

Abstract
Inflammation is a known precipitator of neuronal death after cerebral ischemia. The mechanisms that promote or curtail the start and spread of inflammation in brain are still being debated. By virtue of their capability to modulate gene expression, several transcription factors induced in the ischemic brain can modulate the post-ischemic inflammation. While the induction of transcription factors such as IRF1, NF-κB, ATF-2, STAT3, Egr1 and C/EBPβ is thought to promote post-ischemic inflammation, activation of transcription factors such as HIF-1, CREB, c-fos, PPARα, PPARγ and p53 is thought to prevent post-ischemic inflammation and neuronal damage. Of these, PPARγ which is a ligand-activated transcription factor was recently shown to prevent inflammatory gene expression in several animal models CNS disorders. This review article discusses some of the molecular mechanisms of PPARγ induction by its agonists following focal cerebral ischemia.
Keywords: transcription factors, PPAR gamma, cerebral ischemia, inflammation
1. Introduction
Stroke (focal cerebral ischemia) is the 3rd leading cause of death in the United States, and continues to be the leading cause of long-term disability, with very few options for effective treatment paradigms. The thrombolytic compound tissue plasminogen activator (t-PA) is the only Food and Drug Administration-approved agent for stroke therapy in humans. However, t-PA is known to have a relatively short therapeutic window and is not suitable for many stroke patients (Maestre-Moreno et al., 2005; Juttler et al., 2006). Recently, the thiazolidinedione (TZD) class of non-steroidal anti-inflammatory compounds such as rosiglitazone or pioglitazone (currently being used to treat type-2 diabetes) have shown efficacy in ameliorating the negative outcome following cerebral ischemia. These drugs act by binding and activating a family of transcription factors called Peroxisome Proliferator-Activated Receptors (PPARs), and have been hypothesized to curtail inflammatory processes subsequent to cerebral ischemia. In this review, we have discussed the functional significance of transcription factors in mediating cerebral inflammatory cascades with special emphasis on the beneficial effects of PPAR agonists in animal models of cerebral ischemia.
2. Putative contributors of ischemic brain damage
In experimental animals as well as humans, focal ischemia leads to an infarct with a core (an area with ∼95 to 100% decreased blood flow) surrounded by a penumbra (an area with ∼85 to 90% decreased blood flow). It is believed that the penumbral neurons could be rescued if the therapeutic interventions are given within the first 6 h after stroke (Lipton, 1999). Acutely after focal ischemia, anoxic depolarization occurs in the core, concomitant with increased extracellular potassium and intracellular calcium, as well as rise in extracellular glutamate. In the penumbra, sporadic transient depolarization (spreading depression) results in further glutamate release. Prolonged rise in the extrasynaptic glutamate exerts excitotoxicity via overstimulation of glutamate receptors. Due to insufficient supply of oxygen and glucose, cells undergo mitochondrial failure, leading to an inability to maintain ionic gradient across the membrane. Signs of cellular death are detected both in the core and the penumbral region, either by necrosis, apoptosis or autophagy (reviewed in Lipton, 1999). Furthermore, generation of free radicals and oxidative stress contribute to the development of infarct, resulting in more damage in that region. Lactic acidosis and lowered pH are also presumed to influence the cellular processes, adding to the overall neuronal damage. The damage continues to grow in the penumbral region and after about 7 days, there is almost a complete loss of cellular elements in the infarcted region (termed pan-necrosis). Thus the infarct takes several days to mature and involves a myriad of processes which, with proper understanding, could be targeted to minimize the lesion development.
In addition to the above excitotoxic events, acute inflammatory reactions that start within hours and persist for days after the insult are currently being thought to contribute to the neuronal death following stroke (Barone and Feuerstein, 1999). A major event that triggers inflammation in the brain is the expression of adhesion molecules like intracellular adhesion molecule-1 (ICAM-1) on capillary endothelial cells and white blood cells (WBCs) which will facilitate the adhesion and transendothelial migration of neutrophils and macrophages. While in capillaries, the accumulation of WBCs impairs blood flow and when extravasated into the brain parenchyma they release a host of neurotoxic substances including pro-inflammatory cytokines, chemokines and oxygen/nitrogen free radicals that are extremely toxic to neurons (Clark et al., 1995; Denton and Dietrich, 2003). Following transient as well as permanent middle cerebral artery occlusion (MCAO) in rodents, neutrophil accumulation can be detected very early in the infarct area (Barone et al., 1992; Kochanek and Hallenback, 1992; Garcia et al., 1994; Braun et al., 1996; Yanaka et al., 1997), and reperfusion seems to hasten and intensify the degree of neurtophil infiltration (Clark et al., 1994). Activation of resident microglia also enhances the inflammatory process and drugs like minocycline reduces the infarct volume after stroke by preventing microglial activation (Yrjenheikki et al., 1998; 1999).
3. Cerebral inflammation following focal ischemia
In the brain, inflammation acts as a double-edged sword. The timing, type of inflammatory mediator and the extent of its stimulation determines the balance between the bad versus good effects of inflammation (Barone and Feuerstein, 1999). Uncontrolled numbers of the infiltrated macrophages and neutrophils increases free radical generation which is detrimental to vascular integrity leading to cell death. However, macrophage and microglial response after ischemia is necessary for scavenging the necrotic debris and to facilitate the plasticity (Danton and Dietrich, 2003). For example, the complement system that gets activated in post-ischemic brain initiates local inflammatory responses that ultimately contribute to neuronal survival and tissue remodeling. However, complement activation to an inappropriate extent promotes inflammatory mediator release and tissue injury. Hence, cerebral inflammation can exert beneficial or detrimental effects depending on the pathophysiologic context (Van Beek et al., 2003).
Many studies over the last decade showed the beneficial effects of anti-inflammatory therapies following focal cerebral ischemia. Therapies that deplete leukocytes, prevent proinflammatory cytokines and adhesion molecules, and inhibit cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) were reported to be neuroprotective after ischemia (Iadecola and Alexander, 2001; del Zoppo et al., 2000; Zhang et al., 1994; Yrjanheikki et al., 1998). Targeting individual inflammatory mediators and transcription factors that induce inflammatory genes also prevents ischemic neuronal damage. Knockout mice lacking interleukin-1beta (IL-1β) converting enzyme, ICAM-1, interferon regulatory factor-1 (IRF-1) and cyclic AMP element binding (CREB) protein develop smaller ischemic infarcts (Iadecola et al., 1999; Kitagawa et al., 1998; Schielke et al., 1998; Hata et al., 1998). Anti-inflammatory drugs like minocycline and indomethacin are known to induce neuroprotection by preventing microglial activation (Yrjanheikki et al., 1998, 1999; Tikka et al., 2001; Tikka and Koistinaho, 2001; Monje et al., 2003; Sasaki et al., 2003). However, conclusions should be drawn cautiously, as it is not evident to what extent pro-inflammatory cascades worsen the outcome after cerebral ischemia. Anti-inflammatory substances might act over a short span of time and hence might only delay the infarct development. The functional recovery/outcome following cerebral ischemia can not be linearly linked to the size of infarct (Dirnagl et al., 1999). These arguments add an additional degree of complexity to the question of the beneficial versus the harmful effect of inflammation, and warrant careful dissection of the role of individual inflammatory cascades.
3.1. Role of adhesion molecules in WBC extravasation after focal ischemia
While extravasation of WBCs into the brain is considered as the starting point of cerebral inflammation, many proteins mediate various steps of this process viz., rolling, adhesion and transendothelial migration. Acutely following cerebral ischemia, cytokines and chemokines are released from the infarcted area, some of which induce the ligands and receptors that mediate leukocyte recruitment and infiltration into brain. Focal ischemia leads to significantly elevated levels of the cytokines tumor necrosis factor alpha (TNFα), interleukins IL-1β and IL-6 and the chemokines, monocyte chemotactic protein-1 (MCP-1) and macrophage inflammatory protein-1 alpha (MIP-1α) (Liu et al., 1994; Wang et al., 1994; Kim et al., 1995; Wang et al., 1995; Soriano et al., 2000; Vemuganti et al., 2002; Lu et al., 2003; Kapadia et al., 2006). All these molecules induce the expression of the adhesion molecules ICAM-1, P-selectin and E-selectin on endothelial cells and WBCs (Dustin et al., 1986; Bevilacqua et al., 1987; Stoll et al., 1998; Abbassi et al., 1993; Eppihimer et al., 1996). Interaction of chemokines induces a conformational change in the β2-integrins on the leukocyte membrane. This will facilitate the interaction between integrins and adhesion molecules on the vessel wall leading to rolling, adhesion and transendothelial migration of WBCs (Penberthy et al., 1995; Gerszten et al., 1999). These events considerably slow down the blood flow through the cerebral capillaries, leading to their occlusion by activated neutrophlils and platelets (Mori et al., 1992), clogging the vessel and lowering blood perfusion. In addition to ICAM-1, P-selectin plays a significant role in post-ischemic leukocyte recruitment (Weyrich et al., 1993; Okada et al., 1994; Davenpect et al., 1994; Garcia-Criado et al., 1995; Kubes et al., 1995). It was demonstrated that fewer neutrophils accumulate in the brains of P-selectin-deficient mice following cerebral ischemia (Connolly et al., 1996). Similarly, blocking the ligand site on endothelial cells of ICAM-1 (Clark et al., 1995; Zhang et al., 1995) and antisense knockdown of ICAM-1 (Vemuganti et al., 2004) resulted in significant reduction of infarct volume. Furthermore, ICAM-1-deficient mice develop smaller infarcts (Soriano et al., 1996), supporting the beneficial effect of targeting adhesion molecules in post-ischemic brain. In addition, treatment with antibodies directed against the integrins CD11b/CD18 complex on granulocytes and macrophages led to a significant reduction in infarct volume in rats subjected to transient MCAO (Clark et al., 1991; Chen et al., 1994; Chopp et al., 1994). Depleting circulating neutrophils also reduce infarct volume and edema in various experimental models of cerebral ischemia (Shiga et al., 1991; Matsuo et al., 1994).
Diapedesis and dissolution of vascular integrity also contribute to the cerebral injury. Following MCAO, degradation of the basal lamina occurs, owing to increased expression and activation of metalloproteinases (MMPs), plasminogen activators and serine proteases (del Zoppo et al., 2000). MMP2 and MMP9 induction was thought to be spatio-temporally associated with blood-brain barrier damage and neuronal injury following MCAO (Heo et al., 1999; del Zoppo et al., 2000). Although, leukocytes infiltration into the post-ischemic brain parenchyma and the production of proteases, nitric oxide (NO), hydrogen peroxide and superoxides precipitates the secondary neuronal damage after focal ischemia, the exact contribution of these processes in relation to the neuronal death needs further assessment (Danton and Dietrich, 2003).
