

Summary
α chain of T-cell receptor (TCR) is a typical ERAD (ER-associated degradation) substrate degraded in the absence of other TCR subunits. Depletion of derlin1 fails to induce accumulation of αTCR despite inducing accumulation of α1-antitrypsin, another ERAD substrate. Furthermore, while depletion of VCP does not affect levels of α1-antitrypsin, it induces an increase in levels of αTCR. RNAi of VCP induces preferential accumulation of αTCR with less mannose residues, suggesting its retention within the ER. Mass spectrometric analysis of cellular N-linked glycans revealed that depletion of VCP decreases the level of high mannose glycoproteins, increases the levels of truncated low-mannose glycoproteins and induces changes in the abundance of complex glycans assembled in post-ER compartments. Since proteasome inhibition was unable to mimic those changes, they can not be regarded as a simple consequence of inhibited ERAD but represent a complex effect of VCP on the function of the ER.
Keywords: T-cell receptor, ER-associated degradation (ERAD), valosin-containing protein (VCP), derlin, retrotranslocation, ubiquitin, proteasome, protein degradation
Introduction
The T cell receptor (TCR) is a complex consisting of six polypeptides (TCRα, β, CD3 γ, δ, ε, ζ) which must be properly assembled in the ER in order to reach cell surface [1, 2]. Subunits not incorporated into the complex are retained in a pre-Golgi compartment and subsequently degraded, providing a useful model for investigation of the mechanisms controlling the degradation of individual proteins in the absence of a properly assembled complex [3, 4]. αTCR is a type I transmembrane protein with a short cytoplasmic tail of 5 amino acids, which is dislocated from the ER, deglycosylated, ubiquitinated and degraded in the cytosol by proteasomes in a process known as ER-associated degradation (ERAD) [5-7] (Fig. 1). Glycoproteins which fail to assemble into mature complexes, such as αTCR, or misfolded glycoproteins, such as the Hong Kong variant of α1-antitrypsin [8], are targeted for degradation through a complex series of quality control reactions, known as the calnexin/calreticulin cycle [9]. Calnexin and calreticulin are lectins capable of recognizing the terminal glucose residue of the associated N-glycan, which can be removed by ER-resident glucosidase II. Deglycosylation allows fully folded and/or assembled proteins to exit the cycle, while folding/assembly intermediates are reglucosylated and rebind calnexin/calreticulin. When glycoproteins remain in the ER for a prolonged time, ER-resident mannosidase I removes one terminal mannose from the N-glycan, preventing their effective reglucosylation. Truncated polymannose oligosaccharides are then recognized by specific lectins which target the associated glycoprotein for ERAD [9, 10].
Fig. 1.
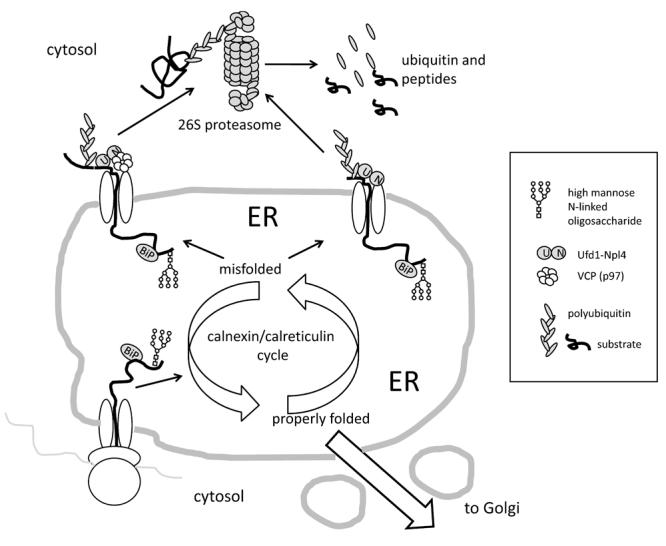
Proteins are co-translationally inserted into the ER, where they associate with chaperones such as BiP and undergo core N-glycosylation. Once in the ER, those proteins enter successive rounds of folding/refolding associated with deglucosylation/glucosylation reactions, known collectively as the calnexin/calreticulin cycle. If properly folded, proteins traffic to the Golgi and beyond, to other compartments of the secretory pathway, the plasma membrane and/or for secretion. If, however, the proteins fail to fold after several attempts, they are demannosylated, recognized by specific lectins and retrotranslocated to the cytosol, where they are ubiquitinated and degraded by the 26S proteasomes, a process known as ER-associated degradation (ERAD). While the classic model of ERAD assumes the need for VCP associated with the Ufd1-Npl4 dimer as the core retrotranslocation motor, many proteins are efficiently degraded without the requirement for VCP. On the other side, VCP controls – either directly or indirectly – multiple aspects of ER structure and function (not shown) and therefore may affect degradation rates of proteins without directly interacting with them. Co-translational insertion of nascent proteins into the ER as well as their retrotranslocation to the cytosol may proceed through the same channel composed of Sec61 or through different channels.
It is not known what specific structural feature or association with what protein triggers αTCR ubiquitination, but it is required not only for its targeting to the 26S proteasomes, but also for its retrotranslocation from the ER [11, 12]. Ubiquitination of αTCR is mediated by at least two different ubiquitin ligases (E3s): cytosolic SCFFbs1,2 which recognizes N-linked oligosaccharides attached to dislocated αTCR chain[13] and ER-membrane associated HRD1 [14]. Degradation of other TCR subunits bears important mechanistic differences from the degradation of αTCR. For example, inhibition of mannosidase does not affect ERAD of αTCR, while it induces retention of δCD3 in the ER. Moreover, when proteasome is inhibited, significant amounts of αTCR are exported to the cytosol, while δCD3 remains in the ER [6]. Studies in yeast have identified Cdc48p, whose mammalian homolog is valosin-containing protein (VCP), as a crucial component of ERAD, involved in retrotranslocation of emerging substrates and their delivery to the proteasome [15-19].
Retrotranslocation of αTCR and other ERAD substrates from the ER is a rate limiting step in their degradation [20] proceeding either through the Sec61 translocone or through alternative channels composed of derlin1 [21-24]. A widely accepted model proposes that ERAD substrates are bound on the cytosolic side by the VCP ATP-ase (a.k.a. Cdc48 in yeast) associated with the Ufd1-Npl4 dimer both prior to and after ubiquitination. Once bound, the ATP-ase activity of VCP supposedly provides the driving force which pulls substrates out of the ER, accompanied by recruitment of ubiquitin ligases, and targeting to the 26S proteasome [10, 16, 19, 25-28]. However, while in yeast most ERAD appears to be Cdc48-mediated, in mammalian cells multiple different ERAD pathways co-exist and at least some of them do not require VCP [10]. According to this, we have shown previously that a ∼90% depletion of VCP does not affect ERAD of δCD3 or the luminal misfolded α1-antitrypsin, while it causes a dramatic buildup of cytosolic VCP-dependent substrates R-GFP and Ub-G76V-GFP [29]. Moreover, under the same conditions there was a discrete accumulation of αTCR, demonstrating that degradation of this subunit of TCR depends on VCP, while the degradation of δCD3 subunit of the TCR does not. However, in contrast to a five-fold increase in the levels of cytosolic VCP-dependent substrates, the increase of αTCR levels was very modest, in the order of 20-30%. Moreover, depletion of either Ufd1 or Npl4 did not induce αTCR accumulation, suggesting that the effect of VCP on αTCR degradation is independent from the intact VCPUfd1-Npl4 complex [30]. Since RNAi of VCP induces profound changes in ER structure and function associated with induction of unfolded protein response (UPR) and specific upregulation of 30 different gene products it is possible that VCP controls αTCR indirectly [29]. In the present paper, we have therefore decided to further explore the possible involvement of VCP in the degradation of αTCR.
