

Abstract
West Nile virus (WNV) has emerged as a significant cause of epidemic viral encephalitis and flaccid limb paralysis, yet the mechanism by which it enters the CNS remains uncertain. We used compartmentalized neuron cultures to demonstrate that WNV spreads in both retrograde and anterograde directions via axonal transport. Transneuronal spread of WNV required axonal release of viral particles and was blocked by addition of a therapeutic neutralizing antibody. To test the physiologic significance of axonal transport in vivo, we directly inoculated the sciatic nerve of hamsters with WNV. Intrasciatic infection resulted in paralysis of the hind limb ipsilateral but not contralateral to the injection site. Limb paralysis was blocked either by surgical transection of the sciatic nerve or treatment with the therapeutic neutralizing antibody. Collectively, these studies establish that WNV undergoes bidirectional spread in neurons and that axonal transport promotes viral entry into the CNS and acute limb paralysis. Moreover, antibody therapeutics directly inhibit transneuronal spread of WNV infection and prevent the development of paralysis in vivo.
Keywords: flavivirus, neuron, retrograde, transneuronal spread
West Nile virus (WNV) is a neurotropic member of the Flaviviridae family of RNA viruses and is related to other important arthropod-borne human pathogens. WNV is maintained in an enzootic cycle between mosquitoes and birds and has become an important global cause of epidemic encephalitis. Since its emergence in the United States in 1999, ≈26,000 cases of symptomatic WNV infection have been confirmed (www.cdc.gov/ncidod/dvbid/westnile/surv&control.htm#maps), and seroprevalence studies suggest that several million people have been infected (1).
Rodent models have provided insight into the mechanisms of WNV spread to the CNS. After s.c. inoculation, WNV-infected dendritic cells traffic to the draining lymph node, resulting in a primary viremia and infection of peripheral tissues. Within 6 days, WNV is cleared from the serum and peripheral organs and enters the CNS and induces neurological disease (reviewed in ref. 2). Nonetheless, the specific mechanisms by which WNV or other neurotropic flaviviruses enter into the CNS are largely unknown. CNS infection may occur in part via hematogenous spread, as increased viremia in immunodeficient mice (2) and TNF-α-mediated changes in blood-brain-barrier permeability correlate with earlier CNS entry (3).
Axonal transport from infected peripheral neurons mediates CNS entry and pathogenesis of viruses in the Herpesviridae, Rhabdoviridae, and Picornaviridae families (4–6). Viral spread in neurons is generally mediated by fast axonal transport, a microtubule-associated, anterograde and retrograde transport system. In classical studies, CNS infection of rabies virus or poliovirus was prevented by axonal ligation or degeneration (7, 8). Insights into the biology of axonal spread have been facilitated by the development of compartmentalized, or Campenot, chambers for culturing neurons (9). These systems allow axons from cells in one chamber to cross barriers and enter distal chambers, permitting separate analysis of cell bodies and their axons.
As mosquito inoculation of WNV occurs in the highly innervated dermis, CNS entry through peripheral neurons could contribute to WNV neuroinvasion (2, 10, 11). Severe human cases of WNV disease most frequently manifest as encephalitis, meningitis, or acute flaccid paralysis. Interestingly, while increased susceptibility to encephalitis or meningitis correlates with depressed immunity and increased age, humans of all age and immune status groups are at risk for developing acute flaccid paralysis (12). Acute flaccid paralysis or associated muscle weakness may occur in 10–50% of patients with neuroinvasive WNV disease and is caused by viral infection and injury of anterior horn motor neurons in the spinal cord (12).
Despite suggestive evidence that axonal transport contributes to WNV entry into the CNS and pathogenesis of the CNS (10), it has yet to be experimentally demonstrated. We used compartmentalized neuronal cultures to show definitively that WNV undergoes axonal transport in both anterograde and retrograde directions to infect neuronal and non-neuronal cells. In vivo, direct infection of the sciatic nerve resulted in viral transport to the spinal cord, neuronal injury, and acute flaccid paralysis. These findings establish that axonal transport can mediate WNV entry into the spinal cord and cause a specific disease phenotype, acute flaccid limb paralysis.
Results
WNV Spreads Via Axons in the Retrograde and Anterograde Directions.
To determine whether WNV can be transported within axons, we used compartmentalized neuronal cultures. This trichamber system was used previously to establish transneuronal spread of pseudorabies virus (13). The culture is composed of three sections, the soma chamber where neurons are plated, the middle chamber, which contains a methylcellulose barrier, and the neurite chamber where distal axons emerge and target cells are plated [supporting information (SI) Fig. 5]. Superior cervical ganglia (SCG) neurons seeded in the soma chamber extend axons through the barriers into the neurite chamber within ≈3 weeks.
