

Abstract
The hippocampus is essential for episodic memory, which requires single-trial learning. Although long-term potentiation (LTP) of synaptic strength is a candidate mechanism for learning, it is typically induced by using repeated synaptic activation to produce precisely timed, high-frequency, or rhythmic firing. Here we show that hippocampal synapses potentiate robustly in response to strong activation by a single burst. The induction mechanism of this single-burst LTP requires activation of NMDA receptors, L-type voltage-gated calcium channels, and dendritic spikes. Thus, dendritic spikes are a critical trigger for a form of LTP that is consistent with the function of the hippocampus in episodic memory.
Keywords: episodic memory, hippocampus, single-trial learning, dendrite
Long-term potentiation (LTP) is widely acknowledged as the primary candidate mechanism for the cellular basis of learning and memory (1). It is therefore important to understand what types of stimuli can induce LTP, in the hopes of determining whether patterns of activity capable of inducing LTP in reduced preparations such as a slice might also induce LTP in behaving animals.
Most forms of LTP follow a Hebbian rule, requiring coincident presynaptic and postsynaptic activity. The presynaptic signal for LTP induction is thought to be glutamate release, whereas the postsynaptic signal is depolarization, sufficient to allow influx of calcium through NMDA receptors and/or voltage-gated calcium channels (2). Another feature of most LTP induction protocols used to date is repetition. Tetanic stimulation (e.g., 100 Hz for 1 s) and θ-burst stimulation (100 Hz bursts repeated at 5 Hz for 1 s) both use tens of stimuli, usually repeated multiple times to induce LTP lasting for hours. Spike-timing dependent plasticity (STDP) is induced by pairing presynaptic stimulation and postsynaptic action potential firing (3); although the number and frequency of such pairings required to induce STDP depend on several factors (4), repeated stimulation (usually ≈100 events) is a universal feature of all published studies on STDP. Primed bursts and high-strength synaptic stimuli have also been used to induce LTP. In most cases, however, these stimuli were repeated several times, albeit at relatively low frequency (5, 6).
Although repeated stimulation is typically used to induce LTP, it may not be required for all forms of LTP (7, 8), nor is it clear what kinds of transmembrane potentials might be required to induce LTP without repetition of the induction pattern. We therefore tested whether LTP induced by dendritic spikes (9) requires repetition of the stimulus or whether a single dendritic spike is capable of inducing LTP. We found that a single burst of action potentials in CA3 axons (Schaffer collaterals) can induce LTP at synapses on CA1 pyramidal neurons, provided that the postsynaptic depolarization triggers a dendritically initiated spike. These findings have important implications for predicting how in vivo activity might lead to LTP at these synapses.
Results
Whole-cell recordings were obtained from CA1 pyramidal neurons in rat hippocampal slices and excitatory postsynaptic potentials (EPSPs) were evoked by stimulation of the Schaffer collaterals (SCs) using a stimulating electrode positioned in the stratum radiatum (SR) near the CA3/CA1 border (for details, see Materials and Methods). Except where noted, GABA-mediated inhibition was blocked by including GABAA and GABAB receptor blockers in the bath solution. In some experiments, field EPSPs were also recorded (Fig. 1A; see Materials and Methods). Stimulus intensity was set to elicit large intracellular EPSPs (see Table 1). After monitoring the EPSP amplitude for 5 min, SCs were activated with a single burst (five pulses at 100 Hz). Temporal summation during the burst resulted in the firing of 2–5 action potentials (4.1 ± 0.2 action potentials; Fig. 1A). After this single-burst conditioning stimulus, EPSPs were potentiated robustly for at least 20 min (Fig. 1B; potentiation ratio: 1.66 ± 0.07, n = 21). In recordings held for longer times, this potentiation lasted >1 h (1.63 ± 0.04, n = 6), and potentiation of field EPSPs lasted >2 h (Fig. 1C; potentiation ratio: 1.60 ± 0.07, 2 h after burst, n = 4), indicating that activation of synaptic inputs with a single burst is sufficient to induce robust and stable LTP (Fig. 1D).
Fig. 1.