3.2. Role of free radicals in post-ischemic inflammation
NO and COX products contributes to the inflammatory cascades following an ischemic insult. NO is produced by the enzyme NO synthase (NOS), which exists in at least 3 isoforms viz., neuronal NOS (nNOS), endothelial NOS (eNOS) and iNOS. Of these, iNOS is expressed at a very low level by brain cells, but induced significantly following inflammatory conditions like focal ischemia. Unlike the other two isoforms, iNOS is not regulated by intracellular calcium and continuously produces large amounts of NO (Vodovotz et al., 1994; Iadecola et al., 1995). NO being a free radical, its excess production exerts cytotoxicity by inhibiting the ATP producing enzymes and by the production of peroxinitrite, which nitrosylates and thus inactivates many proteins (Iadecola, 1997; Iadecola and Alexander, 2001). While eNOS activation is thought to be neuroprotective after focal ischemia, as the NO produced by this isoform could dilate the capillaries by acting on the endothelial cells in its microenvironment (Huang et al., 1996), nNOS activation due to heightened calcium availability is also known to be toxic and together with iNOS seems to play a negative role in the pathophysiology of ischemic injury, as shown by studies with NOS inhibitors and iNOS knockout mice (Iadecola et al., 1997; Willmot et al., 2005). The 3 COX isoforms, COX-1, 2 and 3 generate prostaglandins and thromboxanes which are known to promote inflammatory reactions (Minghetti, 2004). Furthermore, COX-2 upregulation to an inappropriate level after focal ischemia promotes reactive oxygen species (ROS) generation that is thought to contribute to ischemic brain damage (Nogawa et al., 1998). However, recent findings suggest that COX-2 induced brain damage may not involve ROS as COX-2 inhibitors as well as COX-2 knockout mice failed to result in reduced ROS following MCAO (Kunz et al., 2006). In addition, the 5'-flanking region of the COX-2 gene has been shown to contain several potential regulatory sequences for the transcription factors, activating protein-2 (AP-2), nuclear factor kappa-B (NF-κB), CCAAT/enhancer binding protein (C/EBP), CREB, and Sp1, which are known to be upstream to many inflammatory genes (Lee et al., 2006).
4. Role of transcription factors in cerebral inflammation
Transcription factors play a pivotal role in controlling inflammatory gene expression. Microarray studies from our laboratory and other groups showed the induction of many transcription factors after focal ischemia (Soriano et al., 2000; Vemuganti et al., 2002; Lu et al., 2003). Of these, activation of hypoxia inducible factor-1 (HIF-1), CREB, c-fos, PPARα, PPARγ and p53 is known to prevent ischemic neuronal damage and/or promote ischemic tolerance (Tanaka et al., 2000a; 2000b; Cho et al., 2001; Maeda et al., 2001), whereas the induction of IRF-1, activating transcription factor-2 (ATF-2), signal transducer and activator of transcription 3 (STAT3), NF-κB, early growth response-1 (Egr1) and C/EBPβ promotes inflammation and neuronal death after cerebral ischemia (Table 1)(Planas et al., 1996; Bergeron et al., 1999; Iadecola et al., 1999; Johansson et al., 2000; Stephenson et al., 2000; Tanaka et al., 2000; Yan et al., 2000; Hu et al., 2000; Williams et al., 2003; Nurmi et al., 2004; Kapadia et al., 2006; Tureyen et al., 2007).
Table 1.
Transcription factors (TFs) involved in controlling inflammation and brain damage following cerebral ischemia
| TF | Role | Observed actions | References |
|---|---|---|---|
| IRF1 | Pro-inflammatory | IRF-1 null mice showed reduced infarct volume and neurological deficits |
Iadecola et al., 1999 |
| NF-KB | Pro-inflammatory | Inhibiting NF-KB activity resulted in smaller infarcts; mice deficient in p50 develop smaller infarcts |
Nurmi et al., 2004; Schneider et al., 1999 |
| Egr1 | Pro-inflammatory | Egr1 null mice showed smaller infarcts, improved neurological function, and curtailed induction of many inflammatory genes |
Vemuganti et al., 2007 |
| STAT3 | Pro-inflammatory | STAT3 siRNA prevented inflammation and brain damage in rats |
Satriotomo et al., 2006 |
| C/EBPβ | Pro-inflammatory | C/EBPβ null mice developed smaller infarcts, reduced neurological deficits and decreased apoptosis |
Kapadia et al., 2006 |
| PPARα | Anti-inflammatory | PPARα agonists decreased oxidative stress, inflammation and reduced infarction after focal ischemia |
Deplanque et al., 2003; Collino et al., 2006b |
| PPARβ/δ | Anti-inflammatory |
agonists decreased ischemic brain damage |
Iwashita et al., 2007 |
| PPARγ | Anti-inflammatory | PPARγ agonists prevented post- ischemic inflammatory gene expression, induced anti-oxidant gene expression and decreased infarction and neurologic dysfunction; The neuroprotection was observed in hypertensive as well as diabetic rodents subjected to focal ischemia |
Zhao Y et al., 2005, 2006; Sundararajan et al., 2005; Shimazu et al., 2005; Luo et al., 2006; Zhao X et al., 2006; Pereira et al., 2005, 2006; Collino et al., 2006; Tureyen et al., 2007 |
4.1. Nuclear factor kappa B (NF-κB)
Degradation of IκB induces NF-κB activation leading to the coordinated induction of multiple genes is involved in many inflammatory and immune cascades. Genes induced by NF-κB include pro-inflammatory cytokines IL-1β, TNF-α and granulocyte-macrophage colony stimulating factor (GM-CSF), and the chemokines IL-8, MIP-1α and MCP-1, that are largely responsible for attracting inflammatory cells into sites of inflammation (Nelson et al., 1993; Siebenlist et al., 1994; Mukaida et al., 1994; Ueda et al., 1994). Furthermore, NF-κB regulates the expression of adhesion molecules ICAM-1, VCAM1 and E-selectin on endothelial cells, promoting the adherence of inflammatory cells (Van De Stolpe et al., 1994; Iademarco et al., 1995). Many NF-κB down-stream gene products like IL-1β and TNF-α also re-activate NF-κB itself, resulting in a positive regulatory loop that amplifies and perpetuates the inflammatory responses (Barnes and Adcock, 1998). In rodents, activation of NF-κB occurs after MCAO and inhibiting NF-κB activity results in smaller infarcts (Nurmi et al., 2004). Furthermore, mice deficient in p50 develop smaller infarcts following focal ischemia, suggesting detrimental consequence of NF-κB activation (Schneider et al., 1999; Nurmi et al., 2004).
4.2. AP-1
AP-1 is a heterodimer of fos and jun oncoproteins, a collection of related transcription factors belonging to the Fos (cFos, FosB, Fra1, Fra2) and Jun (c-Jun, JunB, JunD) families that dimerize in various combinations through their leucine zipper regions (Barnes and Adcock, 1998). AP-1 will be activated by various cytokines, including TNFα and IL-1β via several protein tyrosine kinases and MAP kinases. Transcription factors Fos, c-Jun and JunB have been shown to be upregulated following cerebral ischemia (Kindy et al., 1991; Woodburn et al., 1993; Dragunow et al., 1994), and c-Jun was thought to play a role in the apoptotic neuronal death (Raivich and Behrenes, 2006).
4.3. HIF-1
Hypoxic conditions induce the oxygen-sensitive transcription factor HIF-1 (Bergeron et al., 1999; Lee and Anderson, 2006). HIF-1 is a heterodimer composed of HIF-1α and HIF-1β, and plays an essential role in hypoxia-induced transcriptional changes (Sharp and Bernaudin, 2004). For example, upregulation of HIF-1 subsequent to stroke induces iNOS and promotes neuronal death (Iadecola et al., 1995) as well as glucose transporter GLUT1 which may contribute to the adaptive mechanism to the oxygen-deprived condition resulting from ischemia (Bergeron et al., 1999).
4.4. C/EBPβ
C/EBPβ is a leucine-zipper transcription factor that regulates cell growth and differentiation in mammals. Expression of many proinflammatory genes including the cytokines and chemokines IL-6, IL-1β, IL-8, IL-12, TNFα, and MCP-1 is known to be controlled by C/EBPβ (Bradley et al., 2003). In addition, iNOS and COX-2 have C/EBPβ recognition sequences in their promoter regions and many of the IL-6 effects are mediated through the activation of C/EBPβ. Our studies showed that transient focal cerebral ischemia significantly upregulated C/EBPβ gene expression in rodent brain (Vemuganti et al., 2002), and focal ischemia in C/EBPβ null mice induced significantly smaller infarcts, reduced neurological deficits and decreased apoptosis (Kapadia et al., 2006). Furthermore, we observed curtailed inflammatory responses such as decreased ICAM-1 immunopositive vessels and less numbers of extravasated neutrophils and macrophages in C/EBPβ null mice following transient MCAO (Kapadia et al., 2006). In addition, GeneChip analysis showed that the post-ischemic induction of many inflammatory and neuronal damage inducing-genes was less pronounced in the brains of C/EBPβ null mice, suggesting a significant role for C/EBPβ in post-ischemic inflammation and brain damage (Kapadia et al., 2006).
4.5. IL6-JAK-STAT signaling
Increased levels of cytokines like IL-6 and TNF-α immediately after focal ischemia initiate cerebral inflammation and thereby secondary neuronal damage (Iadecola and Alexander, 2001; Stoll et al., 1999). The effects of these cytokines are propagated and amplified by the Janus kinase (JAK) and STAT signaling pathways (O'Shea et al., 2002). Binding of cytokines to their receptors induces transphosphorylation of the receptor-associated JAKs, which in turn phosphorylates the down-stream STAT family of transcription factors. Phosphorylated STAT dimerizes and binds to conserved genomic regulatory sequences on DNA to induce the expression of many genes (Darnell et al., 2005). Of the various isoforms of JAKs and STATs, JAK2 and STAT3 are the most-conserved isoforms and mice that lack either JAK2 or STAT3 die En Uterus (O'Shea et al., 2002). Transient focal ischemia was shown to stimulate the phosphorylation of both STAT1 and STAT3 (Planas et al., 1996; Justicia et al., 2000; Suzuki et al., 2001; Satriotomo et al., 2006). Increased STAT1 phosphorylation in neurons is known to contribute to the ischemic neuronal damage and STAT1 knockout mice show smaller infarcts (Takagi et al., 2002). Recent studies from our laboratory showed that following transient focal ischemia, JAK2 and STAT3 phosphorylation increases in the microglia/macrophages and treating ischemic rats with either a JAK2 phosphorylation inhibitor (AG490) or a STAT3 siRNA prevents inflammation as well as brain damage (Satriotomo et al., 2006). These studies indicate that JAK2-STAT3 activation might play a significant role in inducing secondary brain damage after focal ischemia. Furthermore, in the post-ischemic brain JAK-STAT activation leads to increased expression of suppressor of cytokine signaling-3 (SOCS3), which is a negative feed-back regulator of IL6 signaling. Our previous studies demonstrated that preventing SOCS3 expression with an antisense oligonucleotide infusion exacerbates inflammatory brain damage after focal ischemia in rats (Vemuganti et al., 2002).
4.6. Egr1
Egr1 is a sequence-specific transcription factor known to be induced by hypoxia (Yan et al., 1999). In peripheral organs, several inflammatory genes are induced down-stream to Egr1 (Yan et al., 2000). Our laboratory recently observed that focal ischemia increases cerebral Egr1 expression, and Egr1 null mice subjected to transient MCAO show significantly smaller infarcts, improved neurological function, and curtailed induction of many inflammatory genes (Vemuganti et al., 2007). Furthermore, we observed that adenoviral-mediated cerebral Egr1 overexpression exacerbates the focal ischemic brain damage (Vemuganti et al., 2007). These studies for the first time show Egr1 induction as a significant contributor of the inflammation and neuronal damage after stroke.
5. PPARs
Recently, much interest has been focused on the transcription factor PPAR isoforms α and γ, as their activation by specific ligands was observed to control inflammatory gene expression under various pathological conditions including focal ischemia.