Material and Methods
Antibodies, reagents and plasmids
Anti-actin and anti-α1-antitrypsin rabbit polyclonal antisera were from Sigma (St. Louis, MO), anti-HA11 mouse monoclonal antibody was from Covance (Princeton, NJ), anti-polyubiquitin rabbit polyclonal antiserum was from Dako (Carpinteria, CA), anti-TGN46 sheep polyclonal antiserum was from Serotec (Raleigh, NC), anti-VCP mouse monoclonal antibody was from BD Transduction Laboratories (Franklin Lakes, NJ), while anti-BiP polyclonal rabbit antiserum, anti-Hsp70 mouse monoclonal antibody and anti-Grp94 rat monoclonal antibody were from Stressgen (Victoria, Canada). The pcDNA3.1 encoding HA-tagged α-TCR 2B4 [20, 29-31] used to derive the cell line stably expressing αTCR was obtained from Dr. Ron Kopito (Stanford University, CA). The pCMV plasmid encoding the Hong Kong variant of α1-antitrypsin was obtained from Dr. Nobuko Hosokawa (Kyoto University, Kyoto, Japan) [8, 29]. 2,5-Dihydroxybenzoic acid (DHB), sodium hydroxide (NaOH), and methyl iodide were purchased from Aldrich (Milwaukee, WI). MG132 (Calbiochem, La Jolla, CA) was prepared as a 10 mM stock in DMSO and used at a final 10 μM concentration. Brefledin A (BFA, Calbiochem) was prepared as a 5 mM stock in methanol and used at a final 5 μM concentration. Tunicamycin (Calbiochem) was prepared as a 10 mg/ml stock in DMSO and used at a final 10 μg/ml concentration. 1-deoxymannojirimycin (DMJ, Calbiochem) was prepared as a 100 mM stock in DMSO and used at a final 1 mM concentration. Cycloheximide (CHX, Sigma) was prepared as a 10 mg/ml stock in water and used at a final 200 μg/ml concentration. Unless otherwise stated all the remaining reagents were from Sigma (St. Louis, MO).
Cell culture and RNA interference (RNAi)
HeLa cells obtained from American Type Culture Collection (Manassas, VA) were used to derive the stable cell lines as described [29, 30]. Stable lines overexpressing αTCR or α1-antitrypsin were grown in Advanced DMEM supplemented with Gluta-MAX™, antibiotic/antimycotic solution, Geneticin (all from Invitrogen, Carlsbad, CA) and 2% fetal bovine serum (Gemini Bioproducts, Woodland, CA). Small interfering RNAs (siRNAs) were obtained by chemical synthesis from Dharmacon (Lafayette, CO). siRNAs targeting VCP corresponded to positions 811-831 (vcp2) and 480-500 (vcp6) of human VCP sequence (NM_007126) as published previously [29, 30, 32, 33]. siRNA targeting derlin 1 corresponded to positions 345-365 (der1-1) and 440-460 (der1-2) of human derlin 1 sequence (NM_024295), while siRNA targeting derlin 2 corresponded to positions 232-249 of the human derlin 2 sequence (NM_016041). RNAi was performed using X-tremeGENE™ (Roche Applied Science, Penzberg, Germany) transfection reagent according to manufacturer's instructions. Briefly, cells were seeded onto 6-well plates 24 h prior transfection, to reach 70% confluence on the day of transfection. 2.5 μl of the siRNA stock in RNA-se free water was mixed with 3 μl of the transfection reagent in 200 μl of serum-free Advanced DMEM. After a 20 min preincubation this solution was added to 500 μl of serum-free media to a final siRNA concentration of 20 nM. After 4 hrs 4 ml of full culture media was added without removal of the transfection mixture. Cells were collected either 24 or 48 h after the transfection. HeLa cells mock transfected with with a siRNA targeting a non-relevant protein such as EGFP served as control.
RT-PCR
The effectiveness of RNAi in decreasing mRNA levels of the targeted genes was verified by semi quantitative RT-PCR. RNA was isolated from cells collected in Trizol (Invitrogen, Carlsbad, CA) [34]. RT-PCR was performed with the OneStep kit (Qiagen, Valencia, CA) using the following pairs of primers: TCTACGCGACTTGAAACAGGA and CCAACCAGATTTCCAATAAGC to detect derlin 1 (NM_024295), TGTCGAATGCTAGAAGAAGGC and ATTTGGATCCTCATCTGGTGT to detect derlin 2 (NM_016041) and TTCCTTCCTGGGCATGGAGT and ATCCACATCTGCTGGAAGGT to detect actin (NM_00110). DNA electrophoresis was performed on standard 1% agarose gels, DNA was labedled with ethidium bromide and images were acquired using Kodak Image Station 4000MM (Eastman Kodak, Rochester, NY).
SDS-PAGE and Western blotting
Whole cell lysates were obtained in RIPA buffer (150 mM NaCl, 1% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tric-HCl, pH 7.5 @ 25 °C) supplemented with Complete Mini™ protease inhibitors (Roche, Mannheim, Germany) and sodium orthovanadate. All samples were normalized to the same protein concentration determined using Bradford reagent (Biorad, Hercules, CA) [35]. Samples were resolved either by standard SDS-PAGE as previously described [29, 30] or on a modified SDS-PAGE system, where the separating buffer of pH 9.4 was obtained by inverting the proportions of Tris-base and Tris-HCl (i.e. 153.9 g Tris-HCl, 36.9 g Tris-base and 5 g of SDS per 1 l of the 4x solution). Western blotting was performed on Immobilon-P PVDF membrane (Millipore, Billirica, MA). Primary antibodies were detected with appropriate secondary HRP-conjugated antibodies (Jackson Immunoresearch, West Grove, PA). HRP was detected using the Amersham ECL™ Advance kit (GE Healthcare, Piscataway, NJ) and images were acquired using Kodak Image Station 4000MM (Eastman Kodak, Rochester, NY).
Immunoprecipitation and pulse chase experiments
For immunoprecipitation, cells collected from 1 six-well plate 48 hrs after RNAi were lysed in 1 ml lysis buffer (50 mM HEPES-KOH, pH 7.4 @ 4 °C, 100 mM NaCl, 1.5 MgCl2 and 0.1% NP-40) using 10-15 strokes with Dounce homogenizer on ice. After centrifugation (15 min, 16 000g @ 4°C), protein concentration in the supernatants was determined and all lysates were equalized to 1 mg/ml protein concentration, precleared, incubated with 5 μl of the HA11 antibody for 1 hr @ 4°C on a rotary wheel, and then 40 μl of 50% ImmunoPure™ immobilized Protein A/G bead slurry (Pierce, Rockford, IL) was added for overnight incubation @ 4°C on a rotary wheel. Afterwards, beads were washed 5 times in lysis buffer before resuspension and boiling in SDS-PAGE sample buffer. For radioactive labeling, cells before lysis where starved for 1 h in Met-deficient media, labeled for 30 min in Met-deficient media supplemented with 500 μCi/ml of 35S Met (Easytag™ Express Protein Labeling Mix, PerkinElmer, Boston, MA), washed and chased for various times before lysis. Super RX™ X-ray film (Fuji Photo Film, Tokyo, Japan) was exposed to the dried gels. Densitometric analysis of the autoradiograms and Western blots was performed using the Image Quant 5.2 software (Amersham Bioscience, Piscataway, NJ).