EPSPs in CA1 hippocampal neurons robustly potentiate after a single-burst stimulation of the Schaffer collateral pathway. (A) (Left) Recording configuration with whole-cell somatic patch-clamp recording, simultaneous extracellular field-potential recording, and stimulation electrode in stratum radiatum (SR, close to CA3/CA1 border). (Right) Intracellular (upper trace, resting membrane potential: −66 mV) and simultaneous extracellular (lower trace) recording of a single-burst response to a SR stimulus consisting of five pulses at 100 Hz (middle trace). (Insets) Representative EPSPs (upper) and field EPSPs (fEPSPs, lower) before (pre) and 20 min after (post) a single-burst SR stimulus. (B) Mean EPSP amplitude recorded in whole-cell patch-clamp configuration plotted vs. time. Time 0 indicates delivery of the stimulus. (C) Normalized fEPSP amplitude recorded before and after stimulation. All fEPSPs were recorded for at least 120 min (n = 4). In two of these experiments, LTP was recorded for up to 210 min (data not shown). (D) Potentiation ratio for whole-cell recordings (average EPSP 15–20 min after stimulation divided by the average of the 5-min baseline) and field potential recordings (average fEPSP 20–30 min and average fEPSP 110–120 min after stimulation divided by the average of the 10-min baseline). The potentiation ratio was not different between intracellular and extracellular recordings (ANOVA, P > 0.4). The number of experiments in each condition is indicated at bottom of the respective bars. Open circles represent the distribution of potentiation ratios in intracellular recordings (20 min after stimulation). Data are presented as mean ± SEM.
Table 1.
Comparison of baseline EPSP amplitudes and integrals of single-burst responses in different experimental conditions
| Single EPSP, mV | Burst EPSP integral, mV·s | |
|---|---|---|
| Control | 7.19 ± 0.27 (21) | 9.787 ± 0.114 (21) |
| NMDA receptor block | 7.52 ± 0.53 (9) | 10.582 ± 0.130 (9) |
| L-type VGCC block | 7.03 ± 0.55 (10) | 10.220 ± 0.140 (10) |
| TTX spikelets | 7.09 ± 0.18 (7) | 9.845 ± 0.375 (7) |
| TTX no spikelets | 6.94 ± 0.32 (4) | 9.610 ± 0.168 (4) |
VGCC, voltage-gated calcium channel. Numbers in parentheses indicate the number of experiments.
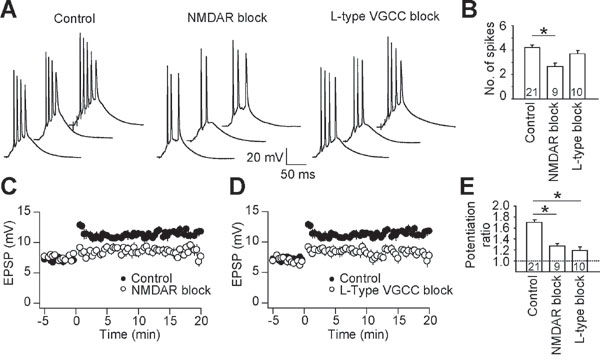
Because most forms of LTP require Ca2+ entry as a trigger, we tested the ability of NMDA-type glutamate receptor antagonists or L-type Ca2+ channel blockers to inhibit single-burst LTP (see Materials and Methods). Blocking each of these channels individually resulted in significant inhibition, but not complete block, of single-burst LTP [supporting information (SI) Fig. 6; potentiation ratio: control, 1.66 ± 0.07, n = 21; NMDA receptor block, 1.26 ± 0.05, n = 9; L-type channel block, 1.19 ± 0.07, n = 10]. The effects of NMDA receptor and L-type Ca2+ channel blockers on LTP were of similar magnitude, suggesting that depolarization and/or Ca2+ entry through each of these classes of channels contributes approximately equally to induction of single-burst LTP.
Several possible sources of depolarization contribute to induction of single-burst LTP. One is the synaptic depolarization itself, mediated by α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)- and NMDA-type glutamate receptors. Strong synaptic depolarization may also trigger locally generated dendritic spikes, a second possible source of depolarization. Finally, somatic or axonally initiated action potentials can propagate back to the synapses, providing a third possible source of postsynaptic depolarization. To determine the contribution of action potential backpropagation, we applied tetrodotoxin (TTX) locally to the soma, proximal axon, and proximal dendrites (i.e., perisomatic) during the LTP-inducing burst. TTX was applied at a concentration sufficient to prevent action potential firing in response to large somatic current injections (see Materials and Methods). Local TTX application also prevented the firing of full-amplitude action potentials during the synaptic burst (Fig. 2A). In most cases, however, small-amplitude spikes (spikelets) were triggered by the bursts. When experiments with and without spikelets were considered separately, LTP was only observed in the experiments with spikelets (Fig. 2 B and C).
Fig. 2.