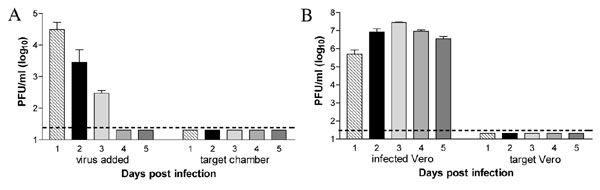
To determine whether WNV could spread via retrograde axonal transport from neuron to neuron, compartmentalized cultures were generated. Two days before infection, target SCG neurons were plated in the axon-containing neurite chamber so that axons from the target SCG neurons did not have time to extend back through the chamber barriers. Neurite chamber SCG neurons were infected, and supernatants from the soma and neurite chambers were collected daily and analyzed by viral plaque assay. As expected, WNV was readily detected from SCG neurons in the neurite chamber throughout the time course. Retrograde transport of WNV into the soma chamber was initially detected on day 3 after infection and subsequently spread within the soma SCG neurons, generating high levels of infectious virus (≈104 to 105 pfu/ml) (Fig. 1A).
Fig. 1.

WNV undergoes both retrograde and anterograde spread. (A and B) SCG neurons in the neurite chamber (A) or soma chamber (B) of compartmentalized cultures were infected with WNV. Spread to target populations was monitored by assaying viral pfu in supernatants harvested from days 1–6 after infection. (C and D) To examine spread to non-neuronal cells, Vero cells in the neurite chamber (C) or SCG neurons in the soma chamber (D) were infected at a moi of 0.1 and 1, respectively, and viral spread was monitored over time. Data represent the average of four to eight independent experiments (mean values ± SEM; dashed line is the limit of detection; ∗, P < 0.05).
To establish whether WNV undergoes axonal transport from neuron to neuron in the anterograde direction, compartmentalized cultures were generated with target SCG neurons plated in the neurite chamber as above, but the soma chamber was infected instead. Anterograde spread of WNV was first detected in target SCG neurons on day 2 after infection, followed by increased viral production in target cells on subsequent days (≈105 pfu/ml) (Fig. 1B).
To assess whether WNV undergoes directional spread to non-neuronal cells, Vero cells were plated in the axon containing neurite chamber of compartmentalized cultures. Infection of Vero cells resulted in efficient retrograde spread to SCG neurons in the soma chamber by day 3 after infection (Fig. 1C). Correspondingly, WNV also spread in an anterograde direction from infected SCG neurons to Vero cells by day 2 (Fig. 1D). Axons and target Vero cell populations were visualized on day 3 after SCG neuron infection to examine the localization of WNV infection after transneuronal spread. As expected, virtually all SCG neurons in the infected soma chamber showed high levels of WNV antigen. WNV envelope protein was also readily observed in target Vero cells both in close proximity to and more distant from axons (Fig. 2, arrows). Overall, these experiments demonstrate that WNV spreads efficiently in both retrograde and anterograde directions between neuronal and non-neuronal cells. As expected, based on the slower speed of retrograde transport (14), anterograde spread occurred more rapidly, resulting in WNV infection of target cells ≈24 h earlier.
Fig. 2.

WNV antigen localizes to target cells after transneuronal spread. Compartmentalized cultures were analyzed by immunoflorescence on day 3 after infection of the soma chamber. Cultures were costained for WNV antigen (red), the neuronal marker neurofilament M (green), and the nuclear stain TO-PRO-3 (blue). Vero cells in the neurite chamber that show WNV antigen in close proximity to axons (yellow arrows) and apart from axons (white arrows) are indicated. Arrows indicate that WNV envelope protein was also readily observed in target Vero cells both in close proximity to and more distant from axons. (Magnification: ×95.)
WNV Neuronal Spread Requires Intact Axons.
To verify that axons extended by SCG neurons to the neurite chamber were required for viral spread, we performed several controls to test the integrity of the cultures. Compartmentalized cultures were assembled in the absence of cells, WNV (105 pfu) was added to the soma chamber, and supernatants from the soma and neurite chamber were harvested daily to assess whether passive diffusion of virus occurred (SI Fig. 6A and SI Text). The level of virus in the soma chamber declined over time as it was diluted after repeated sampling and media replacement. Importantly, infectious virus was never detected in the neurite chamber, indicating that the barriers prevented passive movement of WNV between chambers. To exclude the possibility that infectious virus penetrated the barriers at levels below the limit of plaque assay detection, Vero cells were plated in the absence of neurons as these highly permissive cells should amplify low levels of passively transported virus. Infection of Vero cells on one side of the chamber did not spread to uninfected Vero cells on the other side even after 6 days (SI Fig. 6B). To directly test whether intact axons were required for neuron-to-cell spread, compartmentalized cultures were generated, and 1 h before infection, the axons in the middle chamber were severed. Vero cells in the neurite chamber or the SCG neurons in the soma chamber were then infected, and WNV production in both compartments was monitored. WNV spread in either direction was blocked completely by axotomy (SI Fig. 7). Thus, WNV spread in the compartmentalized chamber system required intact axons and did not occur through passive diffusion or leakage.