Enzymatic digestion of immunoprecipitated αTCR
10 μl of radioactively labeled immunoprecipitate was supplemented with 6 μl of 0.5 M sodium citrate buffer pH 7.5, 20 μl of Milli-Q water, 2 μl of 1% PMSF stock solution in methanol and either left without additives as a control or supplemented with 1 μl of a 0.5 U/ ml solution of recombinant endoglycosidase H isolated from Streptomyces plicatus (Calbiochem) or 1 μl of ammonium sulfate suspension of jack bean mannosidase (Sigma) at 20 U/ml. After an incubation for 16 h @ 37°C the reaction was stopped by addition of 10 μl of 5xSDS PAGE sample buffer. After 5 min of boiling the samples were resolved by SDS PAGE. Super RX™ X-ray film (Fuji) was exposed to the dried gel.
Immunofluorescence microscopy
Cells were grown in Labtek two-chamber slides (Nunc Nalgene, Naperville, IL). After 72 h of RNAi targeting VCP and/or 6 h treatment with 10 μM MG132, treated cells, as well as control cells were fixed in ice cold methanol. After fixation, cells were 3x washed with TBS, pH 7.6, supplemented with 0.1% bovine serum albumin and 0.1% fish gelatin, and incubated with primary antibodies diluted in the same buffer containing Tween-20 for 2 hours. After three 15 minute washes in TBS with 0.1% bovine serum albumin and 0.1% fish gelatin, the cells were incubated with secondary Cy2, Cy5 or TRITC-conjugated anti-rabbit, anti-rat, anti-sheep and/or anti-mouse F(ab')2 fragments (Jackson Immunoresearch, West Grove, PA). After 3 washes in TBS, cells were mounted using Gel/Mount (Biomeda, Foster City, CA). Slides were observed using the 60x Plan Apo objective of a Nikon Eclipse TE2000-U epifluorescence microscope. Images were acquired using the CoolSNAP ES CCD camera operated by the Metamorph 6.3 software (Fryer Company, Cincinnati, OH) and optically deconvoluted with the Autodeblur software (Media Cybernetics, Silver Spring, MD).
Release of N-glycans
72 h following RNAi of VCP with either vcp2 or vcp6 or following a 16 h treatment with 10 μM MG132, treated and control cells were collected, washed in PBS and frozen @ −80°C until further processing. N-glycans were enzymatically released using peptide N-glycanase F (PNGaseF). This enzymatic release was performed according to our previously published procedure [36]. Briefly, samples were suspended in 10 mM sodium phosphate buffer, pH 7.5, containing 0.1% mercaptoethanol. The samples were then thermally denatured by incubation at 95°C for 5 min. Next, the samples were cooled to room temperature prior to the addition of 5 mU of PNGase F. Finally, the reaction mixtures were subsequently incubated for 3 hrs at 37 °C.
Permethylation
Permethylation of released N-glycans was performed according to our recently published procedure [37]. All N-glycans derived from glycoproteins were permethylated using a sodium hydroxide bead column. To protect the packing material from moisture, sodium hydroxide beads were immediately suspended in acetonitrile. The sodium hydroxide beads, suspended in acetonitrile, were then packed in a 1.0-mm i.d. peak tubing, using stainless steel frits, unions, nuts, and ferrules (Upchurch Scientific, Oak Harbor, WA). The packing was accomplished pneumatically, while the columns were conditioned with DMSO prior to use. Samples were prepared in DMSO containing a trace amount of water and methyl iodide or deuteromethyl iodide. These sample solutions were infused through the packed sodium hydroxide column, using a 100-μL Hamilton syringe, and a syringe pump from KD Scientific, Inc. (Holliston, MA). Samples were infused through the sodium hydroxide-packed column at an optimum flow rate of 2 ml/min 12, and collected into microtubes. Permethylated samples were extracted with chloroform and washed several times with water prior to drying under vacuum.
MALDI Spotting
The dried permethylated samples were resuspended in 50:50 methanol/water solution containing 1 mM sodium acetate. Samples were then spotted directly on the MALDI plate and mixed with an equal volume of the DHB matrix. The DHB matrix was prepared by suspending a 10-mg aliquot in 1 ml of water to produce 10mg/ml matrix solution. The resulting spots were dried under vacuum prior to MALDI MS analyses. An Applied Biosystems 4800 Proteomic Analyzer (Applied Biosystems, Framingham, MA) was utilized for this study. This MALDI/TOF/TOF instrument is equipped with an Nd:YAG laser (355-nm wavelength). The MALDI mass spectra were acquired at 1kV accelerating voltage in the positive-ion mode. MS data were further processed using DataExplorer 4.0 (Applied Biosystems).
Results
Overexpressed HA-tagged αTCR is a true ERAD substrate
HA-tagged αTCR is easily detected in a stable cell line [29, 30], where it co-localizes with BiP and Grp94, while it is absent from compartments labeled with the TGN46 antibody, indicating expression and retention of αTCR within the ER (Fig. 2 A). The dominant species of αTCR present under normal growth conditions has a MR of ∼38 kDa (Fig. 2 B, arrowhead). Upon treatment with different proteasome inhibitors, such as 10 μM MG132 (Fig. 2 B), epoxomycin or bortezomib (not shown), there is a gradual decrease of the 38 kDa form accompanied by accumulation of a species of ∼29 kDa (Fig. 2 B, arrow), likely corresponding to deglycosylated αTCR, as well as some high molecular weight smear, likely corresponding to polyubiquitinated αTCR. Upon proteasome inhibition HA-immunoreactivity is present not only within the ER but also in cytosolic, well defined, ubiquitin-positive aggresomes (Fig. 3 A, arrowheads). Treatment of cells overexpressing αTCR with two different agents causing ER stress, 10 μg/ml tunicamycin and 5 μM BFA, induced a rapid disappearance of αTCR below detection levels, which was mediated by ERAD, as indicated by accumulation of αTCR when those drugs were combined with the proteasome inhibitor MG132 (Fig. 2 B). In particular, tunicamycin, which inhibits N-linked glycosylation, induced the accumulation of the 29 kDa αTCR form. Combination of BFA with MG132 produced multiple bands, many of them of a size greater than ∼38 kDa. RNAi of VCP induces the formation of dispersed aggregates of polyubiquitinated proteins [29], however they are not enriched in αTCR, which is distributed like in control, untreated cells. However, when cells submitted to RNAi of VCP are co-treated with MG132, αTCR localizes to dispersed polyubiquitin aggregates. We have immunoprecipitated αTCR using the anti-epitope tag HA11 antibody and blotted the immunoprecipitates with an anti-KDEL antibody detecting two ER chaperones, BiP and Grp94 (Fig. 3B). While they are undetectable in control immunoprecipitates, they are associated with αTCR in cells treated with MG132 and in cells submitted to RNAi of VCP. However, only cells submitted to MG132 treatment show an association of αTCR with the cytoplasmic chaperone Hsp70. Under none of those conditions we were able to detect associated VCP immunoreactivity (not shown).
Fig. 2.