5. 1. PPAR isoforms and ligands
PPARs are members of the nuclear hormone receptor super-family that include the receptors for vitamin D, thyroid hormone, retinoic acid, ecdysone, and some orphan receptors (Laudet et al., 1992). In mammals, PPAR exists as 3 isoforms PPARα, PPARβ/δ, and PPARγ, each encoded by a distinct gene. Naturally occurring fatty acids and eicosanoids bind to and activate PPARs, but the relative need for high concentration for these ligands suggests that PPARs are evolved to be activated by low-affinity ligands (Daynes and Jones, 2002). Fibrates are known to activate PPARα, although, at higher concentrations, PPARγ can be activated as well. NSAIDs such as ibuprofen, indomethacin or fenofibrate have been shown to activate PPARα 5 to 10 fold better than PPARγ (Lehmann et al., 1997). The prostaglandin J2 derivative 15-deoxy-delta12,14-PGJ2 (15d-PGJ2) is an endogenous PPARγ agonist; although as it is not present in sufficient concentrations to activate PPARγ in mammalian cells (Bell-Parich et al., 2003), it might influence other pathways, such as directly acting on IκB and the DNA binding domain of NF-κB (Straus et al., 2000). On the other hand, thiazolidinediones (TZD) like troglitazone, pioglitazone, and rosiglitazone which are synthetic insulin-sensitizing anti-diabetic ligands have emerged as potent PPARγ agonists (Lehmann et al., 1995). Among the TZDs, rosiglitazone and pioglitazone are currently approved by the US FDA for human use (Sood et al., 2000).
5.2. PPAR structure
Considerable amount of information is now available on the various domains of PPAR protein and their function. The PPAR domains include (a) the N-terminal A/B domain called activation function-1 (AF-1) which comprises the ligand-independent transactivation function, (b) DNA-binding domain (DBD), (c) ligand-binding domain (LBD), (d) D-domain linking the LBD and DBD (Escher and Wahli, 2000, Fig. 1a). Retinoic acid-X-receptor (RXR) contains a highly conserved central DNA-binding domain and a less conserved ligand-binding domain and serves as common heterodimeric, permissive partner for many nuclear receptors, including PPAR. When a ligand binds to PPAR, it dimerizes with RXR to form a heterodimeric complex that binds to the cis-acting sequences (peroxisome proliferator response elements; PPREs) on DNA to initiate transcription of target genes (Escher and Wahli, 2000; Chang and Szabo, 2000; Blanquart et al., 2003).
Figure 1.
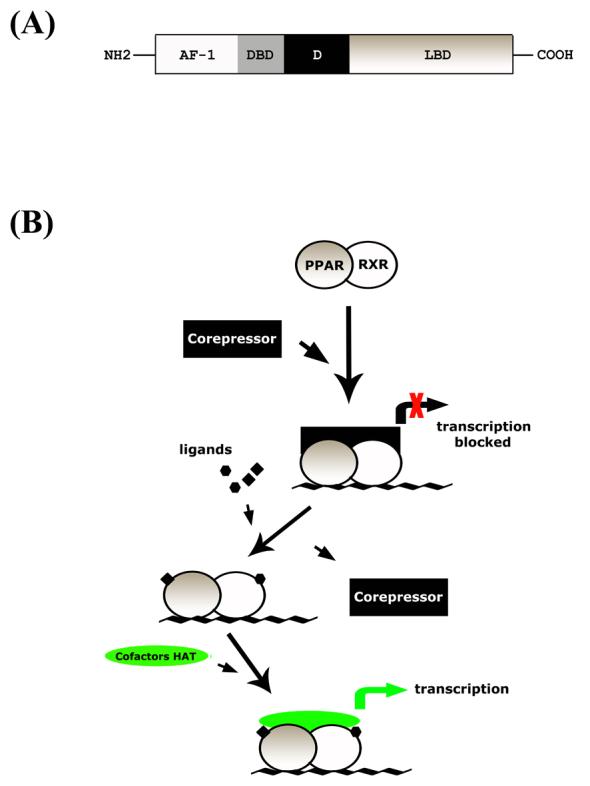
PPARγ gene organization (A) and a simplified representation of action of PPAR:RXR heterodimer and ligands in transcriptional process (B). Under the presence of ligands, corepressor is released from PPAR:RXR heterodimer, enabling it to bind to to cofactor HAT complex. With further recruitment of general transcription complexes, transcription process can resume. AF-1; activation function-1. DBD; DNA binding domain. LBD; ligand binding domain. D; hinge domain linking DBD and LBD. PPRE; peroxisome proliferator activated receptor responseive element. HAT; histone acetyltransferase.
5.3. Mechanism of action of PPAR
Upon ligand binding, PPARs are liberated from inhibitory nuclear receptor co-repressor/silencing factor and, with recruitment of co-activators, act as transcription factors of target genes. Binding of ligand on either receptor of the PPAR:RXR heterodimer complex can activate the complex, but the simultaneous binding of ligands on both PPAR and RXR is known to be more potent (Desvergene and Wahli, 1999). Action of nuclear factors in regulating gene expression is quite complex. PPARs can influence the expression of various genes either positively (by transactivation) or negatively (by transrepression). Upon ligand binding, PPAR:RXR complexes with cofactor complexes and transactivates target genes that are under PPAR control (Fig 1b). For example, TZD binding to PPARγ results in the recruitment of cofactors and increased transcription activity. In the absence of the ligand, the PPAR/RXR complex can bind to the PPRE element with a co-repressor complex and hinder target gene transcription. PPAR can also transrepress certain genes that are under the control of other transcription factors such as NF-κB, AP-1 and Egr1, by inhibiting the ubiquitylation/degradation of co-repressor proteins via ligand-dependent addition of small ubiquitin-like modifier (SUMO) (Pascual et al., 2005). In addition to these, the PPAR:RXR heterodimeric complex competes with the co-activator complexes for other transcription factors, as well as directly interacts with transcription factors to regulate their function. To complicate matters further, in the case of PPARγ, TZDs can have PPARγ-independent actions that include mitochondrial dysfunction-related stress-response, involvement of proteins such as MitoNeet and increased astrocyte glucose uptake and lactate production (Dello Russo et al., 2003; Feinstein et al., 2005).
5.4. Regional and cellular distribution of PPAR
PPARα is expressed in cells with high catabolic rates of fatty acids and high peroxisome-dependent activities such as hepatocytes, cardiomyocytes, proximal tubules of kidney cortex, and intestinal mucosa. This explains its mRNA being most abundant in the liver, heart and kidney (Escher and Wahli, 2000). PPARβ/δ is expressed in relatively high levels in the adrenal gland, heart, intestine and kidney (Kliewer et al., 1994). Widespread expression of this isoform in both embryo and in adult has been shown (Kliewer et al., 1994; Cullingford et al., 1998), and its expression is comparatively higher than the other two isoforms in many tissues (Kliewer et al., 1994; Braissant et al., 1996; Cullingford et al., 1998). PPARγ shows the most restricted pattern of expression; high expression is observed in the adipose tissue, and modest expression is observed in the large intestine and immune system (Braissant et al., 1996). In the adult brain, PPARs show relatively low abundance with the exception of PPARβ/δ form, suggesting limited function of PPARs in the CNS under normal physiological conditions (Kliewer et al., 1994; Braissant et al., 1996).
Immunohistochemical studies demonstrated the distribution of PPARs in the adult rodent CNS (Braissant et al., 1996; Moreno et al., 2004). Except for the inner/outer layer of the retina where all the 3 isoforms are abundantly present, PPARβ/δ was shown to be localized in a relatively uniform fashion in the CNS, whereas PPARα and γ showed a low level of protein throughout the brain. PPARα transcript was expressed in the CA1 area and granule cells of the dentate gyrus, but not in the CA3 area of the hippocampus. PPARγ signal was weak and limited to granule cells of the hippocampal dentate gyrus (Braissant et al., 1996). PPARβ/δ expression was detected in the dentate gyrus, CA1 and CA3, as well as hilus. In the cerebellum, PPARβ/δ was present in Purkinje cells, granule cells, golgi cells and interneurons of the molecular layer, whereas PPARα and γ were scarcely present. Reticular formation was the exception where both α, β/δ and γ were present in similar levels. Moreno et al (2004) showed that PPARγ was also present in caudate putamen and globus pallidus of the basal ganglia as well as thalamic nuclei and piriform cortex. In murine brain, PPARβ/δ was shown to be high in the entorhinal cortex, hippocampus and hypothalamus (Woods et al., 2003), which was not in complete agreement with the other reports (Braissant et al., 1996; Moreno et al., 2004).
With respect to cell types of the CNS, all PPAR isoforms are known to be present in both neurons and astrocytes (Braissant et al., 1996; Moreno et al., 2004). Primary cultures of cortical neurons and astrocytes show mRNA of all isoforms of PPARs, where different isoforms seem to contribute to neuronal and astrocytic differentiation (Cimini et al., 2005; Cristiano et al., 2005). A recent study showed that exposing oligodendrocytes to PPARβ/δ agonists leads to stimulation of oligodendrocyte differentiation in cultured oligodendrocytes, where PPARβ/δ expression has also been reported (Saluja et al., 2001; Woods et al., 2003). The subcellular localization of PPARs seems to be mostly nuclear, but some cytoplasmic localization for PPARα and γ has been reported in subsets of neurons (Moreno et al., 2004).
6. Role of PPARs in brain inflammation
The major function of PPARs seems to be to act as regulators of glucose and lipid metabolism and adipocyte differentiation in several subtypes of cells throughout the body. But PPARs also influence immune and inflammatory functions in macrophages, T cells, B cells, dendritic cells and endothelial cells (Gosset et al., 2001; Jones et al., 2002; Glass and Ogawa, 2006). PPARα and γ agonists have been shown to display anti-inflammatory effects (Devchand et al., 1996; Setoguchi et al., 2001), though it is not yet clear whether all the anti-inflammatory effects are receptor-dependent (Daynes and Jones, 2002).
TZDs inhibit the expression of various inflammatory proteins such as iNOS, TNFα, and MMP9 in macrophages (Li et al., 2000), and prevent the induction of pro-inflammatory transcription factors such as NF-κB, AP-1, and STATs (Ricote et al., 1998; Welch et al., 2003). In macrophages, 15d-PGJ2 or synthetic PPARγ ligand BRL49653 was shown to induce a dose dependent inhibition of iNOS, MMP9, and scavenger receptor-A expression (Recote et al., 1998). Whereas, neither 15d-PGJ2 nor troglitazone was effective in inhibiting phorbol 12-myristate13-acetate (PMA)-induced TNFα release as well as IL-1β and IL-16 expression in human monocytes (Jiang et al., 1998).
In primary mouse microglia and astrocytes, 15d-PGJ2 as well as TZDs inhibited the production of NO, cytokines IL-1β, IL-6, TNF-α and the chemokine MCP-1 (Storer et al., 2005). Furthermore, in activated astrocytes and microglia, 15d-PGJ2 and rosiglitazone inhibited the phosphorylation of STAT1, STAT3, JAK1 and JAK2 (Park et al., 2003), suggesting a role as a negative modulator of cytokine signaling. Similarly, in LPS or IFN-γ-stimulated primary microglia, 15d-PGJ2 attenuated iNOS expression and NO production, and donwregulated TNF-α and MHC class II expression (Bernardo et al., 2000). These studies provide evidence for the potential involvement of PPARγ in inflammatory processes in CNS.
7. PPAR agonists and focal ischemia
As already mentioned, pioglitazone and rosiglitazone (TZD class of PPARγ agonists) have proven to be beneficial in type-2 diabetes mellitus patients. Diabetics are at an increased risk of stroke incidence and stroke causes more damage in diabetics compared to normoglycemic individuals (Kagansky et al., 2001). Recently, enhanced functional recovery was reported in a small group of stroke patients with type-2 diabetes treated with pioglitazone or rosiglitazone (Lee and Reding, 2006). In another clinical study, increased plasma level of 15d-PGJ2 (the natural ligand for PPARγ) was correlated with smaller infarcts and good neurological outcome (Blanco et al., 2005). In addition, the beneficial role of PPAR agonist treatment has been demonstrated in many animal models of focal cerebral ischemia. Table 2 shows a summary of the findings (highlighting the animal model, drug treatment, route of delivery and the effects of PPAR agonist treatment on infarction) which are discussed below.
Table 2.
