

Abstract
Two major DNA repair pathways, nonhomologous end-joining (NHEJ) and homologous recombination (HR), repair double-stranded DNA breaks (DSBs) in all eukaryotes. Additionally, several alternative end-joining pathways (or subpathways) have been reported that characteristically use short-sequence homologies at the DNA ends to facilitate joining. How a cell chooses which DNA repair pathway to use (at any particular DSB) is a central and largely unanswered question. For one type of DSB, there is apparently no choice. DSBs mediated by the lymphocyte-specific recombination activating gene (RAG) endonuclease are repaired virtually exclusively by NHEJ. Here we demonstrate that non-RAG-mediated DSBs can be similarly forced into the NHEJ pathway by physical association with the RAG endonuclease.
Keywords: VDJ recombination, DNA-dependent protein kinase
VDJ recombination is the molecular mechanism that provides for limitless antigen receptor diversity in developing lymphocytes by assembling immune receptor genes from their disparate component gene segments during lymphocyte development (1). Recombination is initiated by lymphocyte-specific recombination activating genes (RAG1/RAG2) (2, 3) generating double-stranded DNA breaks (DSBs) adjacent to immune receptor coding segments (3). Because introducing DSBs into one's genome can have dire consequences, VDJ recombination is highly regulated. Regulation is achieved by several mechanisms: (i) cell stage-specific expression of RAG mRNA, (ii) targeted degradation of RAG2 at the G1/S border mediated by RAG2's C terminus, and (iii) limited access of recombination signal sequences (RSSs) in RAG-expressing cells (4–9). Although two major double-stranded break repair pathways exist in higher eukaryotes, homologous recombination (HR) (error-free) and nonhomologous end-joining (NHEJ) pathways (error-prone), RAG-mediated breaks are resolved exclusively by NHEJ (10). The error-prone nature of NHEJ is beneficial during VDJ recombination because its inherent imprecision contributes substantially to the diversification of antigen receptor exons generated by VDJ recombination.
It is unclear how RAG breaks are restricted to the NHEJ pathway. Several mechanisms may contribute to this restriction. Limiting RAG to G0/G1 may facilitate repair by NHEJ because it is the most active pathway during G0/G1 (4–6). This mechanism alone does not explain the exclusive repair of VDJ DSBs by NHEJ because RAG mutants that lack cell cycle regulation are still absolutely dependent on intact NHEJ for repair. Previous work has suggested that the RAG complex might shepherd recombination intermediates into the NHEJ pathway (11), although a specific mechanism to provide for this RAG function has not been elucidated.
A useful method to study VDJ recombination is to assess recombination of plasmid substrates introduced into cultured cells (the Gellert assay), a method described almost two decades ago (12). This assay (still widely used) is the approach used by Taccioli et al. (10) to make their groundbreaking observation that VDJ recombination is absolutely dependent on intact NHEJ.
In yeast, similar assays can be used to study DSBR, in that recircularization of linearized plasmids is highly dependent on intact DNA repair pathways in these lower eukaryotes. Curiously, numerous investigators have documented that recircularization of linearized plasmids introduced into mammalian cells (to mimic a simple DSB) is not dependent on intact NHEJ or HR (13, 14), although the fidelity of the joint in recircularized plasmids is affected by the cell's NHEJ apparatus. More specifically, plasmids rejoined in the absence of NHEJ display more extensive nucleotide loss at the joint and also prefer to join at sites of short-sequence homologies, suggesting that the cell's NHEJ apparatus affects end processing of the plasmids. Still, joining rates are not affected by the disruption of either NHEJ or HR. Recovery of recircularized plasmids is efficient regardless of the DNA repair capacity of the transfected cell strain. This paradox (i.e., dependence on NHEJ for end processing, but not for joining) has not been clarified.
Here we demonstrate that NHEJ dependence of plasmid end joining can be recapitulated by targeting DSBs (not RAG-mediated) to the RAG complex. This goal was accomplished by generating RAG1 or RAG2–I-Sce1 fusion proteins that specifically cleave an I-Sce1 plasmid substrate. Efficient targeting of I-Sce1 breaks into NHEJ requires both RAG1 and RAG2. Furthermore, core RAG proteins can efficiently mediate NHEJ targeting by the RAG complex. Thus, targeting is independent of the RAG's interaction with core histones, mediated by RAG2's C terminus (absent in core RAG2). Similarly, because the core RAG2 protein lacks phosphorylation sites that direct its targeted degradation at the G1/S border, this shunting of breaks into the NHEJ pathway is not explained by limiting breaks to G0/G1. Although the RAG complex can target I-Sce1 breaks into NHEJ in cells deficient in Ku, DNA-PKcs, or XRCC4, using two different RAG–I-Sce1 fusion proteins revealed dependence (for restricting breaks to NHEJ) on DNA-PKcs. Finally, we show that the capacity of the RAG complex to divert I-Sce1 breaks into the NHEJ pathway requires the presence of two I-Sce1 breaks (in cis), suggesting that only a synapsed RAG complex can divert breaks to the NHEJ pathway.
Results
Rejoining of I-Sce1-Cleaved Plasmid Substrates in Living Cells Is Not Dependent on NHEJ.