WNV Transneuronal Spread Is Mediated by Viral Release from Distal Axons.
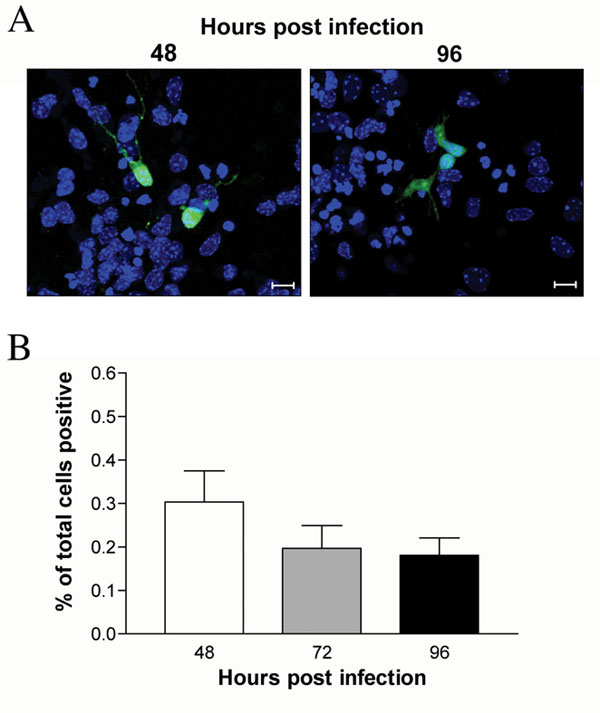
For some neurotropic viruses, transneuronal spread occurs across synapses (4), whereas for others, including pseudorabies virus, it may occur through direct cell-to-cell transfer without release of infectious particles (13). To test whether WNV was released from axon terminals, SCG neurons in the soma chamber of compartmentalized cultures were infected in the absence of target cells in the neurite chamber, and supernatants were harvested over 6 days. As expected, the SCG neuron cell bodies in the soma chamber released significant levels of infectious virus (104 to 105 pfu/ml). Notably, low levels (102 to 103 pfu/ml) of infectious virus were consistently detected in the neurite chamber, suggesting that WNV particles were transported axonally and secreted at distal axon sites (Fig. 3A). Formally, it remained possible that packaging-independent pathways could also contribute to neuron-to-target-cell spread. For some alphaherpes viruses, structural glycoproteins are dispensable for neuronal spread (15). To test whether WNV neuron-to-target-cell spread occurred in the absence of viral packing and egress, primary neurons were infected with pseudotyped WNV reporter virus particles (RVP). RVP deliver WNV replicons that encode GFP but lack structural proteins and thus are only capable of a single round of cellular infection (16). Notably, no increase in the numbers of GFP-positive cells was observed in RVP-infected cortical neuron cultures over time (SI Fig. 8). Similarly, live cell imaging of RVP-infected neurons showed no evidence of spread of GFP signal among neurons (data not shown). Thus, WNV does not undergo transneuronal spread in the absence of structural proteins. These data are consistent with a model in which WNV spread to target cells is mediated through viral release from axon terminals into the supernatant.
Fig. 3.

WNV transneuronal spread occurs via release of virus from distal axons. (A) Compartmentalized cultures were generated, and SCG neurons in the soma chamber were infected in the absence of target cells in the neurite chamber. Viral release from the soma and neurite chambers was monitored over time by viral plaque assay (8–11 independent experiments; mean values ± SEM; dashed line is the limit of detection). (B) To determine the effect of the humanized, neutralizing WNV antibody Hu-E16 on retrograde viral spread, Vero cells in the neurite chamber were infected, and Hu-E16 (20 μg/ml) or an isotype control mAb was added to the soma chamber SCG neurons. Directional spread was monitored over time by quantitative RT-PCR for WNV RNA in supernatants collected from the soma and neurite chambers (three independent experiments; mean values ± SEM; dashed line is the limit of detection). (C) Transmission electron microscopy was performed on compartmentalized cultures after infection of SCG neurons in the soma chamber. Micrographs were generated from infected neurons in the soma chamber and from distal axons in the middle chamber. Intact viral particles were localized in vesicles in distal axons (arrows). (Magnifications: ×22,000, Left and Center; ×8,200, Right.)
To confirm that release of infectious particles was required for transneuronal transmission of WNV, retrograde spread experiments were performed in the presence of Hu-E16, a humanized, neutralizing WNV antibody with therapeutic activity in vivo (17). Vero cells in the neurite chamber were infected, and antibody was added to the soma chamber with target SCG neurons. As Hu-E16 efficiently neutralizes infectious WNV released into the supernatant (17), infection of target SCG neurons was monitored by using both quantitative RT-PCR and a capture ELISA for the secreted nonstructural protein NS1 (18). The presence of Hu-E16 in the soma chamber completely blocked viral spread (Fig. 3B). In corresponding anterograde experiments (data not shown), Hu-E16 was added to target Vero cells after SCG infection. As expected, low levels of virus were still secreted from axons in the neurite chamber (103 to 104 genomic equivalents per ml). However, this virus did not productively infect target Vero cells: whereas high levels of NS1 (0.2 μg/ml) were detected in supernatants from infected SGC neurons and Hu-IgG-treated Vero cells on day 6, NS1 was undetectable in supernatants from Hu-E16-treated target cells. Thus, the spread of WNV from neurons to target cells is mediated by extracellular release of virus, and a neutralizing therapeutic mAb can directly disrupt this process.