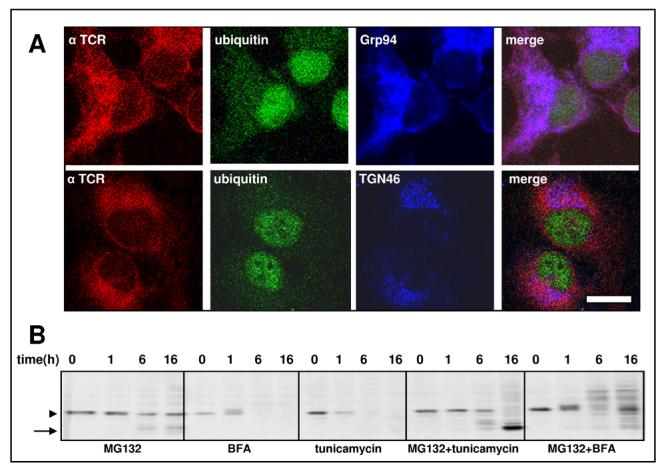
HeLa cells have been stably transfected with HA-tagged α-TCR. (A) Immunofluorescent labeling with HA11 antibody reveals that α-TCR is expressed mostly in the ER as seen by the colocalization with the ER marker Grp94 (upper row), while it does not colocalize with the Golgi marker TGN46 (lower row).Ubiquitin labels nuclei and cytosol, but not the ER, therefore it does not colocalize with α-TCR. The bar indicates 5 μm. (B) Western blotting showing the effects of a time course of different drugs on cellular levels of α-TCR; While MG132 induces an accumulation of αTCR, treatment with either tunicamycin or brefeldin A induces depletion of αTCR from cells, which is mediated by a proteasome-dependent process as evidenced by the effect of MG132.
Fig. 3.
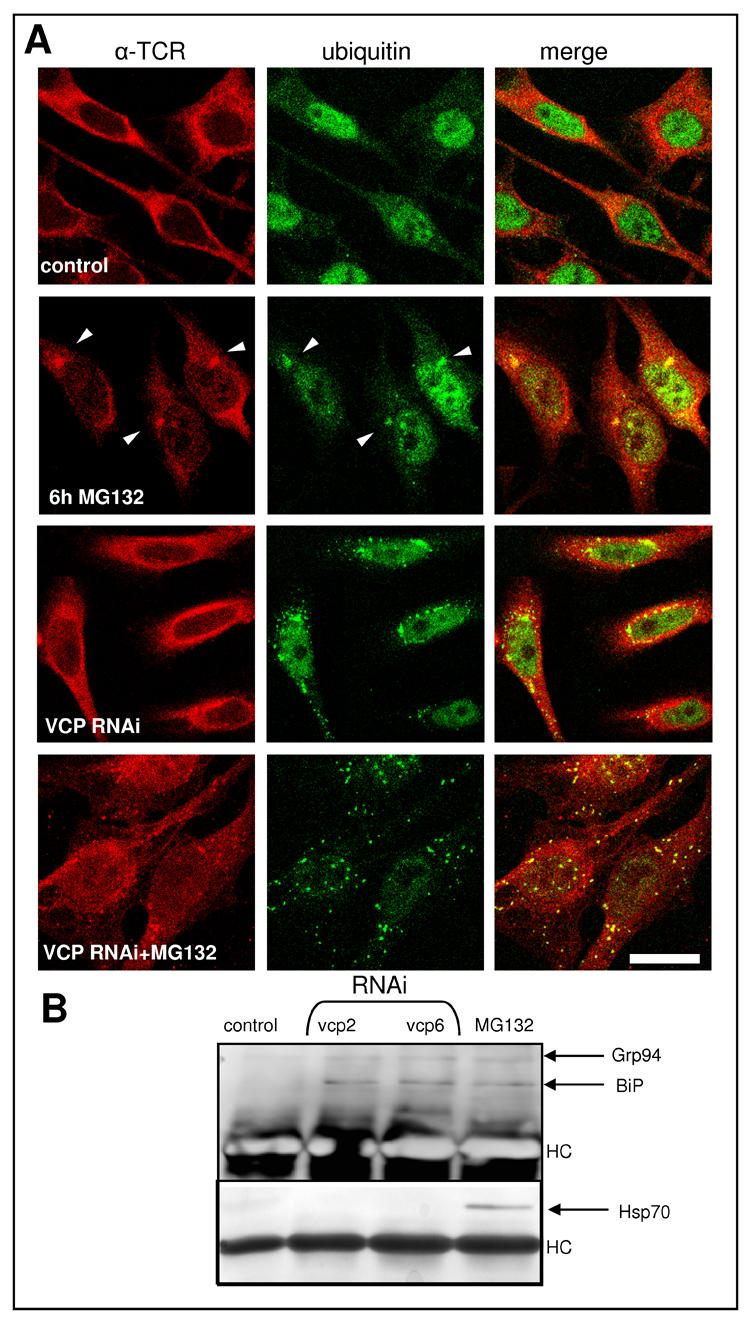
Intracellular distribution of αTCR following inhibition of its degradation. (A) While in control cells αTCR is dispersed in the ER, following a 6h treatment with 10 μM MG132 most αTCR forms single ubiquitin-positive aggresomes. RNAi of VCP induces the formation of dispersed ubiquitin-positive aggregates, which however contain little αTCR, most of which remains dispersed in the cell. However, treatment of cells submitted to RNAi of VCP with MG132 induces a redistribution of most αTCR into dispersed aggregates of polyubiquitinated proteins. The bar indicates 5 μm. (B) Immunoprecipitates obtained using the HA11 antibody from αTCR-expressing cells submitted to RNAi of VCP using the two different sequences (vcp2 and vcp6) have been blotted with antibodies detecting the cytosolic hsp70 or the ER chaperone BiP. While after RNAi of VCP αTCR interacts with BiP, after proteasome inhibition BiP interacts with both hsp70 and BiP.
Derlin 1 is dispensable for retrotranslocation of αTCR
To further investigate the factors controlling retrotranslocation of αTCR we have knocked down derlin 1 using two different siRNAs (der1-1 and der1-2, Fig. 4A) targeting this gene product. To our surprise, we have not observed an accumulation of αTCR. Moreover, it appeared that the levels of αTCR slightly decreased in cells submitted to the knockdown of derlin 1. The effectiveness of the knockdown was measured by semiquantitative RT-PCR (Fig. 4B). To rule out any non-specific effects of RNAi, we performed RNAi of derlin 2, a closely related protein from the derlin family, which did not change αTCR levels despite specific depletion of the derlin 2 mRNA. In order to prove that depletion of derlin 1 by RNAi was sufficient to induce accumulation of an ERAD substrate we have tested the effects of RNAi of derlin 1 in a cell line stably expressing the misfolded Hong Kong variant of the lumenal protein α1-antitrypsin [29, 30]. Indeed, knockdown of derlin 1 induced a marked accumulation of glycosylated α1-antitrypsin (Fig. 4A, arrowhead), while proteasome inhibition with MG132 induced accumulation of a deglycosylated form of this protein (Fig. 4A, ⁎).
Fig. 4.

Depletion of derlin 1 induces accumulation of α1-antitrypsin but not of αTCR. (A) RNAi of derlin 1 with two different siRNAs (der1-1 and der1-2) induces a decrease of the levels of glycosylated αTCR (arrow) while at the same time induces accumulation of glycosylated α1-antitrypsin (arrowhead). RNAi of derlin 2 does not affect the levels of neither αTCR nor α1-antitrypsin, while treatment with MG132 induces accumulation of both substrates, mostly in the deglycosylated form (stars). Equal protein loading is shown by the actin blot. (B) The effectiveness of the knockdown of derlins is demonstrated by semiquantitative RT-PCR, with actin as a control of equal RNA loading.