Plasmid-based VDJ recombination assays have been widely used to characterize joining deficits in NHEJ-deficient cells (10, 15–17). Similarly, plasmid-based assays have been used to study rejoining of breaks by cellular DNA repair systems (13, 14). DSBs are mimicked by transfecting linearized plasmids into cultured cells. However, recovery of recircularized plasmids is efficient regardless of the DNA repair capacity of the transfected cell strain. We considered that mimicking authentic NHEJ in mammalian cells (with plasmid assays) might require creation (of the break) in vivo. To that end, we developed a transient, plasmid-based assay to assess rejoining of I-Sce1-mediated breaks.
Briefly, the RSSs in the VDJ substrate plasmid pJH290 were replaced with two restriction sites for the endonuclease I-Sce1 to generate the I-Sce1 substrate, p28-7 (Fig. 1). This plasmid was cotransfected with an expression construct for the I-Sce1 endonuclease. Plasmids that delete the small oop sequence between the restriction sites were recovered by chloramphenicol selection. Deletion of oop depends on cotransfection of the I-Sce1 expression construct. Furthermore, no recombination of the I-Sce1 substrate (p28-7) is detected when expression constructs encoding the RAG endonuclease are cotransfected instead of I-Sce1. Although RAG expression induces recombination of the pJH290 substrate, I-Sce1 expression does not (Fig. 1A). Rejoining of RAG-induced DSBs is highly dependent on intact NHEJ. Thus, few recombinants are recovered from V3 cells lacking DNA-PKcs, but recombinants are readily isolated from V3 cells expressing DNA-PKcs. In contrast (and similar to other plasmid end-joining assays in mammalian cells), rejoining of plasmids with I-Sce1 breaks does not depend on NHEJ. V3, XR-1, and xrs6 control cell strains support recombination of the I-Sce1 substrate similarly to their isogenic, paired cell strains expressing DNA-PKcs, XRCC4, and Ku80, respectively (Fig. 1B). Plasmid DNA was sequenced from rejoined I-Sce1 breaks from each cell strain [supporting information (SI) Table 1]. Consistent with previous studies transfecting linearized plasmids into NHEJ-deficient cells (13, 14), rejoined I-Sce1 breaks recovered from NHEJ-deficient cells have greater nucleotide loss at the site of joining, and joints are more often mediated by short-sequence homologies.
Fig. 1.

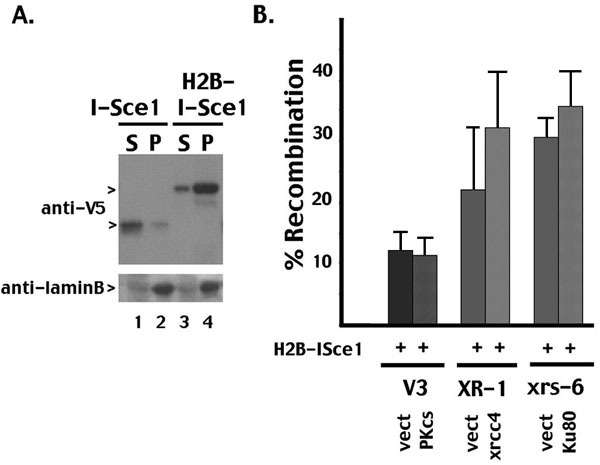
Rejoining of I-Sce1-cleaved plasmid substrates is not dependent on NHEJ. (A) Transient VDJ recombination assays were performed in V3 transfectants expressing either wild-type DNA-PKcs or vector only. The VDJ substrate (pJH290) was transfected into both transfectants with either an expression plasmid encoding the I-Sce1 nuclease or the RAG1/RAG2 proteins as indicated. (B) The I-Sce1 substrate (p28-7) was transfected into V3 transfectants expressing either wild-type DNA-PKcs or vector only with either an expression plasmid encoding the I-Sce1 nuclease or the RAG1/RAG2 proteins as indicated. XR-1 and xrs6 transfectants were similarly analyzed for their capacity to support rejoining of I-Sce1-mediated breaks. (C) The I-Sce1 substrate (p28-7) was transfected into V3 transfectants expressing either wild-type DNA-PKcs or vector only with varying amounts of the expression plasmid encoding the I-Sce1 nuclease as indicated. In A–C, results show the average and average deviation of four independent experiments.
We considered that the number of DSBs introduced by the I-Sce1 nuclease might overwhelm the NHEJ capacity of the cell, which might result in more efficient use of the alternative end-joining pathway. A dilution experiment was performed, limiting the amount of I-Sce1 expression. Although joining rates correlate well with I-Sce1 expression, joining is independent of NHEJ. (The data are presented on a log scale to adequately display the low recombination rates observed with limiting I-Sce1.) We also tested V3 transfectants expressing a DNA-PKcs mutant that is not only defective in NHEJ, but also strongly inhibits HR. Rejoining of I-Sce1 breaks was similarly robust in these cells, suggesting that this alternative end-joining pathway is not dependent on either NHEJ or HR factors (data not shown).
An emerging consensus is that DNA repair occurs in specialized nuclear compartments. We considered that authentic NHEJ requires a chromatinized environment. To test this possibility, I-Sce1 was targeted directly to chromatin by fusion with the core histone, H2B. This strategy has been used recently to study the importance of monoubiquitinated FANCD2's association with chromatin (18). Although the I-Sce1–H2B fusion protein segregates with laminB in nuclear matrix fractions, (robust) rejoining of breaks generated by the I-Sce1–H2B fusion protein is completely independent of NHEJ (SI Fig. 6). Although we have not formally shown that the I-Sce1 breaks in this experiment are chromatinized, these data suggest that targeting breaks to a chromatin compartment does not ensure repair by NHEJ. In summary, we suggest, consistent with previous reports (13, 14), that alternative end-joining pathways exist, but to date are not well characterized (19–23) and can efficiently rejoin I-Sce1 breaks in the absence of NHEJ, albeit with reduced fidelity. A previous report, studying rejoining of a chromosomal I-Sce1 substrate in Ku-deficient cells, made essentially the same conclusion (24).