Intact Viral Particles Are Localized in Distal Axons.
In cell lines, WNV assembles at the rough endoplasmic reticulum and is released by exocytosis (19). While infectious particles were released from distal axons, it was unknown whether intact viral particles were transported through the axon or assembled distally. To examine this, we analyzed compartmentalized cultures by electron microscopy. SCG cell bodies showed clear evidence of virion production in characteristic membranous web structures (20). Assembled viral particles were also visualized within proximal and distal axons and were enclosed in vesicles often containing more than one viral particle (Fig. 3C). These data suggest that WNV undergoes axonal transport and is released to infect target cells.
WNV Undergoes Retrograde Axonal Spread in Vivo and Induces Acute Flaccid Paralysis.
To test whether axonal transport of WNV occurred in vivo and evaluate its contribution to pathogenesis, we performed direct infection and ligation experiments of the sciatic nerve in hamsters (SI Fig. 9). Viral dissemination and clinical disease were compared in the presence or absence of sciatic nerve ligation and transection. Within 7–13 days after sciatic nerve inoculation, animals with intact nerves showed overt disease. By 8–16 days after infection, 21% (4 of 19) of these animals developed acute flaccid paralysis of the limb ipsilateral but not contralateral to the inoculation site (Table 1). Paralysis was induced specifically by viral infection, as nerve ligation alone caused a characteristic foot drop but no limb paralysis.
Table 1.
Axonal spread of WNV in vivo induces acute flaccid paralysis
| Viral challenge | Sciatic transection | Antibody administration | Ipsilateral limb paralysis, % | Contralateral limb paralysis, % | Mean time to death, days | Percent death |
|---|---|---|---|---|---|---|
| WNV | None | None | (21 (4/19)* | 0 (0/19) | 13 ± 2.4 | 85 (11/13) |
| WNV | Below infection | None | 40 (4/10)** | 10 (1/10) | 14.3 ± 3.2 | 86 (6/7) |
| WNV | Above infection | None | 0 (0/15) | 6.6 (1/15) | 14.5 ± 2.1 | 67 (6/9) |
| None | Transect | None | 0 (0/4) | 0 (0/4) | None | None |
| WNV | None | 0.1 mg/kg | 0 (0/6) | 0 (0/6) | None | None |
| WNV | Below infection | 0.1 mg/kg | 0 (0/6) | 0 (0/6) | None | None |
| WNV | Above infection | 0.1 mg/kg | 0 (0/6) | 0 (0/6) | None | None |
| None | Transect | 0.1 mg/kg | 0 (0/3) | 0 (0/3) | None | None |
Hamsters were injected in the sciatic nerve with 101.8 pfu of WNV and monitored for paralysis and lethality. In some groups, sciatic nerve transection was performed by cutting the sciatic nerve above or below the site of WNV infection, and a humanized, neutralizing WNV antibody Hu-E16 was administered i.p. in a single dose (0.1 mg/kg) 20 h after infection. *, P < 0.05; **, P < 0.01 compared with nerve transection above WNV infection. Numbers in parentheses indicate numbers of hamsters.
Flaccid limb paralysis resulted from WNV infection and neuronal injury, as it was associated with high levels of infection and death in the anterior horn motor neurons of the lumbosacral region (L5-S1) of the spinal cord (Fig. 4 E–H, 20.1 ± 8.8% and 52.9 ± 6% of neurons were positive for WNV antigen and TUNEL, respectively). A similar level of ipsilateral paralysis (4 of 10 animals, Table 1) was seen when the sciatic nerve was transected below the infection site. In contrast, when the sciatic nerve was transected above the injection site, paralysis of the ipsilateral limb or associated infection of the L5-S1 segment of the spinal cord did not develop (Fig. 4 I–L). Rather, these animals developed systemic disease, and only one animal developed limb paralysis, which was on the side contralateral to virus inoculation. Regardless of the injection scheme, most animals eventually succumbed to WNV infection with similar kinetics (Table 1, P = 0.2). Thus, sciatic nerve transection prevented rapid spread to the spinal cord and associated paralysis resulting from infection and injury of motor neurons but did not alter WNV-induced lethality. These experiments demonstrate WNV undergoes retrograde axonal transport in vivo and that this pathway of CNS entry induces a specific clinical feature of WNV neuroinvasive disease, acute flaccid paralysis. These data also suggest that WNV likely uses multiple routes to enter the CNS, as transection of the sciatic nerve did not affect the development of encephalitis.
