 | 5.68 ± 0.89 | <0.1 | <0.1 |
|
| ||||
| Soluble epoxide hydrolase metabolism | ||||
| 14,15-DHET | 205 ± 44 | 23.4 ± 8a | 58.9 ±2.6 | 11 ± 7.6a |
| 11,12-DHET | 223 ± 43 | 43.4 ± 11a | 58.6 ±5.4 | 15.7 ± 8.5a |
| 8,9-DHET | <0.1 | <0.1 | 35.7 ±6.8 | 6.9 ± 0.5a |
| 5,6-DHET | 49.7 ± 8.6 | 10.8 ± 0.77a | 11.3 ±3.4 | 8.61 ± 0.7 |
|
| ||||
| ω-Hydroxylase metabolism | ||||
| 20-HETE | 297 ± 63 | 331 ± 30 | 194 ±130 | 827 ± 54a |
|
| ||||
| Lipoxygenase metabolism | ||||
| 15-HETE | 41.8 ± 9.3 | 35.8 ± 6.3 | 269 ±46 | 508 ± 40a |
| 12-HETE | 44 ± 10 | 27.5 ± 8.4 | 93.5 ±23 | 150 ± 14a |
| 11-HETE | 43.5 ± 9.1 | 26.5 ± 4.1 | 104 ±2.8 | 186 ± 26a |
| 8-HETE | 66.2 ± 14 | 42.7 ± 7.2 | 47.1 ±12 | 117 ± 3.4a |
| 5-HETE | 21.7 ± 3 | 20.1 ± 3.8 | 115 ±18 | 306 ± 22a |
|
| ||||
| Cyclooxygenase metabolism | ||||
| 6-keto-PGF1a | 10.3 ± 0.83 | 8.94 ± 1.6 | 13 ±3 | 124 ± 32a |
| PGF2a | 6.56 ± 1.6 | 5.93 ± 0.93 | 63.9 ±7.6 | 330 ± 72a |
| PGE2 | 4.19 ± 0.26 | 4.54 ± 0.093 | 9.78 ±1.2 | 33 ± 7.5a |
|
| ||||
| Autooxidation marker | ||||
| 9-HETE | 8.76 ± 1.1 | 8.66 ± 2 | NAb | NA |
p ≤ 0.05. <0.1 = below the 0.1 nM limit of detection.
NA, not analyzed.
Liver homogenates from Ephx2-null mice generally produced higher concentrations of EETs and reduced DHETs as compared with liver homogenates from their wild type counterparts (Table 3). Specifically the 14(15)- and 8(9)-EETs were elevated, whereas, the 14,15- and 11,12-DHETs were reduced. The low affinity sEH regioisomer-substrate, 5(6)-EET and its corresponding diol are also reduced in the Ephx2-null liver homogenate. Whole kidney oxylipin production shows a pattern that is different from the liver (Table 3).
Whereas lowered DHETs and elevated EET production was observed in Ephx2-null kidney homogenates, the ω-hydroxylase metabolite, 20-HETE was enhanced 4-fold. Moreover, the lipoxygenase-dependent metabolites (mid-chain HETEs) and the cyclooxygenase-dependent prostaglandins PGF2α, PGE2, and 6-keto-PGF1α were also increased 2–10-fold in the Ephx2-null kidney homogenates, suggesting up-regulation of these enzymes (Table 3).
Renal Feedback
Considerable evidence has accumulated indicating that expression of CYP enzymes and the production of EETs and 20-HETE in the kidney and peripheral vasculature are strong regulators of peripheral vascular tone, renal function, and blood pressure. Cellular expression of CYP 4A was measured in liver and kidney tissue homogenates. CYP 4A protein expression was 4-fold higher in whole kidney tissue homogenate from the Ephx2-null mice compared with wild type, and depressed in liver tissue homogenates of both genotypes (Fig. 5).