Stabilization of αTCR by RNAi of VCP
In order to dissect the role played by VCP in αTCR turnover we have treated cells depleted of VCP with drugs affecting different aspects of ER structure and function (Fig. 5A). Tunicamycin induced in control cells an almost complete disappearance of glycosylated αTCR (arrow) – the remaining signal corresponded to a glycanless form of lower molecular mass (★). When combined with RNAi of VCP, tunicamycin failed to completely deplete the glycosylated form of αTCR, while it induced an accumulation of the deglycosylated form. In accordance with that, proteasome inhibition with MG132 in cells subjected to RNAi of VCP induced an accumulation of both glycosylated and deglycosylated forms of αTCR, while in control cells MG132 induced an accumulation of the deglycosylated form alone. Surprisingly, RNAi of VCP did not prevent depletion of αTCR induced by BFA treatment. We further confirmed the role of VCP in the degradation of αTCR by chasing the levels of αTCR in the presence of cycloheximide (CHX) (Fig. 5B,C). As calculated from densitometric measurements, RNAi of VCP extended the t½ of αTCR from ∼50 min to ∼200 min. We have also used the radioactive pulse-chase method (Fig. 4D, E) which allowed as to calculate a t½ of control cells to be of ∼60 min and t½ of cells submitted to RNAi of VCP to be of ∼270 min.
Fig. 5.
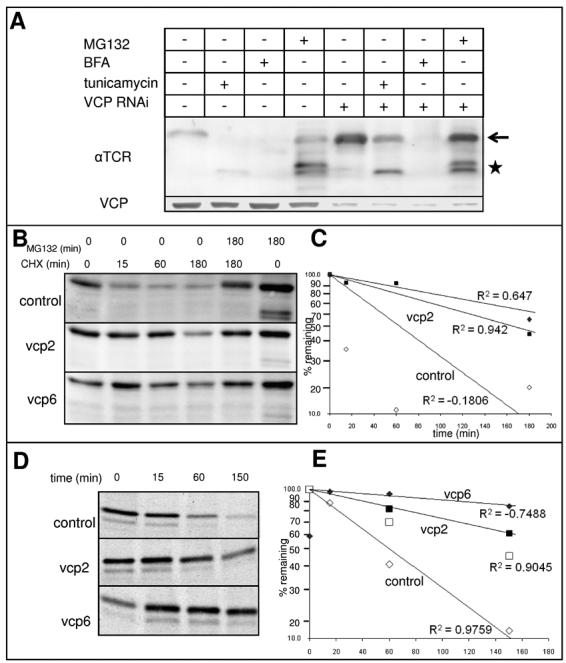
Stabilization of αTCR in HeLa cells following RNAi of VCP. (A) HeLa cells stably overexpressing αTCR were subject to RNAi of VCP or mock transfected; 66 h after transfection, cells were either left untreated or treated with 10 μM MG132, 5 μM brefledin A or 10 μg/ml tunicamycin for additional 6 h before harvesting. Cell lysates were resolved by SDS-PAGE and blotted with the VCP antibody to assess the effectiveness of RNAi or with the HA11 antibody to assess the levels of αTCR. RNAi of VCP induces a mild accumulation of glycosylated αTCR, which is enhanced by tunicamycin and MG132 treatment, but not by BFA. (B) Treatment of cells overexpressing αTCR with cycloheximide depletes αTCR levels in a proteasome dependent process. RNAi of VCP with either vcp2 or vcp6 stabilizes αTCR in the presence of cycloheximide. (C) Determination of the rates of αTCR degradation using quantification of the glycosylated αTCR band. The t ½ of αTCR in control cells is 51 min, while after RNAi of VCP 136 min and 239 min depending on whether vcp2 or vcp6 was used for the knockdown. (D) HeLa cells stably overexpressing αTCR were subject to RNAi of VCP with two different siRNAs (vcp2 and vcp6) or left untreated (control) before labeling with 35S-Met followed by a chase as described in Materials and Methods. Cells were collected at 0, 15, 60 and 150 min after the pulse, lysed, and immunoprecipitated with the HA11 antibody. Immunoprecipitates were resolved on SDS-PAGE and exposed to an autoradiography film (E) Determination of the rates of αTCR degradation using quantification of the glycosylated αTCR band. The t ½ of αTCR was of ∼58 min in control cells, ∼150 min in cells transfected with vcp2 and ∼400 min in cells transfected with vcp6.
RNAi of VCP induces changes in the glycosylation pattern of αTCR
When lysates obtained from cells expressing αTCR were separated on a modified 15% SDS-PAGE (pH 9.4) the band corresponding to the glycosylated species of αTCR (Fig. 6A, arrowhead) separated into two different bands (arrows). Either proteasome inhibition or knockdown of VCP induced preferentially an increase of intensity of the lower band, as quantified by an increase of the ratio of the lower to the upper band (Fig. 6B). Treatment with 1-deoxymannojirymycin (DMJ) induced a complete disappearance of the lower band, suggesting that it is generated by α-mannosidase I, an ER-resident enzyme inhibited by this mannose analog (Fig. 6C). Digestion of immunoprecipitated αTCR with jack bean mannosidase collapses the upper band of the doublet into the lower band (Fig. 6D, arrows), while endoglycosidase H cleaves the core chitiobiose only in the αTCR form detected as the upper band, without affecting the lower band of the doublet. Global changes in glycosylation pattern were detected by a mass spectrometric analysis of cellular N-linked glycans following RNAi of VCP using two different siRNAs, vcp2 and vcp6 (Fig. 6E). Since depletion of VCP induced an accumulation of αTCR, we have used as a positive control the proteasome inhibitor MG132, which inhibits ERAD of αTCR and other ER proteins. While proteasome inhibition did not affect the pattern of cellular N-glycans, RNAi of VCP induced a series of consistent changes, which were observed independently of the siRNA sequence used. In particular, RNAi of VCP increased levels of glycans with core Man3GlcNAc2 (MW=1171.7), associated with a decrease in Man6GlcNAc2 (MW=1784.0), Man7GlcNAc2 (MW=1988.2), Man8GlcNAc2 (MW=2192.3) and Man9GlcNAc2 (MW=2396.4). The decrease in polymannosylated species associated with an increase in oligomannosylated species follows the observed increase in the ratio of the lower (endo H - resistant) to the upper (endo H - sensitive) band of glycosylated αTCR (Fig. 6A, B). In addition to that, RNAi of VCP also induced changes in the abundance of the more complex N-linked glycans. In particular, we have observed a decrease in monosialylated species, regardless of whether they were fucosylated (MW=2605.8) or not (MW=2432.4) as well as an increase of the complex glycan Gal3GlcNAc3Man3GlcNAc2 (MW=2519.4) synthesized in the trans-Golgi stacks.
Fig. 6.