Fig. 4.

WNV undergoes retrograde spread in vivo and induces neuronal injury. Hamsters were injected in the sciatic nerve with 101.8 pfu of WNV and necropsied on the day of initial paralysis (days 8–16). Characteristic confocal images are shown after review of six independent lumbar spinal cords harvested from uninfected animals (A–D), paralyzed animals injected with WNV with intact sciatic nerves (E–H), and nonparalyzed animals with sciatic nerve transection above the WNV injection site (I–L). Spinal cords sections were costained for the neuron-specific enolase (NSE) marker and WNV (C, G, and K) or TUNEL (D, H, and L). Arrows indicate double positive cells. (Scale bar: 20 μm.)
As viral spread in compartmentalized cultures was blocked by neutralizing antibody, we evaluated whether therapeutic administration of Hu-E16 would prevent transneuronal spread and paralysis in vivo. Hu-E16 (0.1 mg/kg) was administered i.p. 20 h after intrasciatic nerve infection with and without sciatic nerve ligation. Notably, antibody treatment completely prevented acute flaccid paralysis and mortality of animals infected via the sciatic nerve (Table 1). Thus, neutralizing antibody blocked both transneuronal spread of WNV and paralysis, while also restricting viral entry into the CNS by other possibly hematogenous routes.
Discussion
Although WNV is the most common cause of epidemic viral encephalitis in the United States (21), the mechanism by which it invades the CNS remains controversial. In this study, we demonstrate that WNV undergoes axonal transport in vitro in compartmentalized cultures and in vivo after intrasciatic nerve infection. Axonal transport of WNV in vivo resulted in spinal cord infection, neuronal injury, and acute flaccid paralysis. Axotomy blocked WNV transneuronal spread in vitro and in vivo and protected animals from paralysis on the side ipsilateral to the injection site. Our data also suggest that neuronal spread occurs via extracellular secretion of WNV from axons: infectious virus was detected in distal axon compartments and axonal spread was blocked by the addition of a neutralizing antibody. These experiments establish transneuronal spread of a flavivirus. Moreover, they demonstrate that neutralizing antibody therapy can disrupt transneuronal spread and thus may explain its efficacy in preventing or limiting WNV replication in the CNS (17).
WNV entered the CNS via axonal transport at rates consistent with the kinetics of neuroinvasive disease development. In rodent models, WNV is detected in the CNS beginning on days 4–5 after infection (2). In humans, the initial onset of WNV disease symptoms occurs after an average 6-day incubation period (22). This timing of CNS entry is similar to retrograde transport rates of other neurotropic viruses, including herpes simplex virus (HSV) (23). Once in the CNS, both retrograde and anterograde neuronal transport likely contributes to centrifugal spread of WNV among neuronal cells in the brain. In the compartmentalized cultures, WNV spreads in both directions to infect neuronal and non-neuronal cells. Although axonal spread of WNV contributes to a distinct disease phenotype, the relative roles of retrograde and anterograde spread through the motor and sensory neurons within the sciatic nerve remain less clear. Although further studies are necessary, we speculate that retrograde transport through peripheral motor nerves directly results in anterior horn neuron injury and paralysis, whereas anterograde transport contributes to regional spread of WNV infection within the CNS.
The mechanisms of neuronal entry and spread likely differ among viruses. Some, including Borna virus, enter neurons directly without evidence of peripheral replication (24). For others, local amplification at the site of infection in non-neuronal cells precedes viral entry into peripheral nerves. Initial replication of rabies virus in myocytes (4), HSV in the corneal epithelium (25), and reovirus in Peyer's patches (26) occurs before axonal transport and CNS infection. Analogously, Langerhans or other skin dendritic cells may be the initial site of WNV replication after skin inoculation. In this context, efficient transneuronal spread may require transport of WNV between neurons and non-neuronal cells. Once peripheral nerve infection occurs, viruses may use distinct pathways to transmit infection to target cells. For pseudorabies virus, spread involves direct cell-to-cell transmission via a fusion event that does not require encapsidation (13). In contrast, for WNV, HSV, and reovirus spread is inhibited directly by neutralizing antibodies (27, 28).
Local amplification, infection of peripheral nerves, and axonal transport to the spinal cord is likely not the only means of WNV entry into the CNS. The route of infection, viral strain, and the immune status of the host affect the CNS entry pathways used by neurotropic viruses. For example, dermal inoculation of HSV results in transneuronal spread to the CNS, whereas i.p. infection is followed by hematogenous spread (29). In comparison, reovirus enters the CNS by both transneuronal and hematogenous routes (30). Intranasal inoculation of WNV can also result in CNS entry through movement across the olfactory mucosa (31). Studies in rodent models suggest that after s.c. infection, WNV enters the brain in part through hematogenous spread (2). In immunodeficient mice, enhanced viremia correlates with more rapid entry into the CNS (32), and inflammatory cytokines such as TNF-α may modulate blood-brain-barrier permeability and WNV spread (3). Consistent with this finding, immunocompromised humans have a 20-fold greater risk of severe neuroinvasive WNV infection and death (33).