Beneficial role of PPAR agonists treatment in animal models of cerebral ischemia.
| Authors and journal |
Animal model | Route of treatment and doses |
Effects of drug treatments and results | |
|---|---|---|---|---|
| 1 | Shimazu et al., 2005 | Permanent and transient MCAO in S.D. rats |
Pio, oral, 20 mg/kg/day |
↓ infarct volume in transient (but not permanent) ischemia, better neurological score |
| 2 | Pereira et al., 2005 | Permanent MCAO in Fischer rats |
L-796,449, i..p., 1mg/kg |
↓ infarct size, better neurological score, ↓ apoptotic and anti-inflammatory processes |
| 3 | Zhao et al., 2005 | Transient MCAO in Wistar rats |
Pio, 0.16mg/kg/day, i.c.v. |
↑ sensory neurological score, ↓ infarct volume, ↓ edema, ↓ ED-1 positive cells |
| 4 | Sundararajan et al., 2005 | Transient MCAO in Wistar rats |
Tro (>10mg/kg), Pio (1mg/kg), i.p. |
↓ infarct size, ↓ microglia/macrophages, COX-2 mRNA/protein, iNOS and IL-1β mRNA |
| 5 | Zhao et al., 2006 | Transient MCAO in Wistar rats |
Pio, 0.16mg/kg/day, | ↓ infarct size, ↓ TNF-α, COX-2+ cells. ↑PPARγ + cells following ischemia in neurons/microglia |
| 6 | Luo et al., 2006 | Transient MCAO in C57/B6 mice, cell culture |
Rosi, up to 12mg/kg, i.p. |
↓ infarct size, better neurological score, Rosi treatment increased binding, ↓ ICAM-1, MPO, activated microglia |
| 7 | Victor et al., 2006. | Transient MCAO in Wistar rats |
Pio (1.0 mg/kg), Rosi (0.1 mg/kg), T0070907 (1.5 mg/kg), i.p. |
Increased PPARγ + cells following ischemia in neurons, ↑binding by Rosi treatment |
| 8 | Chu et al., 2006 | Transient MCAO in S.D. rats |
Rosi, oral, 3mg/kg | ↓ infarct size, better neurological score, ↓ TUNEL + cells, ↓ Caspase-3+ cells, ↓ MPO/OX-42, ↓ caspase-3 activity, ↑ vasculature |
| 9 | Ou et al., 2006 | Transient MCAO/CCAO in Long Evans rats |
15d-PGJ2, 20 μg, i.c.v. |
↑PPARγ + cells following ischemia in neurons/microglia, and ↓ infarct volume |
| 10 | Lin et al., 2005 | Transient MCAO/CCAO in Long Evans rats |
15d-PGJ2 (1-50pg), i.c.v., Rosi (50ng) |
↓ infarct size, HO-1 increase, ↓ IKBα degradation, ↓ caspase-3 activation, 15-d-PGJ2 and Rosi treatment ↑ PPARγ expression |
| 11 | Collino et al., 2006 | Transient CCAO in Wistar rats and C57/B6 mice |
Rosi, Pio, i.v., 1-6 mg/kg. |
↓ oxidative stress, ROS, and HNE. Restored GSH and SOD level, reduced COX-2 increase, and inhibited NF-kB nuclear translocalization |
| 12 | Turuyen et al., 2007 | Transient MCAO in S.D., SH rats, C57/B6 and db mice |
Rosi, Pio, 9-cis-RA, 15d-PGJ2, i.p., 0.5- 6mg/kg |
↓ infarct size, ↓ ICAM-1 and infiltrating macrophages/microglia, ↑ catalase and Cu/ZnSOD, ↑ SOCS3, ↓ STAT phosphorylation |
| 13 | Deplanque et al., 2003 | transient MCAO in C57/B6 mice. Also used APOE−/−, and PPARα−/− mice |
Fenofibrate, 50- 250mg/kg, oral |
↓ infarct size, better MCA endothelial vasodilation, no change in iNOS, more GSH due to ↑ Cu/ZnSOD, ↓ VCAM-1 and ICAM-1 |
| 14 | Collino et al., 2006 | Transient CCAO in Wistar rats |
WY14643 (0.1- 6mg/kg), MK886 (6mg/kg), i.v. |
↓ ROS, restored glutathione level, ↓ HNE, ↓ HO-1 and p38 MAPK level, ↓ nuclear NF-kB, ↓ iNOS, ↓ ICAM-1, ↓ S100β |
Abbreviations used : S.D, Sprague-dawley; i.p.; intraperitoneal; i.c.v., intracerebroventricular; Rosi, rosiglitazone; Pio, pioglitazone; Tro, troglitazone; SHR, spontaneously hypertensive rat; db, diabetic; MPO, myeloperoxidase; HNE, hydroxynonenal; GSH, N-(N-L-gamma-glutamyl-L-cysteinyl)glycine ; SOD, superoxide dismutase; MAPK, mitogen activated protein kinase; SOCS, suppressors of cytokine signaling ; STAT, signal transducers and activators of transcription
Rats fed on a pioglitazone-fortified diet for 3 days prior to the induction of transient focal ischemia were observed to develop smaller infarcts and better neurological scores compared to pair-fed controls (Shimazu et al., 2005). These authors showed that upregulation of the antioxidant enzyme Cu/ZnSOD might be a potential neuroprotective mechanism underlying the neuroprotective effect of pioglitazone after focal ischemia. However, the same treatment did not show protection when rats were subjected to permanent ischemia, suggesting that PPARγ-dependent protection is specific to the free radical-generating and/or inflammatory nature of the reperfusion injury. Interestingly, when a non-TZD PPARγ agonist L-796,449 was injected (i.p., 1mg/kg) 10 min prior to permanent MCA occlusion, the treatment resulted in reduced infarct size and better neurological outcome (Pereira et al., 2005). The reason for this disparity is still unclear, but may be due to the PPAR-independent actions of the drug. L-796,449 treatment also increased the neuroprotective enzyme heme oxygenase-1 (HO-1) while blocking NF-κB translocation into the nucleus, with decreased caspase-3 activity, reduced levels of inflammatory mediators iNOS and MMP-9, as well as decreased amounts of NO2- and NO3- (Pereira et al., 2005). A later study showed that intravenous injection of either pioglitazone or rosiglitazone reduced oxidative stress, COX-2 expression and activation of MAPK and NF-κB following a transient common carotid artery occlusion (Collino et al., 2006a). Chu et al. (2006) suggested that rosiglitazone-fed rats had better neurological scores, smaller infarcts, reduced number of TUNEL-positive cells, reduced caspase-3 activity, and curtailed infiltration of myeloperoxidase positive (MPO+) neutrophils and OX-42+ macrophages following transient focal ischemia. Interestingly these authors also reported an increased vasculature in the rosiglitazone-treated group with increased number of endothelial cells positive for BrdU, suggesting there may be enhanced angiogenesis following PPARγ activation.
The beneficial effect of TZD treatment may be due to the direct action of TZDs on brain rather than on the peripheral organs, as pioglitazone administered into the brain via osmotic mini-pump also resulted in significant neuroprotection following MCAO (Zhao et al., 2005). When pioglitazone was infused intraventricularly for 7 days (5 days prior and 2 days after the MCAO) there was an increase in the sensory (but not motor) neurologic scores, with reduced infarct volume and brain edema compared to vehicle treated controls (Zhao et al., 2005). Furthermore, macrophage extravasation and microglial activation was also significantly reduced in the pioglitazone-infused group supporting its anti-inflammatory properties in the ischemic brain (Zhao et al., 2005).
Further supporting the notion of enhanced anti-inflammatory influence of TZDs after focal ischemia, Sundararajan et al. (2005) showed the beneficial effect of troglitazone and pioglitazone injected i.p. 1 day prior to the ischemia. Troglitazone as well as pioglitazone treatment reduced infarct volume and resulted in better neurological score, reduced number of microglia/macrophages and decreased expression of COX-2, IL-1β and iNOS mRNA/protein in the ischemic brain. This group recently showed that focal ischemia significantly enhances PPARγ mRNA and protein levels in the neurons within the peri-infarct area of the rats subjected to transient MCAO (Victor et al., 2006).
Using a mouse model of transient MCAO, Luo et al. (2006) reported the beneficial effect of rosiglitazone delivered i.p. at differing concentrations and demonstrated that drug treatment at 3mg/kg and higher reduced infarct volume and improved neurologic score. Rosiglitazone treatment also increased the binding of PPARγ to PPRE, suggesting that the drug enhances the translational activity of PPARγ. When animals were treated with PPARγ antagonist GW9662, this protective effect of rosiglitazone was lost. Rosiglitazone treatment further reduced ICAM-1 expression, MPO levels and activity, amount of activated microglia, and cytokine levels (TNF-α, IL-6, MCP-1), supporting the link between anti-inflammatory effects of TZDs acting via PPARγ (Luo et al., 2006).
Recent studies from our laboratory showed that pre-treatment as well as post-treatment with the TZDs, rosiglitazone and pioglitazone significantly decreases the infarct volume and neurological deficits in normotensive and normoglycemic rodents as well as hypertensive and hyperglycemic rodents (Tureyen et al., 2007). Our studies further showed that rosiglitazone neuroprotection will not be enhanced by RXR agonist 9-cis-retinoic acid, but could be prevented by PPARγ antagonist GW9662. Rosiglitazone significantly decreased the post-ischemic expression of ICAM-1 mRNA and protein leading to decreased extravasation of macrophages and neutrophils into the brain parenchyma. Rosiglitazone treatment also curtailed the post-ischemic expression of the pro-inflammatory cytokines (IL-1β and IL-6), chemokines (MIP-1α and MCP-1) as well as the pro-inflammatory enzymes COX-2 and iNOS. Our studies further showed that PPARγ activation by TZDs prevents the post-ischemic cerebral expression of pro-inflammatory transcription factors Egr1, C/EBPβ and NF-κB and at the same time enhances the expression of the anti-oxidant enzymes catalase and Cu/ZnSOD (Tureyen et al., 2007). Most importantly, our studies showed that rosiglitazone-induced neuroprotection after ischemia involves expression of the cytokine signaling regulator SOCS3 and the prevention of STAT3 phosphorylation. Thus, these recent studies indicate that PPARγ activation with TZDs is a potent therapeutic option for preventing inflammation and neuronal damage after stroke with promise in diabetic as well as hypertensive subjects.
It has been shown that transient focal ischemia alone can increase brain PPARγ protein content in peri-infarct area. Increased PPARγ mRNA was detected in the infarcted brain as early as 6h following focal ischemia (Ou et al., 2006), and PPARγ-immunpositive neurons were detected between 4 h and 14 days (Victor et al., 2006), whereas in neurons and microglia only transiently at 12 hr in the post-ischemic brain (Zhao et al., 2006). This increased PPARγ expression was shown to be associated with reduced infarct volume, TNFα level, and number of COX-1 and -2 positive cells in the peri-infarct cortical regions when the animals were treated intraventricularly with pioglitazone (Zhao et al., 2006). In addition, in cultured neurons pioglitazone treatment was able to inhibit COX-2 increase induced due to H2O2, whereas PPARγ antagonist GW9662 treatment abrogated this effect, suggesting a direct action of PPARγ on COX-2 in neurons (Zhao et al., 2006). Victor et al. (2006) also showed that increased PPARγ mRNA expression and protein levels in neurons induces reduced infarct volume if the animals were treated with rosiglitazone and that the PPARγ antagonist T0070907 treatment abrogates this effect. Interestingly, the antagonist T0070907 alone resulted in significantly more damage, suggesting that the endogenous PPARγ increase seen in the penumbral neurons might be beneficial. Ischemia reduced the PPARγ binding in the ipsilateral hemisphere of the brain, and rosiglitazone treatment increased the binding in general, where it resulted in a much more noticeable increase in binding activity in the contralateral side to the injury (Victor et al., 2006). This increased neuronal PPARγ expression was also supported by Ou et al. (2006), who found PPARγ-positive cells in the ipsilateral hemisphere following transient MCAO/CCAO injury with probes recognizing PPARγ. Furthermore, 15d-PGJ2 treatment resulted in increased binding of PPARγ to PPRE and reduced the area of infarct (Ou et al., 2006). Interestingly, intracerebral infusion of 15d-PGJ2 and rosiglitazone were able to increase PPARγ protein level in the normal brain as well (Lin et al., 2006). These authors showed that infusion of 15d-PGJ2 and rosiglitazone into brain given less than 2h post MCAO reduces infarct volume, decreases caspase-3 activation and apoptotic/necrotic cascades occurring after an ischemic insult.