FIGURE 5. Expression of CYP4A in kidney and liver homogenates of wild type and Ephx2-null mice from NIH colony.
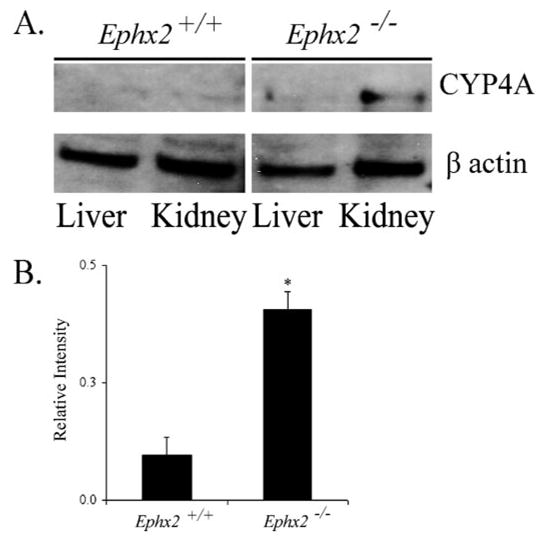
A, representative Western blot showing the expression of CYP4A protein detected with rabbit anti-rat CYP4A1–3 and β-actin to confirm equivalent loading. B, densitometric values of CYP4A expression level in renal extracts from wild type and Ephx2-null mice (mean S.E. of n = 3; t test. *, p < 0.05).
Urine oxylipin analysis from Ephx2-null or wild type male mice from the BI colony is presented in Fig. 6 and Fig. S1C. Whereas concentrations of metabolites among the pooled samples were high, the relative abundance of excreted metabolites showed significant differences between genotypes. Notably, the 12(13)-EpOME and 20-HETE were higher in the Ephx2-null urine, whereas the 12,13-DHOME, 14,15-DHET, and 5,6-DHET were all lower in this genotype. The concentrations of EETs were close to the detection limit in five of the six analyzed samples, likely precluding determination of changes in excretion of these metabolites.
FIGURE 6. Urinary oxylipin analyses showed changes in the relative abundance of excreted linoleate (A) and arachidonate (B) metabolites.
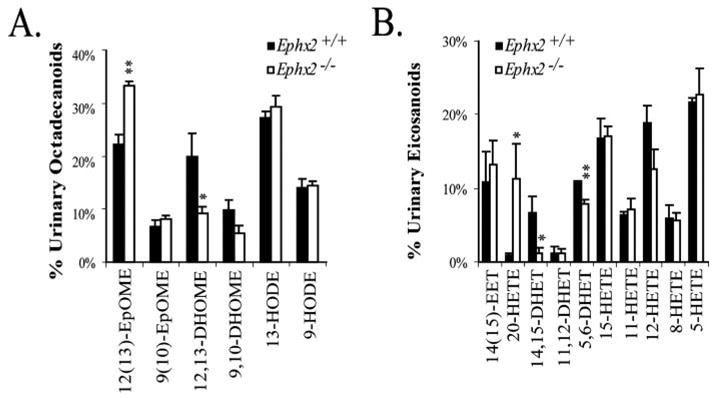
A and B show the mean ± S.E. percentage of each analyte with respect to the quantified lipids of the same carbon number. Differences were determined by two-tailed t-tests and differences are indicated at the **, p < 0.05 and *, p < 0.1 levels.
DISCUSSION
Adaptation to defects in signaling, including genetic disruptions, are natural responses that allow organisms to maintain homeostatic control of critical phenotypic characteristics (36, 37, 38). An inability to compensate for such changes can lead to dire consequences (39). The initial description of the NIH-derived Ephx2-null mouse reported a reduction in male resting blood pressure (10). A number of studies have added additional support to the sEH-blood pressure regulatory link. Pharmacological inhibition of sEH is efficacious in lowering blood pressure in angiotensin-dependent hypertension in rodents (4, 21, 40, 41). Furthermore, several strains of spontaneously hypertensive rats show elevated sEH protein, and only these rats respond to sEH inhibitor therapy (21, 42). Pharmacological inhibition of sEH has also been shown to normalize blood pressure in septic shock induced by LPS in rodents (27). In a recent study it was found that genetic variations of the Ephx2 coding sequence are associated with the risk of coronary heart disease (34, 43). These results clearly suggest that sEH activity plays a role in blood pressure homeostasis.