The RAG Complex Shunts DSBs into the NHEJ Pathway.
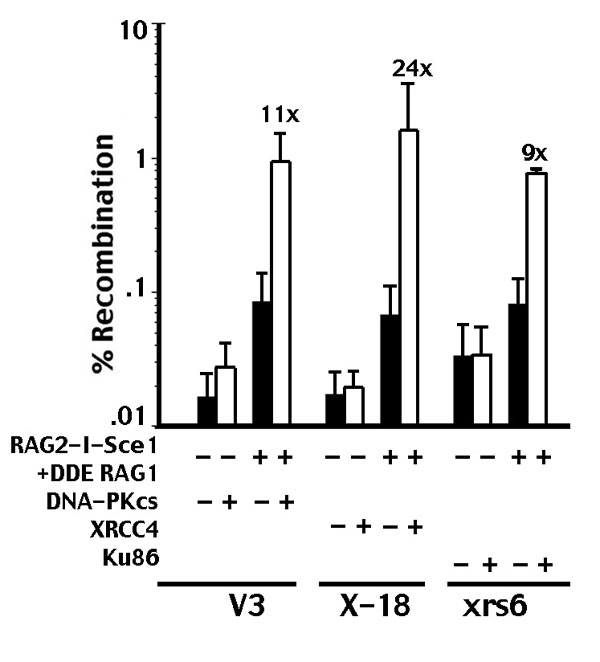
In addition to their role in RSS cleavage, the RAG proteins participate in the joining phase of VDJ recombination (11, 25, 26). It has been suggested that the RAG proteins have the capacity to direct their breaks into the NHEJ pathway. We next investigated whether the RAG proteins could divert I-Sce1 breaks into the NHEJ pathway. To that end, a RAG2–I-Sce1 fusion protein was generated that includes I-Sce1-fused C terminal to core RAG2. The RAG2–I-Sce1 fusion is enzymatically active in both complemented and NHEJ-defective cells (Fig. 2A); oop-deleted plasmids are readily isolated from all cell strains. With all three pairs of isogenic cell strains, there is a modest difference (2- to 4-fold less in NHEJ-defective cells) in rejoining rates between complemented (white bars) and noncomplemented (black bars) cells when the RAG2–I-Sce1 fusion protein alone induces breaks. In NHEJ-proficient cells, coexpression of RAG1 with the RAG2–I-Sce1 fusion protein (Fig. 2A) does not significantly affect rejoining. In contrast, in NHEJ-deficient cells, coexpression of RAG1 dramatically inhibits rejoining. Thus, when DSBs are linked to the RAG complex, recombination rates are 10- to 56-fold lower in NHEJ-defective cells than in NHEJ-proficient cells. (The data are presented on a log scale to adequately display the low recombination rates observed with RAG2–I-Sce1 cotransfected with RAG1.) Full-length RAG1 (as opposed to core RAG1) also was tested with similar results (data not shown). These data suggest that coupling an I-Sce1 break to the RAG complex diverts the break into an NHEJ-specific compartment, where joining is blocked in NHEJ-deficient cells. Rejoined breaks initiated by the RAG2–I-Sce1/RAG1 complex were sequenced. As expected, joints from NHEJ-deficient cells have more nucleotide loss and use short-sequence homologies to a greater extent than joints recovered from NHEJ-proficient cells (SI Table 1). We considered that the difference between complemented and noncomplemented cell strains might be the result of less efficient I-Sce1 cleavage in different cell clones by the RAG2–I-Sce1 fusion protein. To assess cleavage, a ligation-mediated PCR (LMPCR) assay was performed (Fig. 2B). Not surprisingly, wild-type I-Sce1 is a more robust nuclease than the RAG2–I-Sce1 fusion protein. However, there is not a significant difference in the level of I-Sce1 breaks with RAG2–I-Sce1 (that generates DSBs that can be readily joined in both wild-type and NHEJ-defective cells), compared with RAG2–I-Sce1 cotransfected with RAG1 (that generates DSBs that require NHEJ for rejoining). Further, DSBs are similarly generated in both the NHEJ-proficient and NHEJ-deficient cell strains. We conclude that the 10- to 56-fold increase in recombination in NHEJ-proficient cells is not because of more efficient cleavage in those cell strains.
Fig. 2.

The RAG complex shunts DSBs into the NHEJ pathway. (A) Transient recombination assays were performed in V3, XR-1, and xrs6 transfectants expressing vector, DNA-PKcs, XRCC4, or Ku80 as indicated. The I-Sce1 substrate was transfected with core RAG1 alone, RAG2–I-Sce1, or core RAG1 and RAG2–I-Sce1. The results show the average and average deviation of at least six independent experiments. (B) LMPCR experiments were performed to quantify DSBs as described previously (15), except that linkers were designed to anneal to the I-Sce1 overhang. XR-1 cells stably expressing XRCC4 or vector only were transiently transfected with the p28-7 substrate and either no I-Sce1, wild-type I-Sce1, RAG2–I-Sce1, or RAG2–I-Sce1 and RAG1. LMPCR was performed on serial dilutions of Hirt supernatants prepared 48 h after transfection.