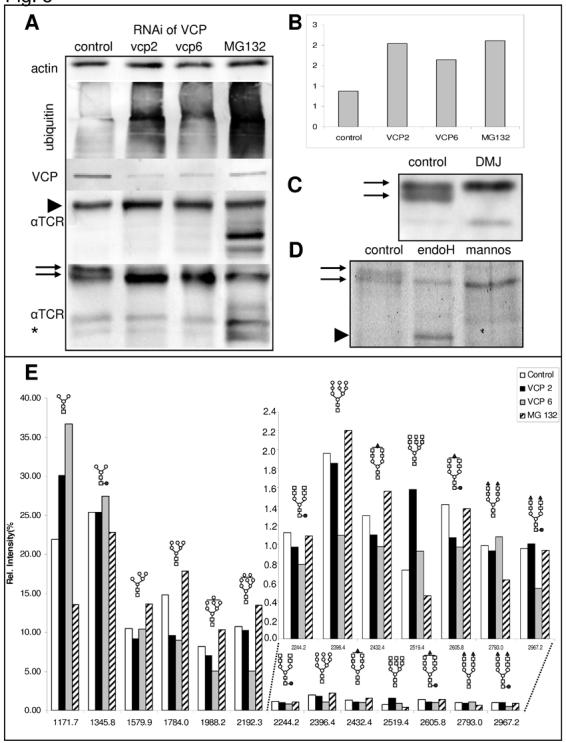
RNAi of VCP affects the glycosylation pattern of multiple glycoproteins, including αTCR. (A) RNAi of VCP using two different siRNAs (vcp2 and vcp6) induces a 20-30% increase of the levels of glycosylated αTCR (arrowhead, A); when the samples are run on a modified 15% SDS-PAGE (pH 9.4) the corresponding band migrates as a doublet and a shift of the ratio between the two adjacent forms of glycosylated αTCR (arrows, A) is apparent as quantified by an increase in the ratio of the lower to the upper band (B). (C) Treatment with 1-deoxymannojirymycin induces the disappearance of the lower band suggesting that it reflects a form of αTCR resulting from the cleavage of a mannose residue. (D) This is confirmed by digestion of the immunoprecipitated αTCR with jack bean mannosidase, which collapses the upper band into a faster migrating form (arrowhead) while the lower band of the doublet is resistant to endoglycosidase H treatment.(E) MALDI of N-linked glycans isolated from cellular glycoproteins reveals that RNAi of VCP with either vcp2 or vcp6 induces a decrease in polymannosylated oligosaccharides and an increase in oligomannosylated oligosaccharides as well as changes in the levels of complex glycans. Symbols used to denote different sugar moieties: open circle – mannose, yellow square – N-acetylglucosamine: red circle – fucose; red triangle – sialic acid; open square – galactose.
Discussion
Retention in the ER and subsequent breakdown of αTCR in the absence of other TCR subunits is one of the best studied examples of ERAD [5-7, 12, 20, 38-45]. ERAD is generally regarded as a UPS-dependent process requiring VCPUfd1-Npl4 for retrotranslocation of substrates from the ER into the cytosol [19, 25, 26, 46]. However, such view is an oversimplification, since multiple evidence exists for UPS-independent ERAD, as well as of UPS-dependent ERAD which does not require VCP [10]. The αTCR transmembrane region contains two charged residues implicated in the rapid turnover of this glycoprotein [38, 47, 48]. While Sec61 channel has been implicated in the retrotranslocation of MHC class I heavy chains [22], involvement of Sec61 in dislocation of αTCR could not be demonstrated using a similar approach [5]. Recently, it was proposed that protein retrotranslocation can proceed through an alternative channel formed by derlin 1, a new VCP-interacting protein [23, 24]. Depletion of derlin 1 by RNAi should therefore induce an accumulation of a protein, for whose dislocation derlin 1 is required. Indeed, upon derlin 1 depletion we have observed an accumulation of a lumenal misfolded glycoprotein α1-antitrypsin Hong Kong variant [8]. Moreover, α1-antitrypsin accumulated upon derlin 1 depletion in a glycosylated form, while upon proteasome inhibition it accumulated in a deglycosylated form, indicating that derlin 1 is specifically involved in a step before it is accessible to the cytosolic peptide-N-glycanase. However, the same approach failed to show an accumulation of αTCR. Moreover, we have shown previously that in contrast to the effects on αTCR, depletion of VCP does not affect levels of α1-antitrypsin [29]. Those results support the view that multiple parallel and/or redundant pathways of ERAD co-exist in mammalian cells. Moreover, a requirement for derlin 1 does not imply a requirement for VCP and vice versa.
We have previously reported that disruption of the VCPUfd1-Np14 complex by RNAi of either Ufd1 or Npl4 not only does not stabilize αTCR, but accelerates its degradation [30]. On the other hand, RNAi of VCP did not affect the levels of two different ERAD substrates, δCD3 and α1-antitrypsin, despite the induction of ER stress evidenced by XBP1 splicing and induction of multiple ER stress genes [29]. At the same time RNAi of VCP induced accumulation of polyubiquitinated proteins [32] and a five-fold increase in the levels of two different cytosolic substrates, R-GFP and UbG76VGFP [29]. The observed modest 20-30% increase in the levels of αTCR following RNAi of VCP was therefore surprising when compared with the five fold increase in the levels of R-GFP and UbG76VGFP. We now report that despite only a 20-30% increase in αTCR levels, there is a four-fold increase in the half-life of αTCR following RNAi of VCP. The discrepancy between a modest increase in αTCR levels and a marked increase in its half-life can be explained by diminished protein synthesis occurring in the setting of continuous ER stress. Diminished synthesis likely counteracts the effects of diminished ERAD, therefore preventing an excessive accumulation of αTCR. In contrast, such homeostatic mechanisms are absent upon inhibition of the degradation of selected cytosolic proteins such as R-GFP and UbG76VGFP. While we have not detected changes in phosphorylation of eIF2α [29], selective degradation of ER-associated mRNAs and co-translational degradation of newly synthesized ER proteins provide additional mechanisms which may account for decreased synthesis of αTCR, which we are currently exploring [49, 50]. The stabilization of αTCR appears to confirm the role of VCP in retrotranslocation of this substrate [19, 25]. Indeed, we have observed that after RNAi of VCP the glycosylated form of αTCR persists 6 h in the presence of tunicamycin, while in control cells it is rapidly degraded, and only the deglycosylated form of αTCR is detected following 6 h of tunicamycin treatment. However, at the same time depletion of VCP did not prevent the rapid, UPS-dependent clearance of αTCR from cells treated with BFA, a drug which disassembles the Golgi mixing its components with the ER. Indeed, when BFA is combined with proteasome inhibitors, αTCR accumulates in form of multiple species of decreased motility on SDS-PAGE, which likely correspond to complex polysaccharides assembled by Golgi enzymes on the core N-glycan structure. Such glycans aberrantly present within the ER are rapidly cleared through UPS-dependent ERAD and are therefore not detected in the absence of proteasome inhibitors. Surprisingly, those adducts are not stabilized in the absence of VCP in contrast to the core-glycosylated αTCR, suggesting mechanistic differences in the ERAD machinery dependent on the glycan moieties attached.