The current study was undertaken to expand the phenotypic and metabolic characterization of the Ephx2-null mouse lines. Genetic analysis confirmed that the sEH gene was successfully disrupted, and that this disruption was associated with loss of sEH expression and activity in all measured tissues (Fig. 1 and Table 1). In contrast to the earlier report by Sinal et al. (10), changes in basal blood pressure in the NIH colony were not observed in our study (Fig. 2). In addition, continuous blood pressure recording via telemetry in an independently derived Ephx2-null mouse colony (BI colony) produced identical findings. In the BI colony, systolic, diastolic and mean blood pressures were recorded continuously over a six-day monitoring period. No sustained changes in blood pressure or heart rate were observed (Fig. 2).
To determine if a blood pressure phenotype could be revealed by a pro-hypotensive treatment, blood pressure and heart rate were recorded in a group of 4 mice from each genotype of the NIH colony after exposure to 10 mg/kg LPS (Fig. 4). Acute LPS administration produces a severe hypotension in mice which can be blunted by prophylactic inhibition of sEH (27). The Ephx2-null mice show a similar resistance (Fig. 4). The recovery was faster and survival was increased in the Ephx2-null mice as compared with wild types. Thus, it appears that an alteration of blood pressure regulation is retained in these mutants. In several hypertensive models, sEH inhibitors lower blood pressure toward normotensive levels, and in LPS-treated animals, sEH inhibitors buffered the fall in blood pressure toward normotensive levels (4, 27). These data indicate a role for epoxyeicosanoids in blood pressure homeostasis.
To further characterize the nature of the adaptive response in the Ephx2-null animals, oxylipin concentrations were measured in plasma and urine of healthy mice. Plasma profiles in sEH-gene disrupted mice revealed elevated epoxy fatty acids and decreases in their corresponding diols. The ratios of these epoxide/diol substrate-product pairs were elevated in the Ephx2-null mice in a structurally specific manner (Fig. 3), consistent with the known substrate specificity of the sEH. Thus, the 12,13-EpOME:DHOME and 14,15-EET:DHET ratios were the most sensitive indicators of sEH activity in these rodents. Interestingly, the epoxy fatty acid degradation was not totally abolished in the Ephx2-null mice (Ref. 10 and Table 2), suggesting an alternative route for their hydrolysis.
The CYP ω-hydroxylase metabolite, 20-HETE, plays a major role in vasoconstriction and renal natriuretic mechanisms (44). Recent reports further strengthen this concept and substantiate the link between CYP4A, 20-HETE, and hypertension (45–47). In the renal vasculature and glomerulus, 20-HETE is a vasocon-strictor that lowers glomerular filtration rate and raises blood pressure (44). In the peripheral microcirculation, 20-HETE also increases vascular tone and raises blood pressure (48). The increase in urine 20-HETE provided evidence for enhanced renal ω-hydroxylase activity in the Ephx2-null mouse. Thus, the elevated 20-HETE production and enhanced CYP4A protein expression in the kidney offers a mechanistic explanation for the stabilization of basal blood pressure and the blunted response to LPS challenge.
Finally, in this study we provide data supporting the hypothesis that the Ephx2-null genotype is associated with a phenotypic adaptive response in renal lipid metabolism. This change results in maintenance of normal basal blood pressure. One possible explanation for the discrepancy between the blood pressure observations made by Sinal et al. (10) and those of this study could be related to the genetic background of the Ephx2 null mice. For example, previous data from Sinal et al. were obtained on a different genetic background and after far fewer generations than either of the two strains reported here. In addition, the earlier observations reported only small changes in blood pressure, which could be missed because of pressure oscillations in the current work. These results support a self-regulating interaction between the epoxygenase and ω-hydroxylase pathways producing a fine-tuned control over blood pressure homeo-stasis in the mouse.