We also tested whether a catalytically inactive version of RAG1 (DDE-RAG1) would have the same effect on targeting breaks mediated by the RAG2–I-Sce1 fusion protein (27). I-Sce1 breaks linked to a catalytically inactive RAG complex are only efficiently rejoined in NHEJ-competent cell strains (SI Fig. 7). We conclude that NHEJ targeting by the RAG complex is independent of RAG's enzymatic activity. Targeting also is independent of RAG's interaction with histone and independent of RAG's restriction to G0/G1 because the C-terminal region of RAG2 that is missing in core RAG2 provides both of those characteristics. Because NHEJ dependence is observed in XR-1, V3, and xrs6 cells, these data suggest that targeting is independent of Ku, DNA-PKcs, and the XRCC4/ligase IV complex. In summary, these data provide substantial support for the model proposed by Lee et al. (11), which reveals that the RAG complex physically shepherds DSBs into the NHEJ pathway.
Rejoining of Single (Versus Paired) DSBs Initiated by I-Sce1 Linked to the RAG Complex Is Not Dependent on NHEJ.
These experiments were initiated to develop another NHEJ-dependent end-joining assay to study DNA end processing in living cells. The initial design of our I-Sce1 substrate (by using the oop transcription terminator) was a design of convenience. However, this design does not accurately reflect rejoining of a simple DSB because there are actually two DSBs. We considered that the NHEJ dependence of RAG-linked I-Sce1 breaks might relate to the fact that the RAGs efficiently compartmentalize four double-strand ends (provided for with this substrate). To address this possibility, another I-Sce1 substrate plasmid was constructed that inserts an I-Sce1 site (that fortuitously contains a stop codon near the cleavage site) so that the initiation codon for the CAT gene is interrupted (Fig. 3A). One-third of joints repaired by NHEJ should restore the appropriated reading frame and induce CAT resistance [minimal end processing (8 bp) of the cleavage site is required to delete the stop codon]. As with the p28-7 substrate, retrieval of chloramphenicol-resistant plasmids depends on the expression of I-Sce1 (Fig. 3B) but is independent of the cell's NHEJ capacity. If the RAG proteins require four DNA ends to perform their targeting function, the prediction would be that joints from this substrate would not be restricted to NHEJ, which is exactly what is observed. When I-Sce1 breaks are linked to the RAG complex (either RAG2–I-Sce1 alone or cotransfected with RAG1), similar levels of chloramphenicol-resistant plasmids are retrieved from both complemented and NHEJ-defective isogenic cell strains. Although other interpretations are possible, these data are consistent with the conclusion that a synapsed RAG complex targets four DNA ends into the NHEJ pathway.
Fig. 3.

Rejoining of single (vs. paired) DSBs initiated by I-Sce1 linked to the RAG complex is not dependent on NHEJ. (A) The single I-Sce1 site substrate (p18) is diagrammed. The deletion of as few as eight nucleotides deletes a stop codon, allowing expression of chloramphenicol if rejoining occurs so that the CAT gene is in frame with the ATG. (B) The I-Sce1 substrate (p18) was transfected with the plasmid encoding the I-Sce1 nuclease as indicated into XR-1 or xrs6 cells stably expressing XRCC4, Ku86, or vector only. (C) The I-Sce1 substrate (p18) was transfected with the RAG2–I-Sce1 fusion protein alone or in combination with RAG1 as indicated into XR-1 or xrs6 cells stably expressing XRCC4, Ku86, or vector only. In B and C, results show the average and average deviation of four independent experiments.
An I-Sce1–RAG1/RAG2 Complex Targets Breaks into NHEJ in XRCC4- and Ku-Deficient Cells, but Not in DNA-PKcs-Deficient Cells.
To further substantiate these findings, an I-Sce1–RAG1 fusion protein was constructed by inserting I-Sce1 between GST and core RAG1 of a previously characterized RAG1 construct. As with breaks generated by the RAG2–I-Sce1 fusion protein, breaks from the I-Sce1–RAG1 fusion protein (without RAG2) are joined somewhat better (3- to 4-fold) in both complemented XR-1 and xrs6 cells, compared with uncomplemented cells (Fig. 4). When core or full-length RAG2 (data not shown) are cotransfected, rejoining rates are 9- to 15-fold higher in complemented XR-1 and xrs6 transfectants, compared with the isogenic vector control cell strains.
Fig. 4.

A RAG1–I-Sce1/RAG2 complex targets breaks into NHEJ in the absence of Ku or XRCC4, but not in the absence of DNA-PKcs. Transient recombination assays were performed in XR-1 and xrs6 transfectants expressing vector alone, XRCC4, Ku80, or DNA-PKcs as indicated. The I-Sce1 p28-7 substrate was transfected with I-Sce1–RAG1 alone or with core RAG2 as indicated. The results show the average and average deviation of four independent experiments.