While upon proteasome inhibition most αTCR accumulated in a deglycosylated, cytoplasmic species, αTCR upon depletion of VCP accumulated as a glycosylated form. However, both upon proteasome inhibition and VCP depletion the glycosylated species of αTCR differed from the glycosylated species present in control cells, since it had an increased proportion of a faster migrating form. The fact that the faster migrating form is absent after DMJ treatment suggests that it is formed through the action of α-mannosidase I, an ER-resident enzyme which catalyzes the removal of mannose residues from Man9GlcNAc2 prior to the transfer of N-glycosylated proteins from ER to cis-Golgi compartments [51]. Moreover, the faster migrating band can be formed from the slower migrating band by the action of jack bean mannosidase, an enzyme usually producing a mix of truncated forms of high-mannose oligosaccharides. Finally, only the slower migrating form is sensitive to endoglycosidase H which cleaves within the core chitiobiose producing the fastest migrating form, where only a single GlcNAc residue remains attached to αTCR. Since endoglycosidase H does not cleave truncated forms of high-mannose oligosaccharides, the faster migrating form of the αTCR doublet is resistant to its action. Truncated high-mannose oligosaccharides are intermediates targeted for ERAD. They are produced in the ER after newly synthesized glycoproteins such as αTCR fail to fold properly within several iterations of the calnexin/calreticulin cycle [9]. Therefore, an increase in the level of αTCR with a truncated high-mannose oligosaccharide moiety is expected in a situation, where retrotranslocation of αTCR is blocked. However, it has been recently reported that ER-resident mannosidase I is regulated by proteolysis mediated by the lysosomal compartment [52]. Since VCP is required for homotypic vesicle fusion in the ER and Golgi [53, 54], VCP depletion likely diminishes the traffic of ER proteins to lysosomes providing an alternative explanation to increased levels of demannosylation of N-glycans in the absence of any direct effect of VCP on ERAD.
Analysis of global patterns of N-glycosylation has shown a generalized increase in the levels of truncated high-mannose glycans associated with a decrease in the levels of full length high-mannose glycans, indicating that upon depletion of VCP multiple glycoproteins behave similar to αTCR. However, while MG132 also induced changes in the pattern of αTCR glycosylation similar to those of RNAi of VCP, it has not affected the global patterns of glycosylation, indicating that such changes are not just a plain consequence of ERAD inhibition. Moreover, RNAi of VCP also induced changes in the levels of different complex glycans synthesized in the post-ER compartments. Those changes may either reflect the different abundance of core-glycosylated precursors trafficked from the ER or – alternatively – they may indicate that VCP controls different aspects of glycosylation, for example the regulated degradation of glycosylation enzymes within a lysosomal compartment [52]. The latter possibility is supported by the fact that VCP is the major ATP-ase associated with transitional ER and the Golgi involved in homotypic membrane fusion within that compartment [53, 54]. Depletion of VCP by RNAi may therefore affect the distribution of different glycosylation enzymes within cis, medial and trans cisternae of the Golgi apparatus.
In summary, we present evidence of a novel cellular function of VCP in mammalian cells, the control of N-linked glycosylation at the level of the ER and post-ER compartments. At this point it is difficult to discern, how much of this effect can be attributed to the inhibition of ERAD versus to an inhibition of membrane fusion within the Golgi. It is likely that both effects may be involved. Upon depletion of VCP by means of RNAi the α chain of TCR is retained within the ER as evidenced by the extension of its half-life and trimming of N-linked high-mannose oligosaccharides, in contrast to α1-antitrypsin and δCD3 whose levels do not change upon VCP depletion [29]. However, significant amounts of αTCR are still retrotranslocated to the cytosol, as indicated by the formation of ubiquitin-positive αTCR aggregates upon combination of RNAi of VCP with proteasome inhibition. While it was shown before that retrotranslocation of αTCR does not proceed through the Sec61 translocon [5], our results suggest that it does not proceed through a the VCP-associated derlin 1 channel as well. Moreover, while in cells submitted to RNAi of VCP dislocation of αTCR is delayed in the presence of tunicamycin, BFA-induced retrotranslocation is not affected by depletion of VCP. This finding indicates that αTCR with complex oligosaccharide modifications may be degraded through an alternative, VCP-independent pathway. Finally, our data do not provide a clear answer whether the retention of αTCR within the ER is a direct effect of VCP depletion or whether it reflects an indirect effect of VCP on ER structure and function caused by formation of ER-derived vacuoles, induction of UPR and changes in the pattern of oligosaccharide modifications [29]. Further studies are required to fully understand the role played by VCP in ERAD of αTCR and other substrates. Unfortunately, knockout of VCP is incompatible with life of mammalian cells [55], therefore unless a pharmacologic inhibitor of VCP becomes available, the in vivo study of VCP is limited to partial depletion via RNAi or overexpression of dominant negative mutants of VCP.
Acknowledgements
*This work was supported by the Biomedical Research Grant from Indiana University School of Medicine 22-812-57 (CW), by the American Cancer Society grant IRG-84-002-22 (CW), and by the NIH/NCRR grant RR018942 as the National Center for Glycomics and Glycoproteomics (MVN and YM). We are highly appreciative of a fellowship from Merck Research Laboratories to one of us (PK). DN was on leave from the Department of Immunology, Center of Biostructure Research, Medical University of Warsaw, Poland. We acknowledge the generous gifts of: pCDNA3.1-HA-α-TCR from Dr. Ron Kopito (Stanford University) and of pCMV-α1-antitrypsin Hong Kong from Dr. Nobuko Hosokawa (Kyoto University, Kyoto, Japan).
Abbreviations
- BFA
brefeldin A
- CHX
cycloheximide
- DMJ
1-deoxymannojirimycin
- MALDI
matrix-assisted laser desorption/ionization
- PNGase
peptide N-glycanase
- UFD
ubiquitin-fusion degradation
- UPS
ubiquitin-proteasome system
- UPR
unfolded protein response
- VCP
valosin-containing protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Call ME, Wucherpfennig KW. Annu. Rev. Immunol. 2005;23:101–125. doi: 10.1146/annurev.immunol.23.021704.115625. [DOI] [PubMed] [Google Scholar]
- 2.Klausner RD, Lippincott-Schwartz J, Bonifacino JS. Annu. Rev. Cell Biol. 1990;6:403–431. doi: 10.1146/annurev.cb.06.110190.002155. [DOI] [PubMed] [Google Scholar]
- 3.Hall C, Berkhout B, Alarcon B, Sancho J, Wileman T, Terhorst C. Int. Immunol. 1991;3:359–368. doi: 10.1093/intimm/3.4.359. [DOI] [PubMed] [Google Scholar]