Supplementary Material
Acknowledgments
We thank C. Williams, D. Cooper, R. Sellati, Mary McFarland, and Lynn Pantages-Torok for technical assistance and Theresa Pedersen for critical review of the manuscript. National Institutes of Health Images is a public domain image processing and analysis program.
The abbreviations used are
- sEH
soluble epoxide hydrolase
- BP
blood pressure
- BPM
beeps per minute
- CYP
cytochrome P450
- DHETs
dihydroxyeicosatrienoic acids
- DHOME
dihydroxyoctadecaenoic acid
- EETs
epoxyeicosatrienoic acids
- EpOMEs
epoxyoctadecenoic acids
- HETE
hydroxyeicosatetraenoic acid
- HODE
hydroxyoctadecadienoic acid
- HR
heart rate
- LPS
lipopolysaccharide
- oxo-ODE
ketooctadeca(10Z,11E)dienoic acid
- PG
prostaglandin
- SBP
systolic blood pressure
- tDPPO
trans-1,3-diphenylpropene oxide
- ANOVA
analysis of variance
Footnotes
This work was supported in part by Grant R37 ES02710 from the NIEHS, National Institutes of Health, the NIEHS Superfund Basic Research Program P42 ES004699, NIEHS P30 ES05707, National Institutes of Health/NIEHS R01 ES013933, and the UCDMC Translational Technology Grant.
The on-line version of this article (available at http://www.jbc.org) contains supplemental data, Tables S1–S3, and Fig. S1.
References
- 1.Enayetallah AE, French RA, Thibodeau MS, Grant DF. J Histochem Cytochem. 2004;52:447–454. doi: 10.1177/002215540405200403. [DOI] [PubMed] [Google Scholar]
- 2.Spector AA, Fang X, Snyder GD, Weintraub NL. Prog Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 3.Imig JD, Navar LG, Roman RJ, Reddy KK, Falck JR. J Am Soc Nephrol. 1996;7:2364–2370. doi: 10.1681/ASN.V7112364. [DOI] [PubMed] [Google Scholar]
- 4.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Hypertension. 2002;39:690–694. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 5.Harder DR, Zhang C, Gebremedhin D. News Physiol Sci. 2002;17:27–31. doi: 10.1152/physiologyonline.2002.17.1.27. [DOI] [PubMed] [Google Scholar]
- 6.Heizer ML, McKinney JS, Ellis EF. Stroke. 1991;22:1389–1393. doi: 10.1161/01.str.22.11.1389. [DOI] [PubMed] [Google Scholar]
- 7.Node K, Ruan XL, Dai J, Yang SX, Graham L, Zeldin DC, Liao JK. J Biol Chem. 2001;276:15983–15989. doi: 10.1074/jbc.M100439200. [DOI] [PubMed] [Google Scholar]
- 8.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith KR, Pinkerton KE, Watanabe T, Pedersen TL, Ma SJ, Hammock BD. Proc Natl Acad Sci U S A. 2005;102:2186–2191. doi: 10.1073/pnas.0409591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ. J Biol Chem. 2000;275:40504–40510. doi: 10.1074/jbc.M008106200. [DOI] [PubMed] [Google Scholar]
- 11.Kroetz DL, Zeldin DC. Curr Opin Lipidol. 2002;13:273–283. doi: 10.1097/00041433-200206000-00007. [DOI] [PubMed] [Google Scholar]
- 12.Sandberg M, Meijer J. Biochem Biophys Res Commun. 1996;221:333–339. doi: 10.1006/bbrc.1996.0596. [DOI] [PubMed] [Google Scholar]
- 13.Beetham JK, Grant D, Arand M, Garbarino J, Kiyosue T, Pinot F, Oesch F, Belknap WR, Shinozaki K, Hammock BD. DNA Cell Biol. 1995;14:61–71. doi: 10.1089/dna.1995.14.61. [DOI] [PubMed] [Google Scholar]