Two unique features are associated with WNV-induced acute flaccid paralysis: it can occur as an isolated disease in the absence of meningitis or encephalitis (34), and patients of all ages and immune status groups are at risk (12). Our experiments suggest that the route of WNV entry into the CNS in part determines pathological outcome. Intrasciatic infection caused acute flaccid paralysis of the ipsilateral but not contralateral limb. Axotomy of the sciatic nerve blocked this paralysis, but did not prevent the spread of WNV to the CNS through other routes. Acute flaccid paralysis may be a stochastic event that reflects virus entry into the peripheral nerves after initial skin infection. Notably, postexposure administration of neutralizing IgG completely blocked transneuronal spread and CNS invasion. These data are consistent with a model in which an effective innate immune response restricts WNV replication in peripheral tissues and hematogenous seeding of the CNS but may not efficiently block axonal spread once WNV has infected neurons in the peripheral nervous system. Instead, inhibition of neuronal spread may require neutralizing IgG, which is not produced until ≈6–8 days after infection when CNS invasion has already occurred (35). Indeed, disruption of transneuronal spread of WNV may be an independent mechanism for the therapeutic actions of neutralizing antibodies. Correspondingly, early therapeutic intervention with neutralizing antibodies could prevent both upper and lower motor neuron disease by blocking CNS entry and amplification.
In summary, our experiments suggest that WNV uses specific pathways to enter the CNS, resulting in distinct disease phenotypes. An improved understanding of the mechanisms by which WNV disseminates to the CNS may facilitate the development of targeted therapies that block specific steps in WNV neuropathogenesis.
Materials and Methods
Cells Lines and Viruses.
Vero T144 and BHK21 were cultured as described (35). The New York 1999 (36) and 2000 (37) strains of WNV were used for in vivo and in vitro experiments, respectively.
Neuron Culture.
Primary neurons were generated from Sprague–Dawley rat SCG as described (38) and maintained in AM50 media [MEM, 10% heat-inactivated FBS, 2 mM l-glutamine, 30 mM fluorodeoxyuridine, 30 mM uridine, and 50 ng/ml nerve growth factor (Harlan Bioproducts, Madison, WI)].
Trichamber Cultures and Infection Experiments.
The design and assembly of trichamber cultures have been described (13). For neuron-to-cell spread experiments, SCG neurons were cultured in the soma chamber for 3–4 weeks, and Vero cells were seeded in the neurite chamber. Methylcellulose (1%) in AM50 media was added to the middle chamber, and either the soma chamber [multiplicity of infection (moi) of 1] or the neurite chamber Vero cells (moi of 0.1) was infected with WNV. For neuron-to-neuron spread experiments, SCG neurons were seeded into the neurite chamber for 2–3 days before infection (moi of 1) to prevent axon growth back into the middle chamber. In all compartmentalized culture experiments, cells were infected for 1 h at 37°C, and free virus was removed by washing. To determine viral spread, supernatants were harvested every 24 h for 6 days. Media were replaced after each harvest, and supernatants were titered on BHK21 cells as described (35). The Mann–Whitney test analyzed differences in viral titers for statistical significance.
Immunofluorescence of Trichamber Cultures.
Cultures were assembled on Aclar (Electron Microscopy Sciences, Hartfield, PA) as described (13). Three days after SCG infection, cells were fixed with 4% paraformaldehyde, permeabilized, blocked (0.2% saponin, 5% normal goat serum, and 1% BSA in PBS), and stained with an antineurofilament M antibody (Chemicon International, Temecula, CA) and anti-WNV immune serum. Cells were counterstained with the nucleic acid stain TO-PRO-3 (Invitrogen, Carlsbad, CA) and visualized with a 510 Meta LSM confocal microscope (Zeiss, Thornwood, NY).
Electron Microscopy.
Trichamber cultures were assembled, and 4 days after SCG neurons in the soma chamber were infected (moi of 1), cultures were fixed and processed for electron microscopy as described (39).
Sciatic Nerve Injection and Transection.
Intrasciatic nerve injection and transection was performed by using adult (6–7 weeks old) Syrian golden hamsters as described (40). The sciatic nerve was injected 1 cm deep at ≈30° angle with 101.8 pfu of WNV in 1–2 μl of MEM by using a 30-gauge needle. Sciatic nerve ligation and transection were performed by cutting the nerve between two constricting sutures. Animals were monitored for paralysis and lethality for 30 days. Animal use was in compliance with the Utah State University Institutional Animal Care and Use Committee.
Immunohistochemistry.