- 4.Lippincott-Schwartz J, Bonifacino JS, Yuan LC, Klausner RD. Cell. 1988;54:209–220. doi: 10.1016/0092-8674(88)90553-3. [DOI] [PubMed] [Google Scholar]
- 5.Huppa JB, Ploegh HL. Immunity. 1997;7:113–122. doi: 10.1016/s1074-7613(00)80514-2. [DOI] [PubMed] [Google Scholar]
- 6.Yang M, Omura S, Bonifacino JS, Weissman AM. J. Exp. Med. 1998;187:835–846. doi: 10.1084/jem.187.6.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu H, Kaung G, Kobayashi S, Kopito RR. J. Biol. Chem. 1997;272:20800–20804. doi: 10.1074/jbc.272.33.20800. [DOI] [PubMed] [Google Scholar]
- 8.Hosokawa N, Tremblay LO, You Z, Herscovics A, Wada I, Nagata K. J. Biol. Chem. 2003;278:26287–26294. doi: 10.1074/jbc.M303395200. [DOI] [PubMed] [Google Scholar]
- 9.Ellgaard L, Helenius A. Nat. Rev. Mol. Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 10.Romisch K. Annu. Rev. Cell Dev. Biol. 2005 doi: 10.1146/annurev.cellbio.21.012704.133250. [DOI] [PubMed] [Google Scholar]
- 11.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. EMBO J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tiwari S, Weissman AM. J. Biol. Chem. 2001;276:16193–16200. doi: 10.1074/jbc.M007640200. [DOI] [PubMed] [Google Scholar]
- 13.Yoshida Y, Tokunaga F, Chiba T, Iwai K, Tanaka K, Tai T. J. Biol. Chem. 2003;278:43877–43884. doi: 10.1074/jbc.M304157200. [DOI] [PubMed] [Google Scholar]
- 14.Kikkert M, Doolman R, Dai M, Avner R, Hassink G, van VS, Thanedar S, Roitelman J, Chau V, Wiertz E. J. Biol. Chem. 2004;279:3525–3534. doi: 10.1074/jbc.M307453200. [DOI] [PubMed] [Google Scholar]
- 15.Ye Y, Meyer HH, Rapoport TA. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- 16.Ye Y, Meyer HH, Rapoport TA. J. Cell Biol. 2003;162:71–84. doi: 10.1083/jcb.200302169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, Sommer T. Nat. Cell Biol. 2002;4:134–139. doi: 10.1038/ncb746. [DOI] [PubMed] [Google Scholar]
- 18.Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S. Mol. Cell Biol. 2002;22:626–634. doi: 10.1128/MCB.22.2.626-634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bar-Nun S. Curr. Top. Microbiol. Immunol. 2005;300:95–125. doi: 10.1007/3-540-28007-3_5. [DOI] [PubMed] [Google Scholar]
- 20.Yu H, Kopito RR. J. Biol. Chem. 1999;274:36852–36858. doi: 10.1074/jbc.274.52.36852. [DOI] [PubMed] [Google Scholar]
- 21.Zhou M, Schekman R. Mol. Cell. 1999;4:925–934. doi: 10.1016/s1097-2765(00)80222-1. [DOI] [PubMed] [Google Scholar]
- 22.Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL. Nature. 1996;384:432–438. doi: 10.1038/384432a0. [DOI] [PubMed] [Google Scholar]
- 23.Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- 24.Lilley BN, Ploegh HL. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- 25.Tsai B, Ye Y, Rapoport TA. Nat. Rev. Mol. Cell Biol. 2002;3:246–255. doi: 10.1038/nrm780. [DOI] [PubMed] [Google Scholar]
- 26.Bays NW, Hampton RY. Curr. Biol. 2002;12:R366–R371. doi: 10.1016/s0960-9822(02)00862-x. [DOI] [PubMed] [Google Scholar]
- 27.Ahner A, Brodsky JL. Trends Cell Biol. 2004;14:474–478. doi: 10.1016/j.tcb.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 28.Kostova Z, Wolf DH. EMBO J. 2003;22:2309–2317. doi: 10.1093/emboj/cdg227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wojcik C, Rowicka M, Kudlicki A, Nowis D, McConnell E, Kujawa M, DeMartino GN. Mol. Biol. Cell. 2006;17:4606–4618. doi: 10.1091/mbc.E06-05-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nowis D, McConnell E, Wojcik C. Exp. Cell Res. 2006;312:2921–2932. doi: 10.1016/j.yexcr.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Saito T, Weiss A, Miller J, Norcross MA, Germain RN. Nature. 1987;325:125–130. doi: 10.1038/325125a0. [DOI] [PubMed] [Google Scholar]
- 32.Wojcik C, Yano M, DeMartino GN. J. Cell Sci. 2004;117:281–292. doi: 10.1242/jcs.00841. [DOI] [PubMed] [Google Scholar]
- 33.Wojcik C, Fabunmi R, DeMartino GN. Hypertension. In: Fennell JP, Baker AH, editors. Methods and Protocols. Humana Press; Totowa: 2004. pp. 381–394. [Google Scholar]
- 34.Chomczynski P, Sacchi N. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 35.Bradford MM. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 36.Mechref Y, Novotny MV. Anal. Chem. 1998;70:455–463. doi: 10.1021/ac970947s. [DOI] [PubMed] [Google Scholar]
- 37.Kang P, Mechref Y, Klouckova I, Novotny MV. Rapid Commun. Mass Spectrom. 2005;19:3421–3428. doi: 10.1002/rcm.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonifacino JS, Suzuki CK, Klausner RD. Science. 1990;247:79–82. doi: 10.1126/science.2294595. [DOI] [PubMed] [Google Scholar]
- 39.Bonifacino JS, Suzuki CK, Lippincott-Schwartz J, Weissman AM, Klausner RD. J. Cell Biol. 1989;109:73–83. doi: 10.1083/jcb.109.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wileman T, Kane LP, Young J, Carson GR, Terhorst C. J. Cell Biol. 1993;122:67–78. doi: 10.1083/jcb.122.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wileman T, Carson GR, Concino M, Ahmed A, Terhorst C. J. Cell Biol. 1990;110:973–986. doi: 10.1083/jcb.110.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang S, Ferrone M, Yang C, Jensen JP, Tiwari S, Weissman AM. Proc. Natl. Acad. Sci. U. S. A. 2001;98:14422–14427. doi: 10.1073/pnas.251401598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 44.Fayadat L, Kopito RR. Mol. Biol. Cell. 2003;14:1268–1278. doi: 10.1091/mbc.E02-06-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lenk U, Yu H, Walter J, Gelman MS, Hartmann E, Kopito RR, Sommer T. J. Cell Sci. 2002;115:3007–3014. doi: 10.1242/jcs.115.14.3007. [DOI] [PubMed] [Google Scholar]
- 46.Meusser B, Hirsch C, Jarosch E, Sommer T. Nat. Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- 47.Bonifacino JS, Cosson P, Klausner RD. Cell. 1990;63:503–513. doi: 10.1016/0092-8674(90)90447-m. [DOI] [PubMed] [Google Scholar]
- 48.Bonifacino JS, Cosson P, Shah N, Klausner RD. EMBO J. 1991;10:2783–2793. doi: 10.1002/j.1460-2075.1991.tb07827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hollien J, Weissman JS. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 50.Oyadomari S, Yun C, Fisher EA, Kreglinger N, Kreibich G, Oyadomari M, Harding HP, Goodman AG, Harant H, Garrison JL, Taunton J, Katze MG, Ron D. Cell. 2006;126:727–739. doi: 10.1016/j.cell.2006.06.051. [DOI] [PubMed] [Google Scholar]
- 51.Gonzalez DS, Karaveg K, Vandersall-Nairn AS, Lal A, Moremen KW. J. Biol. Chem. 1999;274:21375–21386. doi: 10.1074/jbc.274.30.21375. [DOI] [PubMed] [Google Scholar]
- 52.Wu Y, Termine DJ, Swulius MT, Moremen KW, Sifers RN. J. Biol. Chem. 2006 doi: 10.1074/jbc.M607156200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Latterich M, Frohlich KU, Schekman R. Cell. 1995;82:885–893. doi: 10.1016/0092-8674(95)90268-6. [DOI] [PubMed] [Google Scholar]
- 54.Rabouille C, Kondo H, Newman R, Hui N, Freemont P, Warren G. Cell. 1998;92:603–610. doi: 10.1016/s0092-8674(00)81128-9. [DOI] [PubMed] [Google Scholar]
- 55.Muller JM, Deinhardt K, Rosewell I, Warren G, Shima DT. Biochem. Biophys. Res Commun. 2007 doi: 10.1016/j.bbrc.2006.12.206. [DOI] [PubMed] [Google Scholar]