The experiments with the I-Sce1–RAG1 fusion protein recapitulated the findings by using the RAG2–I-Sce1 fusion protein in the XR-1 and xrs6 cell strains, but did not with the DNA-PKcs-deficient V3 cell strain. When the I-Sce1–RAG1/RAG2 complex was used to initiate breaks in the V3 cell strain, joining was similarly proficient in cells with or without DNA-PKcs (Fig. 4). These data are obviously inconsistent with results obtained with the RAG2–I-Sce1/RAG1 complex, and we considered possible explanations for the discrepancy. First, we considered that the position of I-Sce1 in the RAG1 fusion protein interfered with RAG1's ability to complex appropriately with core RAG2, and for some reason this issue was most problematic in V3 cells. However, we tested whether the I-Sce1–RAG1 fusion protein (when cotransfected with RAG2) could induce authentic RAG breaks and thus recombination of the pJH290 substrate. We found that the I-Sce1–RAG1 fusion protein was active in generating RAG breaks when cotransfected with RAG2 in complemented V3 cells (data not shown). Next, we considered that the position of I-Sce1 in the RAG1 fusion protein disrupts an important interaction responsible for restricting breaks in V3 cells to the NHEJ pathway. Thus, it would seem that restriction would be disrupted in all three sets of NHEJ-defective cell strains, and this result is clearly not the case. However, these data could be explained if NHEJ restriction were mediated by an interaction (either directly or indirectly) between the RAGs and either DNA-PKcs or another factor, perhaps Ku. For instance, if limiting breaks to NHEJ (by the RAG complex) required either Ku or DNA-PKcs, NHEJ restriction in Ku-deficient cells could be accomplished by the interaction of RAGs with DNA-PKcs, whereas NHEJ restriction in DNA-PKcs-deficient cells could be accomplished by the interaction of the RAGs with Ku (explaining why all three pairs of NHEJ-defective cells restrict RAG2–I-Sce1 breaks to the NHEJ pathway). If the position of I-Sce1 in the RAG1 fusion protein disrupts a putative interaction (either directly or indirectly) between Ku and the RAG complex, I-Sce1 breaks (initiated by the I-Sce1–RAG1/RAG2 complex) would not be restricted to NHEJ in V3 cells (Fig. 5).
Fig. 5.

Does DNA-PK interact with the RAG complex to direct breaks into the NHEJ pathway? (A) The RAG complex interacts with both Ku and DNA-PKcs. With the RAG2–I-Sce1 fusion protein, I-Sce1 does not interfere with either interaction. Breaks are restricted to NHEJ in both xrs6 and V3 cells. In contrast, the position of I-Sce1 in the I-Sce1–RAG1 fusion protein interferes with the Ku interaction. Without DNA-PKcs, the alternative pathway efficiently joins breaks. In the presence of DNA-PKcs, breaks are NHEJ restricted, but also are efficiently joined because NHEJ is intact. However, in the presence of NHEJ-defective DNA-PKcs, breaks are restricted to NHEJ, but joining is blocked because NHEJ is defective. (B) (Left) Transient recombination assays were performed in V3 cells, including the I-Sce1 substrate p28-7, RAG2–I-Sce1/RAG1, and no DNA-PKcs, wild-type DNA-PKcs, or K>R mutant as indicated. (Right) Transient recombination assays were performed in V3 cells, including the I-Sce1 substrate p28-7, I-Sce1–RAG1/RAG2, and no DNA-PKcs, wild-type DNA-PKcs, or K>R mutant as indicated. The results show the average and average deviation of three independent experiments.
Although hypothesized by many, there are no reports of direct physical interactions between the RAG and DNA-PK complexes. Consistent with a role for the RAG complex in the joining phase of VDJ recombination, Agrawal and Schatz (28) demonstrated an interaction between the RAG postcleavage complex and each of the subunits of DNA-PK. This interaction required DNA ends. Cortes et al. (29) provide indirect support for an interaction between the RAG complex and DNA-PK before RSS cleavage. Although other reports have shown clear coupled 12/23 cleavage without NHEJ factors (30, 31), in their experimental system, Sawchukm et al. (29) suggest that DNA-PK is important in facilitating enforcement of the 12/23 rule in in vitro RAG cleavage assays. This effect required both Ku and DNA-PKcs. These authors suggested an interaction between the RAG complex and both Ku and DNA-PKcs. Because DNA-PK's effect was on enforcement of the 12/23 rule, this interaction likely occurs before cleavage. Thus, the interaction might not involve an assembled DNA-PK complex (i.e., DNA-PKcs recruited to Ku bound to a DNA end) and could involve separate interactions between the RAG complex and each of the two DNA-PK subunits. Still, to date, there is no compelling experimental evidence for a direct physical interaction between the RAG complex and any of the seven NHEJ factors.
We considered the following model to explain our data (Fig. 5). A synapsed RAG complex can divert its breaks to NHEJ by “communication” with DNA-PKcs or another factor, perhaps Ku. In our model, either type of communication would be sufficient to divert breaks into the NHEJ pathway. The position of I-Sce1 in the Rag2–I-Sce1 fusion protein does not interfere with the RAG's ability to shunt its breaks to NHEJ. Thus, joining is restricted to NHEJ and is low in cells deficient in DNA-PKcs and high in complemented cells. However, the position of I-Sce1 in the RAG1 fusion protein disrupts communication between the RAG complex and Ku. The uncomplemented V3 cells join I-Sce1–RAG1/RAG2 breaks proficiently because the breaks are not restricted to NHEJ and are efficiently joined by the alternative pathway. In complemented V3 cells, NHEJ restriction is accomplished because DNA-PKcs is present. However, joining is still proficient because wild-type DNA-PKcs restores NHEJ. To test this model, we repeated these experiments by including an enzymatically inactive version of DNA-PKcs (K>R). We hypothesized that this mutant could restrict breaks to the NHEJ pathway, but joining would be minimal because NHEJ would not be restored. This prediction is exactly the result observed (Fig. 5). When DSBs are initiated by the RAG2–I-Sce1/RAG1 complex, joining rates are similarly low in cells lacking DNA-PKcs and those expressing the K>R mutant. In contrast, rejoining of breaks initiated by I-Sce1–RAG1/RAG2 is similarly proficient in cells either with or without DNA-PKcs, but joining is substantially reduced in cells expressing the K>R mutant. These data are consistent with the hypothesis that the ability of the RAG complex to restrict its breaks to NHEJ depends on DNA-PKcs and a second factor, perhaps Ku.