There are also some studies supporting the beneficial effects of PPARα in cerebral ischemia. The PPARα agonist fenofibrate when given orally for 14 days prior to MCA occlusion reduced cortical infarct volume as opposed to very little effect of acute i.p injected fenofibrate (Deplanque et al., 2003). This discrepancy might be due to the slow rate of penetration of fenofibrate into brain. This protection by fenofibrate is suggested to occur via improved sensitivity of endothelial cells, decreased cerebral oxidative stress by increased activity of Cu/ZnSOD and prevention of pro-inflammatory events such as VCAM-1 and ICAM-1 expression in the post-ischemic brain (Deplanque et al., 2003). In further support to the beneficial effects of stimulating PPARα, treating rats with the PPARα antagonist WY14643 increased the oxidative stress, iNOS, and ICAM-1 expression following transient ischemia (Collino et al., 2006b). Recently, the beneficial role of PPARβ/δ agonists L-165041 and GW501516 in transient MCAO was demonstrated, perhaps via their anti-apoptotic effect as shown using in vitro conditions (Iwashita et al., 2006).
8. Future directions
While more evidences accumulate supporting the significance of transcription factors in promoting and curtailing inflammation thus contributing to brain damage after stroke, the therapeutic targeting of individual transcription factors is still at its infancy. Developing small molecules that can prevent the activation of pro-inflammatory transcription factors such as C/EBPβ, IRF-1 and NF-κB without undesired side effects may be beneficial as future stroke therapeutics. On the other hand, the agonists for anti-inflammatory transcription factors PPARα and PPARγ show much promise in the experimental models of stroke. Large scale clinical trials with thiazolidinediones (PPARγ agonists) and fenofibrate (PPARα agonist) will be the necessary next step to evaluate their safety and effectiveness in humans. In parallel, defining more specific mode of action by identifying the endogenous co-activators and modulators of these transcription factors in animal models will help to device more efficient therapeutic strategy. Studies in these directions are underway in several laboratories.
9. Conclusion
Inflammatory cascades are known to contribute to the pathophysiology of focal cerebral ischemia. Transcription factors induced in the post-ischemic brain plays a significant role in promoting as well as curtailing neuroinflammation; for example, transcription factors like C/EBPβ, Egr1, PPARs and NF-κB might promote and prevent inflammatory brain damage. By virtue of their prevention of many inflammatory reactions, the agonists that bind to the various isoforms of PPAR prevent ischemic brain damage. The neuroprotective effects of PPARγ agonists involve multiple mechanisms including preventing inflammatory gene expression, inducing anti-oxidant enzymes and other neuroprotective transcripts. PPARγ agonists might be acting in brain by stimulating PPARγ as well as directly without involving the PPARγ. The mechanisms of neuroprotection induced by PPARα and β/δ are not very clear and further studies are needed to elucidate them in depth. Overall, PPAR isoform activators might be developed into future stroke therapeutics.
ACKNOWLEDGEMENTS
These studies were funded by grants from the United States National Institute of Health (RO1 NS044173 and RO1 NS049448), the American Heart Association (Grant-in-Aid 0350164N) and the faculty start-up funds from the University of Wisconsin to R. Vemuganti.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbassi O, Kishimoto TK, McIntire LV, Anderson DC, Smith CV. E-selectin supports neutrophil rolling in vitro under conditions of flow. J. Clin. Invest. 1993;92:2719–2730. doi: 10.1172/JCI116889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ, Adcock IM. Transcription factors and asthma. Eur. Respir. J. 1998;12:221–234. doi: 10.1183/09031936.98.12010221. Review. [DOI] [PubMed] [Google Scholar]
- Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J. Cereb. Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Barone FC, Schmidt DB, Hillegass LM. Reperfusion increases neutrophils and leukotriene B4 receptor binding in rat focal ischemia. Stroke. 1992;23:1337–1348. doi: 10.1161/01.str.23.9.1337. [DOI] [PubMed] [Google Scholar]
- Bell-Parikh LC, Ide T, Lawson JA, McNamara P, Reilly M, FitzGerald GA. Biosynthesis of 15-deoxy-delta12,14-PGJ2 and the ligation of PPARgamma. J. Clin. Invest. 2003;112:945–955. doi: 10.1172/JCI18012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron M, Yu AY, Solway KE, Semenza GL, Sharp FR. Induction of hypoxia-inducible factor-1 (HIF-1) and its target genes following focal ischaemia in rat brain. Eur J. Neurosci. 1999;11:4159–4170. doi: 10.1046/j.1460-9568.1999.00845.x. [DOI] [PubMed] [Google Scholar]
- Bernardo A, Levi G, Minghetti L. Role of the peroxisome proliferator-activated receptor-gamma (PPAR-gamma) and its natural ligand 15-deoxy-Delta12, 14-prostaglandin J2 in the regulation of microglial functions. Eur. J. Neurosci. 2000;12:2215–2223. doi: 10.1046/j.1460-9568.2000.00110.x. [DOI] [PubMed] [Google Scholar]
- Bevilacqua MP, Pober JS, Mendrick DL, Cotran RS, Gimbrone MA., Jr. Identification of an inducible endothelial-leukocyte adhesion molecule. Proc. Natl. Acad. Sci. U S A. 1987;84:9238–9242. doi: 10.1073/pnas.84.24.9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanquart C, Barbier O, Fruchart JC, Staels B, Glineur C. Peroxisome proliferator-activated receptors: regulation of transcriptional activities and roles in inflammation. J. Steroid Biochem. Mol. Biol. 2003;85:267–273. doi: 10.1016/s0960-0760(03)00214-0. [DOI] [PubMed] [Google Scholar]
- Blanco M, Moro MA, Davalos A, Leira R, Castellanos M, Serena J, Vivancos J, Rodriguez-Yanez M, Lizasoain I, Castillo J. Increased plasma levels of 15-deoxyDelta prostaglandin J2 are associated with good outcome in acute atherothrombotic ischemic stroke. Stroke. 2005;36:1189–1194. doi: 10.1161/01.STR.0000166054.55993.e5. [DOI] [PubMed] [Google Scholar]
- Bradley MN, Zhou L, Smale ST. C/EBPb regulation in lipopolysaccharide-stimulated macrophages. Mol. Cell. Biol. 2003;23:4841–4858. doi: 10.1128/MCB.23.14.4841-4858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology. 1996;137:354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- Braun JS, Jander S, Schroeter M, Witte OW, Stoll G. Spatiotemporal relationship of apoptotic cell death to lymphomonocytic infiltration in photochemically induced focal ischemia of the rat cerebral cortex. Acta. Neuropathol. 1996;92:255–263. doi: 10.1007/s004010050516. [DOI] [PubMed] [Google Scholar]
- Chang T-H, Szabo E. Induction of differentiation and apoptosis by ligands of peroxisome proliferator-activated receptor-γ in non-small cell lung cancer. Cancer Res. 2000;60:1129–1138. [PubMed] [Google Scholar]
- Chen H, Chopp M, Zhang RL, Bodzin G, Chen Q, Rusche JR, Todd RF., 3rd. Anti-CD11b monoclonal antibody reduces ischemic cell damage after transient focal cerebral ischemia in rat. Ann. Neurol. 1994;35:458–463. doi: 10.1002/ana.410350414. [DOI] [PubMed] [Google Scholar]
- Cho S, Park EM, Kim Y, Liu N, Gal J, Volpe BT, Joh TH. Early c-Fos induction after cerebral ischemia: a possible neuroprotective role. J. Cereb. Blood Flow Metab. 2001;21:550–556. doi: 10.1097/00004647-200105000-00009. [DOI] [PubMed] [Google Scholar]
- Chopp M, Zhang RL, Chen H, Li Y, Jiang N, Rusche JR. Postischemic administration of an anti-Mac-1 antibody reduces ischemic cell damage after transient middle cerebral artery occlusion in rats. Stroke. 1994;25:869–875. doi: 10.1161/01.str.25.4.869. [DOI] [PubMed] [Google Scholar]
- Chu K, Lee ST, Koo JS, Jung KH, Kim EH, Sinn DI, Kim JM, Ko SY, Kim SJ, Song EC, Kim M, Roh JK. Peroxisome proliferator-activated receptor-gamma-agonist, rosiglitazone, promotes angiogenesis after focal cerebral ischemia. Brain Res. 2006;1093:208–218. doi: 10.1016/j.brainres.2006.03.114. [DOI] [PubMed] [Google Scholar]
- Cimini A, Benedetti E, Cristiano L, Sebastiani P, D'Amico MA, D'Angelo B, Di Loreto S. Expression of peroxisome proliferator-activated receptors (PPARs) and retinoic acid receptors (RXRs) in rat cortical neurons. Neuroscience. 2005;130:325–337. doi: 10.1016/j.neuroscience.2004.09.043. [DOI] [PubMed] [Google Scholar]
- Clark RK, Lee EV, White RF, Jonak ZL, Feuerstein GZ, Barone FC. Reperfusion following focal stroke hastens inflammation and resolution of ischemic injured tissue. Brain Res. Bull. 1994;35:387–392. doi: 10.1016/0361-9230(94)90119-8. [DOI] [PubMed] [Google Scholar]
- Clark WM, Lauten JD, Lessov N, Woodward W, Coull BM. The influence of antiadhesion therapies on leukocyte subset accumulation in central nervous system ischemia in rats. J. Mol. Neurosci. 1995;6:43–50. doi: 10.1007/BF02736758. [DOI] [PubMed] [Google Scholar]
- Clark WM, Madden KP, Rothlein R, Zivin JA. Reduction of central nervous system ischemic injury by monoclonal antibody to intercellular adhesion molecule. J. Neurosurg. 1991;75:623–627. doi: 10.3171/jns.1991.75.4.0623. [DOI] [PubMed] [Google Scholar]
- Collino M, Aragno M, Mastrocola R, Gallicchio M, Rosa AC, Dianzani C, Danni O, Thiemermann C, Fantozzi R. Modulation of the oxidative stress and inflammatory response by PPAR-gamma agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur. J. Pharmacol. 2006;530:70–80. doi: 10.1016/j.ejphar.2005.11.049. [DOI] [PubMed] [Google Scholar]
- Collino M, Aragno M, Mastrocola R, Benetti E, Gallicchio M, Dianzani C, Danni O, Thiemermann C, Fantozzi R. Oxidative stress and inflammatory response evoked by transient cerebral ischemia/reperfusion: effects of the PPAR-alpha agonist WY14643. Free Rad. Biol. Med. 2006;41:579–589. doi: 10.1016/j.freeradbiomed.2006.04.030. [DOI] [PubMed] [Google Scholar]
- Connolly ES, Jr., Winfree CJ, Springer TA, Naka Y, Liao H, Yan SD, Stern DM, Solomon RA, Gutierrez-Ramos JC, Pinsky DJ. Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J. Clin. Invest. 1996;97:209–216. doi: 10.1172/JCI118392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristiano L, Cimini A, Moreno S, Ragnelli AM, Paola, Ceru M. Peroxisome proliferator-activated receptors (PPARs) and related transcription factors in differentiating astrocyte cultures. Neuroscience. 2005;131:577–587. doi: 10.1016/j.neuroscience.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Cullingford TE, Bhakoo K, Peuchen S, Dolphin CT, Patel R, Clark JB. Distribution of mRNAs encoding the peroxisome proliferator-activated receptor alpha, beta, and gamma and the retinoid X receptor alpha, beta, and gamma in rat central nervous system. J. Neurochem. 1998;70:1366–1375. doi: 10.1046/j.1471-4159.1998.70041366.x. [DOI] [PubMed] [Google Scholar]