- 14.Argiriadi MA, Morisseau C, Hammock BD, Christianson DW. Proc Natl Acad Sci U S A. 1999;96:10637–10642. doi: 10.1073/pnas.96.19.10637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez GA, Morisseau C, Hammock BD, Christianson DW. Biochemistry. 2004;43:4716–4723. doi: 10.1021/bi036189j. [DOI] [PubMed] [Google Scholar]
- 16.Cronin A, Mowbray S, Durk H, Homburg S, Fleming I, Fisslthaler B, Oesch F, Arand M. Proc Natl Acad Sci U S A. 2003;100:1552–1557. doi: 10.1073/pnas.0437829100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newman JW, Morisseau C, Harris TR, Hammock BD. Proc Natl Acad Sci U S A. 2003;100:1558–1563. doi: 10.1073/pnas.0437724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tran KL, Aronov PA, Tanaka H, Newman JW, Hammock BD, Morisseau C. Biochemistry. 2005;44:12179–12187. doi: 10.1021/bi050842g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Enayetallah AE, Grant DF. Biochem Biophys Res Commun. 2006;341:254–260. doi: 10.1016/j.bbrc.2005.12.180. [DOI] [PubMed] [Google Scholar]
- 20.Su P, Kaushal KM, Kroetz DL. Am J Physiol. 1998;275:R426–R438. doi: 10.1152/ajpregu.1998.275.2.R426. [DOI] [PubMed] [Google Scholar]
- 21.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Circ Res. 2000;87:992–998. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 22.Xu F, Straub WO, Pak W, Su P, Maier KG, Yu M, Roman RJ, Ortiz De Montellano PR, Kroetz DL. Am J Physiol Regul Integr Comp Physiol. 2002;283:R710–R720. doi: 10.1152/ajpregu.00522.2001. [DOI] [PubMed] [Google Scholar]
- 23.Makita K, Takahashi K, Karara A, Jacobson HR, Falck JR, Capdevila JH. J Clin Investig. 1994;94:2414–2420. doi: 10.1172/JCI117608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holla VR, Makita K, Zaphiropoulos PG, Capdevila JH. J Clin Investig. 1999;104:751–760. doi: 10.1172/JCI7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dey A, Williams RS, Pollock DM, Stepp DW, Newman JW, Hammock BD, Imig JD. Obes Res. 2004;12:1278–1289. doi: 10.1038/oby.2004.162. [DOI] [PubMed] [Google Scholar]
- 26.Wang MH, Smith A, Zhou Y, Chang HH, Lin S, Zhao X, Imig JD, Dorrance AM. Hypertension. 2003;42:594–599. doi: 10.1161/01.HYP.0000090123.55365.BA. [DOI] [PubMed] [Google Scholar]
- 27.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Proc Natl Acad Sci U S A. 2005 doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zambrowicz BP, Friedrich GA, Buxton EC, Lilleberg SL, Person C, Sands AT. Nature. 1998;392:608–611. doi: 10.1038/33423. [DOI] [PubMed] [Google Scholar]
- 29.Newman JW, Stok JE, Vidal JD, Corbin CJ, Huang Q, Hammock BD, Conley AJ. Endocrinology. 2004;145:5097–5105. doi: 10.1210/en.2004-0710. [DOI] [PubMed] [Google Scholar]
- 30.Du Teaux SB, Newman JW, Morisseau C, Fairbairn EA, Jelks K, Hammock BD, Miller MG. Toxicol Sci. 2004;78:187–195. doi: 10.1093/toxsci/kfh066. [DOI] [PubMed] [Google Scholar]
- 31.Borhan B, Mebrahtu T, Nazarian S, Kurth MJ, Hammock BD. Anal Biochem. 1995;231:188–200. doi: 10.1006/abio.1995.1520. [DOI] [PubMed] [Google Scholar]