Discussion
Here we demonstrate that the RAG complex can direct non-RAG breaks into the NHEJ pathway by physically linking the RAG proteins to another endonuclease. This finding provides substantial support for the model put forth previously by Roth et al. (11): that the RAG complex shepherds its breaks into the NHEJ pathway. Although our initial experiments with core RAG2–I-Sce1 cotransfected with core RAG1 demonstrate clear joining dependence on Ku, DNA-PKcs, and XRCC4 (suggesting that NHEJ targeting is independent of these factors), results with an I-Sce1–RAG-1 fusion protein cotransfected with core RAG2 show joining independence from DNA-PKcs. To reconcile these apparently discordant results, we propose a model whereby the ability of the RAG complex to restrict breaks to NHEJ requires either DNA-PKcs or Ku. Experiments with NHEJ-defective DNA-PKcs mutants provide support for this model. Finally, if the RAG complex directs its DSBs into the NHEJ pathway, one might predict that RAG mutants would be defective in NHEJ targeting. Recent work from Roth et al. (32), to which we contributed, describes a RAG2 mutant with exactly this phenotype. This RAG mutant allows significant signal and coding joint formation in cells deficient in DNA-PKcs or XRCC4 effectively, bypassing the NHEJ requirement for both coding and signal end joining.
One issue not addressed by these data is how non-RAG-targeted I-Sce1 breaks and other plasmid breaks are rejoined. Several reports have documented a variety of NHEJ subpathways, some which are independent of the DNA-PK and XRCC4/ligase IV complexes (18–22). One of these pathways may mediate the efficient plasmid rejoining observed in NHEJ-defective cells. Still there is no genetic evidence defining factors that function in these pathways. One pathway characterized recently depends on PARP-1 (22). However, PARP inhibitors did not affect rejoining rates of the I-Sce1 substrate after cleavage by wild-type I-Sce1 (data not shown). Emerging data from the Alt laboratory provide additional support for the existence of a (fairly) robust alternative end-joining pathway that functions efficiently in the absence of a classical NHEJ (33).
Although plasmid end-joining assays are not dependent on NHEJ, several plasmid-based transposition assays are dependent, including assays to detect Sleeping Beauty transposon excision sites and Tn5 transposition. Unlike VDJ assays, detection of recombined plasmids is low, generally requiring sensitive PCR assays for detection (34, 35). It is interesting that these two transposons, as well as the RAG endonuclease (an ancestral transposase), all target their breaks to NHEJ. Perhaps the relative inefficiency of Sleeping Beauty and Tn5, which did not evolve in mammalian cells, relates to inefficient interaction with cellular NHEJ.
RAG-mediated transposition is independent of NHEJ and is remarkably difficult to detect in mammalian systems (36). Transposition is estimated to occur only once in every 50,000 VDJ recombination events. Oettinger et al. (37) established VDJ joining and transposition assays in yeast. Although RAG-mediated transposition is detectable (although minimally) in yeast, authentic coding and signal end resolution (which one would predict to be less difficult to detect) also are remarkably inefficient in yeast. A possible explanation is that this inefficiency relates to ineffective interaction of the mammalian RAG complex with the yeast NHEJ machinery.
In summary, these data provide direct evidence for the hypothesis put forth previously (i.e., that the RAG proteins shepherd their breaks directly into the NHEJ pathway) (11). Our current model is that a synapsed RAG complex compartmentalizes four DNA ends and directs them into the NHEJ pathway.
Methods
Oligonucleotides.
Oligonucleotides used in this study include the following. I-SceI site oligonucleotides: SalI site, 5′TCGACTATATTACCCTGTTATCCCTAGCGTAACT; and BamHI site, 5′GATCAGTTACGCTAGGGATAACAGGG T A A T ATAG. Oligonucleotides used to construct V5-His-tagged I-Sce1: 5′ PCMV, ′TATAGCAGAGCTCGTTTA; and KAM425, 5′CCGCTCGAGTTTCAGGAAAGTTTC. Oligonucleotides used to construct H2B-V5-His-tagged I-Sce1: KAM563, 5′CGCCTCGAGATGCCTGAACCGGCA; and KAM564, 5′GCTC T A GACTTGGAGCTGGTGTAC. Oligonucleotides used to construct RAG2core-V5-His-tagged I-Sce1: KAM590, 5′CGCCTCGAGATGTCCCTGCAG; and KAM591, 5′GGCTCTAGATTCCTCTGAGTC. Oligonucleotides used to construct GST–I-Sce1–RAG1: KAM684, 5′CGCGGATCCATG A A AAACATC; and KAM686, 5′CGCGGATCCTTTCAGGAAAGT.