- Danton GH, Dietrich WD. Inflammatory mechanisms after ischemia and stroke. J. Neuropathol. Exp. Neurol. 2003;62:127–136. doi: 10.1093/jnen/62.2.127. Review. [DOI] [PubMed] [Google Scholar]
- Davenpeck KL, Gauthier TW, Albertine KH, Lefer AM. Role of P-selectin in microvascular leukocyte-endothelial interaction in splanchnic ischemia-reperfusion. Am. J. Physiol. 1994;267:H622–630. doi: 10.1152/ajpheart.1994.267.2.H622. [DOI] [PubMed] [Google Scholar]
- Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2002;2:748–759. doi: 10.1038/nri912. Review. [DOI] [PubMed] [Google Scholar]
- Dello Russo C, Gavrilyuk V, Weinberg G, Almeida A, Bolanos JP, Palmer J, Pelligrino D, Galea E, Feinstein DL. Peroxisome proliferator-activated receptor gamma thiazolidinedione agonists increase glucose metabolism in astrocytes. J. Biol. Chem. 2003;278:5828–5836. doi: 10.1074/jbc.M208132200. [DOI] [PubMed] [Google Scholar]
- Deplanque D, Gele P, Petrault O, Six I, Furman C, Bouly M, Nion S, Dupuis B, Leys D, Fruchart JC, Cecchelli R, Staels B, Duriez P, Bordet R. Peroxisome proliferator-activated receptor-alpha activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. J. Neurosci. 2003;23:6264–6271. doi: 10.1523/JNEUROSCI.23-15-06264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr. Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature. 1996;384:39–43. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. Review. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Beilharz E, Sirimanne E, Lawlor P, Williams C, Bravo R, Gluckman P. Immediate-early gene protein expression in neurons undergoing delayed death, but not necrosis, following hypoxic-ischaemic injury to the young rat brain. Brain Res. Mol. Brain Res. 1994;25:19–33. doi: 10.1016/0169-328x(94)90274-7. [DOI] [PubMed] [Google Scholar]
- Dustin ML, Rothlein R, Bhan AK, Dinarello CA, Springer TA. Induction by IL 1 and interferon-gamma: tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1) J. Immunol. 1986;137:245–254. [PubMed] [Google Scholar]
- Eppihimer MJ, Wolitzky B, Anderson DC, Labow MA, Granger DN. Heterogeneity of expression of E- and P-selectins in vivo. Circ. Res. 1996;79:560–569. doi: 10.1161/01.res.79.3.560. [DOI] [PubMed] [Google Scholar]
- Escher P, Wahli W. Peroxisome proliferator-activated receptors: insight into multiple cellular functions. Mutat. Res. 2000;448:121–138. doi: 10.1016/s0027-5107(99)00231-6. Review. [DOI] [PubMed] [Google Scholar]
- Feinstein DL, Spagnolo A, Akar C, Weinberg G, Murphy P, Gavrilyuk V, Dello Russo C. Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem. Pharmacol. 2005;70:177–188. doi: 10.1016/j.bcp.2005.03.033. Review. [DOI] [PubMed] [Google Scholar]
- Garcia JH, Liu KF, Yoshida Y, Lian J, Chen S, Del Zeppo GJ. Influx of leukocytes and platelets in an evolving brain infarct (Wistar Rat) Am. J. Pathol. 1994;144:188–199. [PMC free article] [PubMed] [Google Scholar]
- Garcia-Criado FJ, Toledo-Pereyra LH, Lopez-Neblina F, Phillips ML, Paez-Rollys A, Misawa K. Role of P-selectin in total hepatic ischemia and reperfusion. J. Am. Coll. Surg. 1995;181:327–334. [PubMed] [Google Scholar]
- Gerszten RE, Garcia-Zepeda EA, Lim YC, Yoshida M, Ding HA, Gimbrone MA, Jr., Luster AD, Luscinskas FW, Rosenzweig A. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. 1999;398:718–723. doi: 10.1038/19546. [DOI] [PubMed] [Google Scholar]
- Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat. Rev. Immunol. 2006;6:44–55. doi: 10.1038/nri1748. Review. [DOI] [PubMed] [Google Scholar]
- Gosset P, Charbonnier AS, Delerive P, Fontaine J, Staels B, Pestel J, Tonnel AB, Trottein F. Peroxisome proliferator-activated receptor gamma activators affect the ma,turation of human monocyte-derived dendritic cells. Eur. J. Immunol. 2001;31:2857–2865. doi: 10.1002/1521-4141(2001010)31:10<2857::aid-immu2857>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Hata R, Gass P, Mies G, Wiessner C, Hossmann KA. Attenuated c-fos mRNA induction after middle cerebral artery occlusion in CREB knockout mice does not modulate focal ischemic injury. J. Cereb. Blood Flow Metab. 1998;18:1325–1335. doi: 10.1097/00004647-199812000-00007. [DOI] [PubMed] [Google Scholar]
- Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, del Zoppo GJ. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J. Cereb. Blood. Flow. Metab. 1999;19:624–633. doi: 10.1097/00004647-199906000-00005. [DOI] [PubMed] [Google Scholar]
- Hu BR, Liu CL, Park DJ. Alteration of MAP kinase pathways after transient forebrain ischemia. J. Cereb. Blood Flow Metab. 2000;20:1089–1095. doi: 10.1097/00004647-200007000-00008. [DOI] [PubMed] [Google Scholar]
- Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, Moskowitz MA. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J. Cereb. Blood. Flow. Metab. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 1997;20:132–139. doi: 10.1016/s0166-2236(96)10074-6. Review. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Alexander M. Cerebral ischemia and inflammation. Curr. Opin. Neurol. 2001;14:89–94. doi: 10.1097/00019052-200102000-00014. Review. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Salkowski CA, Zhang F, Aber T, Nagayama M, Vogel SN, Ross ME. The transcription factor interferon regulatory factor 1 is expressed after cerebral ischemia and contributes to ischemic brain injury. J. Exp. Med. 1999;189:719–727. doi: 10.1084/jem.189.4.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Xu S, Casey R, Ross ME. Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J. Cereb. Blood. Flow. Metab. 1995;15:378–384. doi: 10.1038/jcbfm.1995.47. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J. Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iademarco MF, Barks JL, Dean DC. Regulation of vascular cell adhesion molecule-1 expression by IL-4 and TNF-alpha in cultured endothelial cells. J. Clin. Invest. 1995;95:264–271. doi: 10.1172/JCI117650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwashita A, Muramatsu Y, Yamazaki T, Muramoto M, Kita Y, Yamazaki S, Mihara K, Moriguchi A, Matsuoka N. Neuroprotective Efficacy of Peroxisome Proliferator-Activated Receptor delta (PPAR-{delta}) Selective Agonists, L-165041 and GW501516, in Vitro and in Vivo. J. Pharmacol. Exp. Ther. 2006 doi: 10.1124/jpet.106.115758. 2006 Dec 13 Epub. [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- Johansson IM, Wester P, Hakova M, Gu W, Seckl JR, Olsson T. Early and delayed induction of immediate early gene expression in a novel focal cerebral ischemia model in the rat. Eur. J. Neurosci. 2000;12:3615–3625. doi: 10.1046/j.1460-9568.2000.00252.x. [DOI] [PubMed] [Google Scholar]
- Jones DC, Manning BM, Daynes RA. A role for the peroxisome proliferator-activated receptor alpha in T-cell physiology and ageing immunobiology. Proc. Nutr. Soc. 2002;61:363–369. doi: 10.1079/pns2002173. Review. [DOI] [PubMed] [Google Scholar]
- Justicia C, Gabriel C, Planas AM. Activation of the JAK/STAT pathway following transient focal cerebral ischemia: signaling through Jak1 and Stat3 in astrocytes. Glia. 2000;30:253–270. doi: 10.1002/(sici)1098-1136(200005)30:3<253::aid-glia5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Juttler E, Kohrmann M, Schellinger PD. Therapy for early reperfusion after stroke. Nat. Clin. Pract. Cardiovasc. Med. 2006;3:656–663. doi: 10.1038/ncpcardio0721. Review. [DOI] [PubMed] [Google Scholar]
- Kagansky N, Levy S, Knobler H. The role of hyperglycemia in acute stroke. Arch. Neurol. 2001;58:1209–1212. doi: 10.1001/archneur.58.8.1209. Review. [DOI] [PubMed] [Google Scholar]
- Kapadia R, Tureyen K, Bowen KK, Kalluri H, Johnson PF, Vemuganti R. Decreased brain damage and curtailed inflammation in transcription factor CCAAT/enhancer binding protein beta knockout mice following transient focal cerebral ischemia. J. Neurochem. 2006;98:1718–1731. doi: 10.1111/j.1471-4159.2006.04056.x. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Mabuchi T, Yagita Y, Ohtsuki T, Hori M, Yanagihara T. Deficiency of intercellular adhesion molecule 1 attenuates microcirculatory disturbance and infarction size in focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1998;18:1336–1345. doi: 10.1097/00004647-199812000-00008. [DOI] [PubMed] [Google Scholar]
- Kim JS, Gautam SC, Chopp M, Zaloga C, Jones ML, Ward PA, Welch KM. Expression of monocyte chemoattractant protein-1 and macrophage inflammatory protein-1 after focal cerebral ischemia in the rat. J. Neuroimmunol. 1995;56:127–134. doi: 10.1016/0165-5728(94)00138-e. [DOI] [PubMed] [Google Scholar]
- Kindy MS, Carney JP, Dempsey RJ, Carney JM. Ischemic induction of protooncogene expression in gerbil brain. J. Mol. Neurosci. 1991;2:217–228. [PubMed] [Google Scholar]
- Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, Umesono K, Evans RM. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. U S A. 1994;91:7355–7359. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanek PM, Hallenbeck JM. Polymorphonuclear leukocytes and monocytes/macrophages in the pathogenesis of cerebral ischemia and stroke. Stroke. 1992;23:1367–1379. doi: 10.1161/01.str.23.9.1367. [DOI] [PubMed] [Google Scholar]
- Kubes P, Jutila M, Payne D. Therapeutic potential of inhibiting leukocyte rolling in ischemia/reperfusion. J. Clin. Invest. 1995;95:2510–2519. doi: 10.1172/JCI117952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz A, Anrather J, Zhou P, Orio M, Iadecola C. Cyclooxygenase-2 does not contribute to postischemic production of reactive oxygen species. J. Cereb. Blood. Flow. Metab. 2006 doi: 10.1038/sj.jcbfm.9600369. Jul 5. Epub. [DOI] [PubMed] [Google Scholar]
- Laudet V, Hanni C, Coll J, Catzeflis F, Stehelin D. Evolution of the nuclear receptor gene superfamily. EMBO. J. 1992;11:1003–1013. doi: 10.1002/j.1460-2075.1992.tb05139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DW, Andersen JK. Role of HIF-1 in iron regulation: potential therapeutic strategy for neurodegenerative disorders. Curr. Mol. Med. 2006;6:883–893. doi: 10.2174/156652406779010849. Review. [DOI] [PubMed] [Google Scholar]