- 32.Newman JW, Watanabe T, Hammock BD. J Lipid Res. 2002;43:1563–1578. doi: 10.1194/jlr.d200018-jlr200. [DOI] [PubMed] [Google Scholar]
- 33.Newman JW, Morisseau C, Hammock BD. Prog Lipid Res. 2005;44:1–51. doi: 10.1016/j.plipres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 34.Seubert JM, Sinal CJ, Graves J, Degraff LM, Bradbury JA, Lee CR, Goralski K, Carey MA, Luria A, Newman JW, Hammock BD, Falck JR, Roberts H, Rockman HA, Murphy E, Zeldin DC. Circ Res. 2006;99:442–450. doi: 10.1161/01.RES.0000237390.92932.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Donnell VB, Kuhn H. Biochem J. 1997;327:203–208. doi: 10.1042/bj3270203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi N, Brooks HL, Wade JB, Liu W, Kondo Y, Ito S, Knepper MA, Smithies O. J Am Soc Nephrol. 2002;13:604–610. doi: 10.1681/ASN.V133604. [DOI] [PubMed] [Google Scholar]
- 37.Besson V, Nalesso V, Herpin A, Bizot JC, Messaddeq N, Romand R, Puech A, Blanquet V, Herault Y. Biol Cell. 2005;97:787–798. doi: 10.1042/BC20040525. [DOI] [PubMed] [Google Scholar]
- 38.Scarff KL, Ung KS, Nandurkar H, Crack PJ, Bird CH, Bird PI. Mol Cell Biol. 2004;24:4075–4082. doi: 10.1128/MCB.24.9.4075-4082.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parlakian A, Charvet C, Escoubet B, Mericskay M, Molkentin JD, Gary-Bobo G, De Windt LJ, Ludosky MA, Paulin D, Daegelen D, Tuil D, Li Z. Circulation. 2005;112:2930–2939. doi: 10.1161/CIRCULATIONAHA.105.533778. [DOI] [PubMed] [Google Scholar]
- 40.Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. J Am Soc Nephrol. 2004;15:1244–1253. [PubMed] [Google Scholar]
- 41.Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. Hypertension. 2005;46:975–981. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sellers KW, Sun C, Diez-Freire C, Waki H, Morisseau C, Falck JR, Hammock BD, Paton JF, Raizada MK. Faseb J. 2005;19:626–628. doi: 10.1096/fj.04-3128fje. [DOI] [PubMed] [Google Scholar]
- 43.Lee CR, North KE, Bray MS, Fornage M, Seubert JM, Newman JW, Hammock BD, Couper DJ, Heiss G, Zeldin DC. Hum Mol Genet. 2006;15:1640–1649. doi: 10.1093/hmg/ddl085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roman RJ. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 45.Holla VR, Adas F, Imig JD, Zhao X, Price E, Jr, Olsen N, Kovacs WJ, Magnuson MA, Keeney DS, Breyer MD, Falck JR, Waterman MR, Capdevila JH. Proc Natl Acad Sci U S A. 2001;98:5211–5216. doi: 10.1073/pnas.081627898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Imig JD, Falck JR, Inscho EW. Br J Pharmacol. 1999;127:1399–1405. doi: 10.1038/sj.bjp.0702662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Messer-Letienne I, Bernard N, Roman RJ, Sassard J, Benzoni D. Eur J Pharmacol. 1999;378:291–297. doi: 10.1016/s0014-2999(99)00470-7. [DOI] [PubMed] [Google Scholar]
- 48.Sarkis A, Lopez B, Roman RJ. Curr Opin Nephrol Hypertens. 2004;13:205–214. doi: 10.1097/00041552-200403000-00009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.