Construction and Transfection of Expression Plasmids.
An I-Sce1 expression plasmid was a generous gift from J. Nickoloff (University of New Mexico, Albuquerque, NM). PCR mutagenesis (to introduce EcoR1 and XhoI restriction sites) was used to introduce I-Sce1 in frame with the V5-His tag included in pCDNA-6A by using primers 5′pCMV and KAM425. H2B was amplified from cDNA by using primers KAM563 and KAM564 that include XhoI and XbaI sites. The H2B cDNA was subcloned in frame 5′ of I-Sce1-V5-His. Core RAG2 was subcloned in frame 5′ of I-Sce1-V5-His with primers designated previously. I-Sce1 was subcloned in between the GST and RAG1 sequences in a GST-RAG1 fusion construct by using primers KAM684 and KAM686. Membrane-insoluble fractions were prepared from cells transiently transfected with either V5-tagged I-Sce1 or V5-tagged H2B–I-Sce1 as described previously (38).
Cell Lines, VDJ Recombination Assays, Plasmid-Joining Assays, and LMPCR.
A derivation of V3 DNA-PKcs transfectants, XR-1 XRCC4 transfectants, and xrs6 Ku80 transfectants has been described previously (15, 39, 40). The DNA-PKcs, kinase-inactive mutant (K>R) has been described previously (41). Extrachromosomal VDJ recombination assays and LMPCR assays were performed essentially as described previously (16). In our previous studies, we used RAG expression constructs that use the CMV promoter to drive RAG expression. Here RAG expression constructs that use the EF-1α promoter were used [a generous gift from D. Roth (New York University, New York)]. These plasmids induce considerably higher VDJ recombination rates than in our previous studies, significantly increasing the sensitivity of the assay. For plasmid end-joining assays, the RSSs in pJH290 were replaced with oligonucleotides containing I-SceI sites to generate the I-Sce1 substrate, p28-7. Assays were performed by cotransfecting 1 μg of substrate plasmid with 3 μg of expression plasmid encoding the restriction endonuclease.
Supplementary Material
Acknowledgments
This work was supported by Public Health Service Grant AI048758 (to K.M.).
Abbreviations
- NHEJ
nonhomologous end-joining
- HR
homologous recombination
- DSB
double-stranded DNA break
- RAG
recombination-activating gene
- RSS
recombination signal sequence
- LMPCR
ligation-mediated PCR.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0610928104/DC1.
References
- 1.Grawunder U, West RB, Lieber MR. Curr Opin Immunol. 1998;10:172–180. doi: 10.1016/s0952-7915(98)80246-x. [DOI] [PubMed] [Google Scholar]
- 2.Lewis SM. Adv Immunol. 1994;56:27–150. doi: 10.1016/s0065-2776(08)60450-2. [DOI] [PubMed] [Google Scholar]
- 3.Gellert M. Adv Immunol. 1997;64:39–64. doi: 10.1016/s0065-2776(08)60886-x. [DOI] [PubMed] [Google Scholar]
- 4.Li Z, Dordai DI, Lee J, Desiderio S. Immunity. 1996;5:575–589. doi: 10.1016/s1074-7613(00)80272-1. [DOI] [PubMed] [Google Scholar]
- 5.Lin WC, Desiderio S. Proc Natl Acad Sci USA. 1994;91:2733–2737. doi: 10.1073/pnas.91.7.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin WC, Desiderio S. Science. 1993;260:953–959. doi: 10.1126/science.8493533. [DOI] [PubMed] [Google Scholar]
- 7.Schatz DG, Oettinger MA, Baltimore D. Cell. 1989;59:1035–1048. doi: 10.1016/0092-8674(89)90760-5. [DOI] [PubMed] [Google Scholar]
- 8.Oettinger MA, Schatz DG, Gorka C, Baltimore D. Science. 1990;248:1517–1523. doi: 10.1126/science.2360047. [DOI] [PubMed] [Google Scholar]
- 9.Grawunder U, Leu TM, Schatz DG, Werner A, Rolink AG, Melchers F, Winkler TH. Immunity. 1995;3:601–608. doi: 10.1016/1074-7613(95)90131-0. [DOI] [PubMed] [Google Scholar]
- 10.Taccioli GE, Rathbun G, Oltz E, Stamato T, Jeggo PA, Alt FW. Science. 1993;260:207–210. doi: 10.1126/science.8469973. [DOI] [PubMed] [Google Scholar]
- 11.Lee GS, Neiditch MB, Salus SS, Roth DB. Cell. 2004;117:171–184. doi: 10.1016/s0092-8674(04)00301-0. [DOI] [PubMed] [Google Scholar]
- 12.Hesse JE, Lieber MR, Gellert M, Mizuuchi K. Cell. 1987;49:775–783. doi: 10.1016/0092-8674(87)90615-5. [DOI] [PubMed] [Google Scholar]