Immunohistochemistry was performed on tissues as described (41) with the anti-WNV envelope antibody 7H2 (BioReliance, Rockville, MD) and polyclonal anti-neuron specific enolase antibody. TUNEL-mediated BrdUTP labeling was performed per the manufacturer's instructions (Molecular Probes, Eugene, OR). Sections were visualized with a MRC 1024 confocal microscope (Bio-Rad, Hercules, CA).
Viral Spread in the Presence of WNV-Specific Antibody.
Vero cells or SCG neurons in compartmentalized cultures were infected at an moi of 0.1 or 1, respectively, and humanized E16 (Hu-E16, IgG1) (20 μg/ml) or a human IgG1 isotype control (Hu-IgG) was added concurrently to the respective target chamber (17). Supernatants were harvested every 24 h for 6 days. The levels of WNV RNA were measured by quantitative RT-PCR using primers and probes to the E gene (42), and NS1 was measured by using a capture ELISA as described (18). For in vivo experiments, intrasciatic nerve injection was performed, and Hu-E16 was administered i.p. (0.1 mg/kg) 20 h after infection. Animals were monitored for paralysis and lethality for 30 days, and data were analyzed by using the likelihood χ2 test.
Supplementary Material
Acknowledgments
We thank L. Enquist and T. Ch'ng for developing the compartmentalized culture system and their invaluable help in adapting this system for use with WNV; B. Samuel, D. Gill, and W. Beatty for technical assistance; and J. Nordstrom and S. Johnson (MacroGenics, Rockville, MD) for providing the Hu-E16. This work was supported by a Howard Hughes Medical Institute Predoctoral Fellowship (to M.A.S.), the Ellison Medical Foundation Global Infectious Diseases Program (M.S.D.), and National Institutes of Health Grants AI53870 (to M.S.D.) and AI06357-01 and NO1-AI-15435 (to J.D.M.).
Abbreviations
- WNV
West Nile virus
- SCG
superior cervical ganglia
- RVP
reporter virus particles
- HSV
herpes simplex virus
- moi
multiplicity of infection.
Footnotes
Conflict of interest statement: M.S.D. and J.D.M. have consulting agreements with MacroGenics, a company that has licensed the E16 monoclonal antibody used in these studies for possible commercial development. M.A.S., H.W., and V.S. declare no conflict of interest.
This article is a PNAS Direct Submission. M.B.A.O. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/cgi/content/full/0705837104/DC1.
References
- 1.Busch MP, Caglioti S, Robertson EF, McAuley JD, Tobler LH, Kamel H, Linnen JM, Shyamala V, Tomasulo P, Kleinman SH. N Engl J Med. 2005;353:460–467. doi: 10.1056/NEJMoa044029. [DOI] [PubMed] [Google Scholar]
- 2.Samuel MA, Diamond MS. J Virol. 2006;80:9349–9360. doi: 10.1128/JVI.01122-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Nat Med. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 4.Dietzschold B, Schnell M, Koprowski H. Curr Top Microbiol Immunol. 2005;292:45–56. doi: 10.1007/3-540-27485-5_3. [DOI] [PubMed] [Google Scholar]
- 5.Mettenleiter TC. Virus Res. 2003;92:197–206. doi: 10.1016/s0168-1702(02)00352-0. [DOI] [PubMed] [Google Scholar]
- 6.Ohka S, Nomoto A. Trends Microbiol. 2001;9:501–506. doi: 10.1016/s0966-842x(01)02200-4. [DOI] [PubMed] [Google Scholar]
- 7.Nicolau S, Meteiesco E. Comp Rendu Acad Sci. 1928;186:1072–1074. [Google Scholar]
- 8.Bodian D, Howe HA. Bull Johns Hopkins Hosp. 1941;69:79–85. [Google Scholar]
- 9.Campenot RB. Proc Natl Acad Sci USA. 1977;74:4516–4519. doi: 10.1073/pnas.74.10.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hunsperger EA, Roehrig JT. J Neurovirol. 2006;12:129–139. doi: 10.1080/13550280600758341. [DOI] [PubMed] [Google Scholar]
- 11.Monath TP, Cropp CB, Harrison AK. Lab Invest. 1983;48:399–410. [PubMed] [Google Scholar]
- 12.Debiasi RL, Tyler KL. Nat Clin Pract Neurol. 2006;2:264–275. doi: 10.1038/ncpneuro0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ch'ng TH, Enquist LW. J Virol. 2005;79:10875–10889. doi: 10.1128/JVI.79.17.10875-10889.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.von Bartheld CS. J Neurobiol. 2004;58:295–314. doi: 10.1002/neu.10315. [DOI] [PubMed] [Google Scholar]