- Lee J, Kosaras B, Aleyasin H, Han JA, Park DS, Ratan RR, Kowall NW, Ferrante RJ, Lee SW, Ryu H. Role of cyclooxygenase-2 induction by transcription factor Sp1 and Sp3 in neuronal oxidative and DNA damage response. FASEB. J. 2006;20:2375–2377. doi: 10.1096/fj.06-5957fje. [DOI] [PubMed] [Google Scholar]
- Lee J, Reding M. Effects of Thiazolidinediones on Stroke Recovery: A Case-Matched Controlled Study. Neurochem. Res. 2006 doi: 10.1007/s11064-006-9138-3. Sep 8. Epub. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 1997;272:3406–3410. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J. Biol. Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- Li M, Pascual G, Glass CK. Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. Mol. Cell. Biol. 2000;20:4699–4707. doi: 10.1128/mcb.20.13.4699-4707.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TN, Cheung WM, Wu JS, Chen JJ, Lin H, Chen JJ, Liou JY, Shyue SK, Wu KK. 15d-prostaglandin J2 protects brain from ischemia-reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 2006;26:481–487. doi: 10.1161/01.ATV.0000201933.53964.5b. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol. Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. Review. [DOI] [PubMed] [Google Scholar]
- Liu T, Clark RK, McDonnell PC, Young PR, White RF, Barone FC, Feuerstein GZ. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke. 1994;25:1481–1488. doi: 10.1161/01.str.25.7.1481. [DOI] [PubMed] [Google Scholar]
- Lu A, Tang Y, Ran R, Clark JF, Aronow BJ, Sharp FR. Genomics of the periinfarction cortex after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2003;23:786–810. doi: 10.1097/01.WCB.0000062340.80057.06. [DOI] [PubMed] [Google Scholar]
- Luo Y, Yin W, Signore AP, Zhang F, Hong Z, Wang S, Graham SH, Chen J. Neuroprotection against focal ischemic brain injury by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. J. Neurochem. 2006;97:435–448. doi: 10.1111/j.1471-4159.2006.03758.x. [DOI] [PubMed] [Google Scholar]
- Maeda K, Hata R, Gillardon F, Hossmann K. Aggravation of brain injury after transient focal ischemia in p53-deficient mice. Mol. Brain Res. 2001;88:54–61. doi: 10.1016/s0169-328x(01)00017-1. [DOI] [PubMed] [Google Scholar]
- Maestre-Moreno JF, Fernandez-Perez MD, Arnaiz-Urrutia C, Minguez A, Navarrete-Navarro P, Martinez-Bosch J. Thrombolysis in stroke: inappropriate consideration of the bwindow period Q as the time available. Rev. Neurol. 2005;40:274–278. [PubMed] [Google Scholar]
- Matsuo Y, Onodera H, Shiga Y, Nakamura M, Ninomiya M, Kihara T, Kogure K. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke. 1994;25:1469–1475. doi: 10.1161/01.str.25.7.1469. [DOI] [PubMed] [Google Scholar]
- Minghetti L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004;63:901–910. doi: 10.1093/jnen/63.9.901. [DOI] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- Moreno S, Farioli-Vecchioli S, Ceru MP. Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience. 2004;123:131–145. doi: 10.1016/j.neuroscience.2003.08.064. [DOI] [PubMed] [Google Scholar]
- Mori E, del Zoppo GJ, Chambers JD, Copeland BR, Arfors KE. Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons. Stroke. 1992;23:712–718. doi: 10.1161/01.str.23.5.712. [DOI] [PubMed] [Google Scholar]
- Mukaida N, Morita M, Ishikawa Y, Rice N, Okamoto S, Kasahara T, Matsushima K. Novel mechanism of glucocorticoid-mediated gene repression. Nuclear factor-kappa B is target for glucocorticoid-mediated interleukin 8 gene repression. J. Biol. Chem. 1994;269:13289–13295. [PubMed] [Google Scholar]
- Nelson PJ, Kim HT, Manning WC, Goralski TJ, Krensky AM. Genomic organization and transcriptional regulation of the RANTES chemokine gene. J. Immunol. 1993;151:2601–2612. [PubMed] [Google Scholar]
- Nogawa S, Forster C, Zhang F, Nagayama M, Ross ME, Iadecola C. Interaction between inducible nitric oxide synthase and cyclooxygenase-2 after cerebral ischemia. Proc. Natl. Acad. Sci. U S A. 1998;95:10966–10971. doi: 10.1073/pnas.95.18.10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, Karjalainen-Lindsberg ML, Weih F, Frank N, Schwaninger M, Koistinaho J. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke. 2004;35:987–991. doi: 10.1161/01.STR.0000120732.45951.26. [DOI] [PubMed] [Google Scholar]
- Okada Y, Copeland BR, Mori E, Tung MM, Thomas WS, del Zoppo GJ. P-selectin and intercellular adhesion molecule-1 expression after focal brain ischemia and reperfusion. Stroke. 1994;25:202–211. doi: 10.1161/01.str.25.1.202. [DOI] [PubMed] [Google Scholar]
- O'Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109:S121–131. doi: 10.1016/s0092-8674(02)00701-8. Review. [DOI] [PubMed] [Google Scholar]
- Ou Z, Zhao X, Labiche LA, Strong R, Grotta JC, Herrmann O, Aronowski J. Neuronal expression of peroxisome proliferator-activated receptor-gamma (PPARgamma) and 15d-prostaglandin J2--mediated protection of brain after experimental cerebral ischemia in rat. Brain Res. 2006;1096:196–203. doi: 10.1016/j.brainres.2006.04.062. [DOI] [PubMed] [Google Scholar]
- Park EJ, Park SY, Joe EH, Jou I. 15d-PGJ2 and rosiglitazone suppress Janus kinase-STAT inflammatory signaling through induction of suppressor of cytokine signaling 1 (SOCS1) and SOCS3 in glia. J. Biol. Chem. 2003;278:14747–14752. doi: 10.1074/jbc.M210819200. [DOI] [PubMed] [Google Scholar]
- Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penberthy TW, Jiang Y, Luscinskas FW, Graves DT. MCP-1-stimulated monocytes preferentially utilize beta 2-integrins to migrate on laminin and fibronectin. Am. J. Physiol. 1995;269:C60–68. doi: 10.1152/ajpcell.1995.269.1.C60. [DOI] [PubMed] [Google Scholar]
- Pereira MP, Hurtado O, Cardenas A, Alonso-Escolano D, Bosca L, Vivancos J, Nombela F, Leza JC, Lorenzo P, Lizasoain I, Moro MA. The nonthiazolidinedione PPARgamma agonist L-796,449 is neuroprotective in experimental stroke. J. Neuropathol. Exp. Neurol. 2005;64:797–805. doi: 10.1097/01.jnen.0000178852.83680.3c. [DOI] [PubMed] [Google Scholar]
- Pereira MP, Hurtado O, Cardenas A, Bosca L, Castillo J, Davalos A, Vivancos J, Serena J, Lorenzo P, Lizasoain I, Moro MA. Rosiglitazone and 15-deoxy-Delta12,14-prostaglandin J2 cause potent neuroprotection after experimental stroke through noncompletely overlapping mechanisms. J. Cereb. Blood Flow Metab. 2006;26:218–229. doi: 10.1038/sj.jcbfm.9600182. [DOI] [PubMed] [Google Scholar]
- Planas AM, Soriano MA, Berruezo M, Justicia C, Estrada A, Pitarch S, Ferrer I. Induction of Stat3, a signal transducer and transcription factor, in reactive microglia following transient focal cerebral ischaemia. Eur. J. Neurosci. 1996;8:2612–2618. doi: 10.1111/j.1460-9568.1996.tb01556.x. [DOI] [PubMed] [Google Scholar]
- Raivich G, Behrens A. Role of the AP-1 transcription factor c-Jun in developing, adult and injured brain. Prog. Neurobiol. 2006;78:347–363. doi: 10.1016/j.pneurobio.2006.03.006. Review. [DOI] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Saluja I, Granneman JG, Skoff RP. PPAR delta agonists stimulate oligodendrocyte differentiation in tissue culture. Glia. 2001;33:191–204. [PubMed] [Google Scholar]
- Sasaki T, Kitagawa K, Sugiura S, Omura-Matsuoka E, Tanaka S, Yagita Y, Okano H, Matsumoto M, Hori M. Implication of cyclooxygenase-2 on enhanced proliferation of neural progenitor cells in the adult mouse hippocampus after ischemia. J. Neurosci. Res. 2003;72:461–471. doi: 10.1002/jnr.10595. [DOI] [PubMed] [Google Scholar]
- Satriotomo I, Bowen KK, Vemuganti R. JAK2 and STAT3 activation contributes to neuronal damage following transient focal cerebral ischemia. J. Neurochem. 2006;98:1353–1368. doi: 10.1111/j.1471-4159.2006.04051.x. [DOI] [PubMed] [Google Scholar]
- Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- Schielke GP, Yang GY, Shivers BD, Betz AL. Reduced ischemic brain injury in interleukin-1 beta converting enzyme-deficient mice. J. Cereb. Blood Flow Metab. 1998;18:180–185. doi: 10.1097/00004647-199802000-00009. [DOI] [PubMed] [Google Scholar]
- Setoguchi K, Misaki Y, Terauchi Y, Yamauchi T, Kawahata K, Kadowaki T, Yamamoto K. Peroxisome proliferator-activated receptor-gamma haploinsufficiency enhances B cell proliferative responses and exacerbates experimentally induced arthritis. J. Clin. Invest. 2001;108:1667–1675. doi: 10.1172/JCI13202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiga Y, Onodera H, Kogure K, Yamasaki Y, Yashima Y, Syozuhara H, Sendo F. Neutrophil as a mediator of ischemic edema formation in the brain. Neurosci. Lett. 1991;125:110–112. doi: 10.1016/0304-3940(91)90003-c. [DOI] [PubMed] [Google Scholar]
- Shimazu T, Inoue I, Araki N, Asano Y, Sawada M, Furuya D, Nagoya H, Greenberg JH. A peroxisome proliferator-activated receptor-gamma agonist reduces infarct size in transient but not in permanent ischemia. Stroke. 2005;36:353–359. doi: 10.1161/01.STR.0000152271.21943.a2. 2005. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Bernaudin M. HIF1 and oxygen sensing in the brain. Nat. Rev. Neurosci. 2004;5:437–448. doi: 10.1038/nrn1408. Review. [DOI] [PubMed] [Google Scholar]
- Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu. Rev. Cell. Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. Review. [DOI] [PubMed] [Google Scholar]
- Sood V, Colleran K, Burge MR. Thiazolidinediones: a comparative review of approved uses. Diabetes Technol. Ther. 2000;2:429–440. doi: 10.1089/15209150050194297. Review. [DOI] [PubMed] [Google Scholar]
- Soriano SG, Lipton SA, Wang YF, Xiao M, Springer TA, Gutierrez-Ramos JC, Hickey PR. Intercellular adhesion molecule-1-deficient mice are less susceptible to cerebral ischemia-reperfusion injury. Ann. Neurol. 1996;39:618–624. doi: 10.1002/ana.410390511. [DOI] [PubMed] [Google Scholar]
- Soriano MA, Tessier M, Certa U, Gill R. Parallel gene expression monitoring using oligonucleotide probe arrays of multiple transcripts with an animal model of focal ischemia. J. Cereb. Blood Flow Metab. 2000;20:1045–1055. doi: 10.1097/00004647-200007000-00004. [DOI] [PubMed] [Google Scholar]
- Stephenson D, Yin T, Smalstig EB, Hsu MA, Panetta J, Little S, Clemens J. Transcription factor nuclear factor-kappa B is activated in neurons after focal cerebral ischemia. J. Cereb. Blood. Flow. Metab. 2000;20:592–603. doi: 10.1097/00004647-200003000-00017. [DOI] [PubMed] [Google Scholar]