- 13.Kabotyanski EB, Gomelsky L, Han JO, Stamato TD, Roth DB. Nucleic Acids Res. 1998;26:5333–5342. doi: 10.1093/nar/26.23.5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verkaik NS, Esveldt-van Lange RE, van Heemst D, Bruggenwirth HT, Hoeijmakers JH, Zdzienicka MZ, van Gent DC. Eur J Immunol. 2002;32:701–709. doi: 10.1002/1521-4141(200203)32:3<701::AID-IMMU701>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 15.Ding Q, Reddy YV, Wang W, Woods T, Douglas P, Ramsden DA, Lees-Miller SP, Meek K. Mol Cell Biol. 2003;23:5836–5848. doi: 10.1128/MCB.23.16.5836-5848.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cui X, Yu T, Gupta S, Cho YM, Lees-Miller SP, Meek K. Mol Cell Biol. 2005;25:10842–10852. doi: 10.1128/MCB.25.24.10842-10852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nadel B, Feeney AJ. Mol Cell Biol. 1997;17:3768–3778. doi: 10.1128/mcb.17.7.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsushita N, Kitao H, Ishiai M, Nagashima N, Hirano S, Okawa K, Ohta T, Yu DS, McHugh PJ, Hickson ID, et al. Mol Cell. 2005;19:841–847. doi: 10.1016/j.molcel.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 19.Audebert M, Salles B, Calsou P. J Biol Chem. 2004;279:55117–55126. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- 20.Perrault R, Wang H, Wang M, Rosidi B, Iliakis G. J Cell Biochem. 2004;92:781–794. doi: 10.1002/jcb.20104. [DOI] [PubMed] [Google Scholar]
- 21.Udayakumar D, Bladen CL, Hudson FZ, Dynan WS. J Biol Chem. 2003;278:41631–41635. doi: 10.1074/jbc.M306470200. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Perrault AR, Takeda Y, Qin W, Wang H, Iliakis G. Nucleic Acids Res. 2003;31:5377–5388. doi: 10.1093/nar/gkg728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guirouilh-Barbat J, Huck S, Bertrand P, Pirzio L, Desmaze C, Sabatier L, Lopez BS. Mol Cell. 2004;14:611–623. doi: 10.1016/j.molcel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 25.Yarnell Schultz H, Landree MA, Qiu JX, Kale SB, Roth DB. Mol Cell. 2001;7:65–75. doi: 10.1016/s1097-2765(01)00155-1. [DOI] [PubMed] [Google Scholar]
- 26.Qiu JX, Kale SB, Yarnell Schultz H, Roth DB. Mol Cell. 2001;7:77–87. doi: 10.1016/s1097-2765(01)00156-3. [DOI] [PubMed] [Google Scholar]
- 27.Landree MA, Wibbenmeyer JA, Roth DB. Genes Dev. 1999;13:3059–3069. doi: 10.1101/gad.13.23.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agrawal A, Schatz DG. Cell. 1997;89:43–53. doi: 10.1016/s0092-8674(00)80181-6. [DOI] [PubMed] [Google Scholar]
- 29.Sawchukm DJ, Mansilla-Soto J, Alarcon C, Singha NC, Langen H, Bianchi ME, Lees-Miller SP, Nussenzweig MC, Cortes P. J Biol Chem. 2004;279:29821–29831. doi: 10.1074/jbc.M403706200. [DOI] [PubMed] [Google Scholar]
- 30.van Gent DC, Ramsden DA, Gellert M. Cell. 1996;85:107–113. doi: 10.1016/s0092-8674(00)81086-7. [DOI] [PubMed] [Google Scholar]
- 31.Eastman QM, Leu TM, Schatz DG. Nature. 1996;380:85–88. doi: 10.1038/380085a0. [DOI] [PubMed] [Google Scholar]
- 32.Corneo B, Wendland R, Deriano L, Cui X, Klein I, Wong S, Arnal S, Holub AJ, Weller GR, Pancake BA, et al. Nature. 2007;449:478–482. [Google Scholar]
- 33.Yan CT, Boboila C, Souza EK, Franco S, Hickernell T, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, et al. Nature. 2007;449:483–486. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- 34.van Heemst D, Brugmans L, Verkaik NS, van Gent DC. DNA Repair. 2004;3:43–50. doi: 10.1016/j.dnarep.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 35.Izsvak Z, Stuwe EE, Fiedler D, Katzer A, Jeggo PA, Ivics Z. Mol Cell. 2004;13:279–290. doi: 10.1016/s1097-2765(03)00524-0. [DOI] [PubMed] [Google Scholar]
- 36.Reddy YV, Perkins EJ, Ramsden DA. Genes Devel. 2006;20:1575–1582. doi: 10.1101/gad.1432706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clatworthy AE, Valencia MA, Haber JE, Oettinger MA. Mol Cell. 2003;12:489–499. doi: 10.1016/s1097-2765(03)00305-8. [DOI] [PubMed] [Google Scholar]
- 38.Drouet J, Delteil C, Lefrancois J, Concannon P, Salles B, Calsou P. J Biol Chem. 2005;280:7060–7069. doi: 10.1074/jbc.M410746200. [DOI] [PubMed] [Google Scholar]
- 39.Yu Y, Wang W, Ding Q, Ye R, Chen D, Merkle D, Schriemer D, Meek K, Lees-Miller SP. DNA Repair. 2003;2:1239–1252. doi: 10.1016/s1568-7864(03)00143-5. [DOI] [PubMed] [Google Scholar]
- 40.Douglas P, Gupta S, Morice N, Meek K, Lees-Miller SP. DNA Repair. 2005;4:1006–1018. doi: 10.1016/j.dnarep.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 41.Kienker LJ, Shin EK, Meek K. Nucleic Acids Res. 2000;28:752–2761. doi: 10.1093/nar/28.14.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}