- 15.Tomishima MJ, Enquist LW. J Virol. 2002;76:8310–8317. doi: 10.1128/JVI.76.16.8310-8317.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pierson TC, Sanchez MD, Puffer BA, Ahmed AA, Geiss BJ, Valentine LE, Altamura LA, Diamond MS, Doms RW. Virology. 2006;346:53–65. doi: 10.1016/j.virol.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 17.Oliphant T, Engle M, Nybakken GE, Doane C, Johnson S, Huang L, Gorlatov S, Mehlhop E, Marri A, Chung KM, et al. Nat Med. 2005;11:522–530. doi: 10.1038/nm1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noueiry AO, Olivo PD, Slomczynska U, Zhou Y, Buscher B, Geiss B, Engle M, Roth RM, Chung KM, Samuel MA, Diamond MS. J Virol. 2007 doi: 10.1128/JVI.01358-07. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mackenzie JM, Westaway EG. J Virol. 2001;75:10787–10799. doi: 10.1128/JVI.75.22.10787-10799.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westaway EG, Mackenzie JM, Kenney MT, Jones MK, Khromykh AA. J Virol. 1997;71:6650–6661. doi: 10.1128/jvi.71.9.6650-6661.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weaver SC, Barrett AD. Nat Rev Microbiol. 2004;2:789–801. doi: 10.1038/nrmicro1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tyler KL. Arch Neurol. 2004;61:1190–1195. doi: 10.1001/archneur.61.8.1190. [DOI] [PubMed] [Google Scholar]
- 23.Engel JP, Madigan TC, Peterson GM. J Virol. 1997;71:2425–2435. doi: 10.1128/jvi.71.3.2425-2435.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carbone KM, Trapp BD, Griffin JW, Duchala CS, Narayan O. J Neuropathol Exp Neurol. 1989;48:631–644. doi: 10.1097/00005072-198911000-00005. [DOI] [PubMed] [Google Scholar]
- 25.Summers BC, Margolis TP, Leib DA. J Virol. 2001;75:5069–5075. doi: 10.1128/JVI.75.11.5069-5075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morrison LA, Sidman RL, Fields BN. Proc Natl Acad Sci USA. 1991;88:3852–3856. doi: 10.1073/pnas.88.9.3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mikloska Z, Sanna PP, Cunningham AL. J Virol. 1999;73:5934–5944. doi: 10.1128/jvi.73.7.5934-5944.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tyler KL, Virgin HWT, Bassel-Duby R, Fields BN. J Exp Med. 1989;170:887–900. doi: 10.1084/jem.170.3.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson RT. J Exp Med. 1964;119:343–356. doi: 10.1084/jem.119.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tyler KL, McPhee DA, Fields BN. Science. 1986;233:770–774. doi: 10.1126/science.3016895. [DOI] [PubMed] [Google Scholar]
- 31.Nir Y, Beemer A, Goldwasser RA. Br J Exp Pathol. 1965;46:443–449. [PMC free article] [PubMed] [Google Scholar]
- 32.Samuel MA, Diamond MS. J Virol. 2005;79:13350–13361. doi: 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huhn GD, Austin C, Langkop C, Kelly K, Lucht R, Lampman R, Novak R, Haramis L, Boker R, Smith S, et al. Am J Trop Med Hyg. 2005;72:768–776. [PubMed] [Google Scholar]
- 34.Sejvar JJ, Bode AV, Marfin AA, Campbell GL, Ewing D, Mazowiecki M, Pavot PV, Schmitt J, Pape J, Biggerstaff BJ, Petersen LR. Emerg Infect Dis. 2005;11:1021–1027. doi: 10.3201/eid1107.040991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diamond MS, Shrestha B, Marri A, Mahan D, Engle M. J Virol. 2003;77:2578–2586. doi: 10.1128/JVI.77.4.2578-2586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torrence PF, Gupta N, Whitney C, Morrey JD. Antiviral Res. 2006;70:60–65. doi: 10.1016/j.antiviral.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 37.Ebel GD, Dupuis AP, II, Ngo K, Nicholas D, Kauffman E, Jones SA, Young D, Maffei J, Shi PY, Bernard K, Kramer LD. Emerg Infect Dis. 2001;7:650–653. doi: 10.3201/eid0704.010408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Easton RM, Deckwerth TL, Parsadanian AS, Johnson EM., Jr J Neurosci. 1997;17:9656–9666. doi: 10.1523/JNEUROSCI.17-24-09656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shrestha B, Gottlieb D, Diamond MS. J Virol. 2003;77:13203–13213. doi: 10.1128/JVI.77.24.13203-13213.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma QP. Neuroscience. 2001;107:665–673. doi: 10.1016/s0306-4522(01)00387-6. [DOI] [PubMed] [Google Scholar]
- 41.Morrey JD, Siddharthan V, Olsen AL, Roper GY, Wang H, Baldwin TJ, Koenig S, Johnson S, Nordstrom JL, Diamond MS. J Infect Dis. 2006;194:1300–1308. doi: 10.1086/508293. [DOI] [PubMed] [Google Scholar]
- 42.Samuel MA, Whitby K, Keller BC, Marri A, Barchet W, Williams BRG, Silverman RH, Gale M, Jr, Diamond MS. J Virol. 2006;80:7009–7019. doi: 10.1128/JVI.00489-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}