- Stoll G, Jander S, Schroeter M. Inflammation and glial responses in ischemic brain lesions. Prog. Neurobiol. 1998;56:149–171. doi: 10.1016/s0301-0082(98)00034-3. Review. [DOI] [PubMed] [Google Scholar]
- Storer PD, Xu J, Chavis J, Drew PD. Peroxisome proliferator-activated receptor-gamma agonists inhibit the activation of microglia and astrocytes: implications for multiple sclerosis. J. Neuroimmunol. 2005;161:113–122. doi: 10.1016/j.jneuroim.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G, Glass CK. 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc. Natl. Acad. Sci. U S A. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararajan S, Gamboa JL, Victor NA, Wanderi EW, Lust WD, Landreth GE. Peroxisome proliferator-activated receptor-gamma ligands reduce inflammation and infarction size in transient focal ischemia. Neuroscience. 2005;130:685–696. doi: 10.1016/j.neuroscience.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Tanaka K, Nogawa S, Dembo T, Kosakai A, Fukuuchi Y. Phosphorylation of signal transducer and activator of transcription-3 (Stat3) after focal cerebral ischemia in rats. Exp Neurol. 2001;170:63–71. doi: 10.1006/exnr.2001.7701. [DOI] [PubMed] [Google Scholar]
- Takagi Y, Harada J, Chiarugi A, Moskowitz MA. STAT1 is activated in neurons after ischemia and contributes to ischemic brain injury. J. Cereb. Blood Flow Metab. 2002;22:1311–1318. doi: 10.1097/01.WCB.0000034148.72481.F4. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Nogawa S, Ito D, Suzuki S, Dembo T, Kosakai A, Fukuuchi Y. Activated phosphorylation of cyclic AMP response element binding protein is associated with preservation of striatal neurons after focal cerebral ischemia in the rat. Neuroscience. 2000a;100:345–354. doi: 10.1016/s0306-4522(00)00289-x. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Nogawa S, Nagata E, Ito D, Suzuki S, Dembo T, Kosakai A, Fukuuchi Y. Persistent CREB phosphorylation with protection of hippocampal CA1 pyramidal neurons following temporary occlusion of the middle cerebral artery in the rat. Exp. Neurol. 2000b;161:462–471. doi: 10.1006/exnr.1999.7313. [DOI] [PubMed] [Google Scholar]
- Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J. Neurosci. 2001;21:2580–2588. doi: 10.1523/JNEUROSCI.21-08-02580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against n-methyl-d-aspartate neurotoxicity by inhibiting microglia. J. Immunol. 2001;166:7527–7233. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- Tureyen K, Kapadia R, Bowen K, Satriotomo I, Liang J, Feinstein D, Vemuganti R. Peroxisome proliferators-activated receptor-γ agonists induce neuroprotection following transient focal ischemia in normotensive, normoglycemic as well as hypertensive and type-2 diabetic rodents. J. Neurochem. 2007 doi: 10.1111/j.1471-4159.2006.04376.x. In Press. [DOI] [PubMed] [Google Scholar]
- Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T, Matsunaga K, Fukushima J, Kawamoto S, Ishigatsubo Y, Okubo T. NF-kappa B and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J. Immunol. 1994;153:2052–2063. [PubMed] [Google Scholar]
- Van Beek J, Elward K, Gasque P. Activation of complement in the central nervous system: roles in neurodegeneration and neuroprotection. Ann. N. Y. Acad. Sci. 2003;992:56–71. doi: 10.1111/j.1749-6632.2003.tb03138.x. [DOI] [PubMed] [Google Scholar]
- Van de Stolpe A, Caldenhoven E, Stade BG, Koenderman L, Raaijmakers JA, Johnson JP, van der Saag PT. 12-O-tetradecanoylphorbol-13-acetate- and tumor necrosis factor alpha-mediated induction of intercellular adhesion molecule-1 is inhibited by dexamethasone. Functional analysis of the human intercellular adhesion molecular-1 promoter. J. Biol. Chem. 1994;269:6185–6192. [PubMed] [Google Scholar]
- Vemuganti R, Bowen KK, Dhodda VK, Song G, Franklin JL, Gavva NR, Dempsey RJ. Gene expression analysis of spontaneously hypertensive rat cerebral cortex following transient focal ischemia. J. Neurochem. 2002;83:1072–1086. doi: 10.1046/j.1471-4159.2002.01208.x. [DOI] [PubMed] [Google Scholar]
- Vemuganti R, Dempsey RJ, Bowen KK. Inhibition of intercellular adhesion molecule-1 protein expression by antisense oligonucleotides is neuroprotective after transient middle cerebral artery occlusion in rat. Stroke. 2004;35:179–184. doi: 10.1161/01.STR.0000106479.53235.3E. [DOI] [PubMed] [Google Scholar]
- Vemuganti R, Tureyen K, Brooks N, Bowen K, Liang J, Svaren J. Transcription factor NGFI-A/EGR1 induction mediates postischemic inflammatory gene expression and brain damage in rodents. Stroke. 2007;38:556. [Google Scholar]
- Victor NA, Wanderi EW, Gamboa J, Zhao X, Aronowski J, Deininger K, Lust WD, Landreth GE, Sundararajan S. Altered PPARgamma expression and activation after transient focal ischemia in rats. Eur. J. Neurosci. 2006;24:1653–1663. doi: 10.1111/j.1460-9568.2006.05037.x. [DOI] [PubMed] [Google Scholar]
- Vodovotz Y, Kwon NS, Pospischil M, Manning J, Paik J, Nathan C. Inactivation of nitric oxide synthase after prolonged incubation of mouse macrophages with IFN-gamma and bacterial lipopolysaccharide. J. Immunol. 1994;152:4110–4118. [PubMed] [Google Scholar]
- Wang X, Yue TL, Barone FC, White RF, Gagnon RC, Feuerstein GZ. Concomitant cortical expression of TNF-alpha and IL-1 beta mRNAs follows early response gene expression in transient focal ischemia. Mol. Chem. Neuropathol. 1994;23:103–114. doi: 10.1007/BF02815404. [DOI] [PubMed] [Google Scholar]
- Wang X, Yue TL, Barone FC, Feuerstein GZ. Monocyte chemoattractant protein-1 messenger RNA expression in rat ischemic cortex. Stroke. 1995;26:661–665. doi: 10.1161/01.str.26.4.661. [DOI] [PubMed] [Google Scholar]
- Welch JS, Ricote M, Akiyama TE, Gonzalez FJ, Glass CK. PPARgamma and PPARdelta negatively regulate specific subsets of lipopolysaccharide and IFN-gamma target genes in macrophages. Proc. Natl. Acad. Sci. U S A. 100:6712–6717. doi: 10.1073/pnas.1031789100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyrich AS, Ma XY, Lefer DJ, Albertine KH, Lefer AM. In vivo neutralization of P-selectin protects feline heart and endothelium in myocardial ischemia and reperfusion injury. J. Clin. Invest. 1993;91:2620–2629. doi: 10.1172/JCI116501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AJ, Hale SL, Moffett JR, Dave JR, Elliott PJ, Adams J, Tortella FC. Delayed treatment with MLN519 reduces infarction and associated neurologic deficit caused by focal ischemic brain injury in rats via anti-inflammatory mechanisms involving nuclear factor-kappaB activation, gliosis, and leukocyte infiltration. J. Cereb. Blood Flow Metab. 2003;23:75–87. doi: 10.1097/01.WCB.0000039285.37737.C2. [DOI] [PubMed] [Google Scholar]
- Willmot M, Gibson C, Gray L, Murphy S, Bath P. Nitric oxide synthase inhibitors in experimental ischemic stroke and their effects on infarct size and cerebral blood flow: a systematic review. Free Rad. Biol. Med. 2005;39:412–425. doi: 10.1016/j.freeradbiomed.2005.03.028. [DOI] [PubMed] [Google Scholar]
- Woodburn VL, Hayward NJ, Poat JA, Woodruff GN, Hughes J. The effect of dizocilpine and enadoline on immediate early gene expression in the gerbil global ischaemia model. Neuropharmacology. 1993;32:1047–1059. doi: 10.1016/0028-3908(93)90070-j. [DOI] [PubMed] [Google Scholar]
- Woods JW, Tanen M, Figueroa DJ, Biswas C, Zycband E, Moller DE, Austin CP, Berger JP. Localization of PPARdelta in murine central nervous system: expression in oligodendrocytes and neurons. Brain Res. 2003;975:10–21. doi: 10.1016/s0006-8993(03)02515-0. [DOI] [PubMed] [Google Scholar]
- Yan SF, Lu J, Zou YS, Soh-Won J, Cohen DM, Buttrick PM, Cooper DR, Steinberg SF, Mackman N, Pinsky DJ, Stern DM. Hypoxia-associated induction of early growth response-1 gene expression. J. Biol. Chem. 1999;274:15030–15040. doi: 10.1074/jbc.274.21.15030. [DOI] [PubMed] [Google Scholar]
- Yan SF, Fujita T, Lu J, Okada K, Shan Zou Y, Mackman N, Pinsky DJ, Stern DM. Egr-1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nat. Med. 2000;6:1355–1361. doi: 10.1038/82168. [DOI] [PubMed] [Google Scholar]
- Yanaka K, Camarata PJ, Spellman SR, McCarthy JB, Furcht LT, Low WC. Antagonism of leukocyte adherence by synthetic fibronectin peptide V in a rat model of transient focal cerebral ischemia. Neurosurgery. 1997;40:557–563. doi: 10.1097/00006123-199703000-00026. [DOI] [PubMed] [Google Scholar]
- Yrjenheikki J, Keinanen R, Pelikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc. Natl. Acad. Sci. USA. 1998;95:15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc. Natl. Acad. Sci. U.S.A. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RL, Chopp M, Jiang N, Tang WX, Prostak J, Manning AM, Anderson DC. Anti-intercellular adhesion molecule-1 antibody reduces ischemic cell damage after transient but not permanent middle cerebral artery occlusion in the Wistar rat. Stroke. 1995;26:1438–1442. doi: 10.1161/01.str.26.8.1438. [DOI] [PubMed] [Google Scholar]
- Zhang RL, Chopp M, Li Y, Zaloga C, Jiang N, Jones ML, Miyasaka M, Ward PA. Anti-ICAM-1 antibody reduces ischemic cell damage after transient middle cerebral artery occlusion in rat. Neurology. 1994;44:1747–1751. doi: 10.1212/wnl.44.9.1747. [DOI] [PubMed] [Google Scholar]
- Zhao X, Zhang Y, Strong R, Grotta JC, Aronowski J. 15d-Prostaglandin J2 activates peroxisome proliferator-activated receptor-gamma, promotes expression of catalase, and reduces inflammation, behavioral dysfunction, and neuronal loss after intracerebral hemorrhage in rats. J. Cereb. Blood Flow Metab. 2006;26:811–820. doi: 10.1038/sj.jcbfm.9600233. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Patzer A, Gohlke P, Herdegen T, Culman J. The intracerebral application of the PPARgamma-ligand pioglitazone confers neuroprotection against focal ischaemia in the rat brain. Eur. J. Neurosci. 2005;22:278–282. doi: 10.1111/j.1460-9568.2005.04200.x. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Patzer A, Herdegen T, Gohlke P, Culman J. Activation of cerebral peroxisome proliferator-activated receptors gamma promotes neuroprotection by attenuation of neuronal cyclooxygenase-2 overexpression after focal cerebral ischemia in rats. FASEB. J. 2006;20:1162–1175. doi: 10.1096/fj.05-5007com. [DOI] [PubMed] [Google Scholar]
- del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000;10:95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]