Single-burst LTP depends on dendritic spikes and does not require backpropagating action potentials. (A) (Left) Recording configuration with somatic whole-cell recording of a CA1 pyramidal neuron and stimulation of SR. Action potential generation and backpropagation was blocked by local pressure application of TTX (2 μM; see Materials and Methods) to the soma and axon initial segment. (Right) Four example traces of somatic whole-cell responses to single-burst stimulation during somatic TTX application (four different cells; resting membrane potentials from left to right: −64 mV, −65 mV, −66 mV, −65 mV). Somatic spikelets (asterisks), indicating dendritically initiated spikes, are apparent in the three leftmost traces. (B) EPSP amplitude before and after single-burst stimulation of SR with action potential generation and backpropagation blocked by local TTX application; 7 of 11 neurons displayed somatic spikelets, whereas the other 4 did not. The 7 neurons exhibiting spikelets also exhibited potentiation in response to single-burst stimulation, whereas the 4 neurons with no spikelets did not exhibit LTP (P < 0.05). (C) Potentiation ratio for neurons that either displayed somatic spikelets or for neurons where no somatic spikelets were apparent. The number of experiments in each condition is indicated at bottom of the respective bars. Data are presented as mean ± SEM.
We have shown previously that spikelets observed under similar conditions in response to perforant path (PP) stimulation reflect large-amplitude dendritic spikes, which are significantly attenuated in the soma (9). To determine whether this is also the case during SC stimulation, we performed simultaneous somatic and dendritic recordings (recording distance: 180–285 μm from the soma) during single-burst stimulation and perisomatic TTX application (Fig. 3). In six of seven recordings, the peak dendritic depolarization was larger than the peak amplitude of the spikelet observed in the soma, and the dendritic membrane potential had a rapid depolarizing phase characteristic of a dendritically initiated spike.
Fig. 3.

Spikelets observed during single-burst stimulation originate from the dendrites. (A) Recording configuration. Whole-cell current-clamp recordings from the main dendrite of CA1 pyramidal neurons and from the soma were performed simultaneously. TTX was pressure applied to the soma, axon initial segment and proximal dendrite through a patch pipette. The response to strong somatic current injections was routinely monitored to confirm the absence of somatic regenerative events before and during single-burst stimulation (see Materials and Methods). (B) Dual somatic and dendritic whole-cell recordings of responses to single-burst stimulation during somatic TTX application in four different neurons (somatic resting membrane potentials from top to bottom: −65 mV, −67 mV, −65 mV, −64 mV). Six of seven neurons (including the four neurons shown here) exhibited a dendritic depolarization larger than the peak amplitude of the spikelet observed in the soma, and the dendritic membrane potential had a rapid depolarizing phase characteristic of a dendritically initiated spike. The locations of the dendritic recordings were 180–285 μm (median 220 μm) from the soma.
These data suggest that somatic spikelets are indicators of dendritic spikes, which in turn contribute to the induction of single-burst LTP. In addition, because neither the initial EPSP amplitude nor the burst integral was significantly different in experiments with spikelets and LTP compared with those with no spikelets and no LTP (see Table 1; unpaired t test, P > 0.5), synaptic depolarization alone (i.e., without dendritic spikes) appears to be insufficient to trigger LTP. Furthermore, the magnitude of LTP induced during perisomatic TTX application was not different from that induced in control experiments (potentiation ratio: control, 1.66 ± 0.07, n = 21; TTX, 1.65 ± 0.10, n = 7; single-factor ANOVA, P > 0.7). This result suggests that action potential backpropagation does not significantly affect the induction of LTP in response to strong synaptic bursts. Rather, somatic and axonal action potentials are likely triggered by dendritically initiated, forward-propagating spikes, which do not give rise to backpropagating action potentials in CA1 pyramidal neurons (10). Taken together, the data indicate that synaptic depolarization is not sufficient to induce LTP, that dendritic spikes are required for the induction of LTP, and that backpropagating action potentials do not affect the magnitude of LTP induced by dendritic spikes after single-burst synaptic activation.
NMDA receptor blockers, and to a lesser extent L-type Ca2+ channel blockers, reduced the number of action potentials during the single-burst synaptic conditioning stimulus (SI Fig. 6). However, the finding that block of action potentials using perisomatic TTX does not affect the magnitude of LTP makes it unlikely that the reduced number of action potentials itself contributes to the block of LTP by these drugs. Furthermore, the stimulus intensity was set to obtain EPSPs of the same amplitude in drugs as in control (see Table 1), and indeed the integral of the summated EPSPs during the burst was also not smaller in the drugs than in control (see Table 1). Thus, the effect of these drugs may be largely dendritic, influencing synaptic depolarization (by dendritic spiking or otherwise) more than axonal or somatic action potential initiation. Our data do not allow us to determine, however, whether the primary influence of these channels on single-burst LTP induction is dendritic spike-mediated depolarization, Ca2+ entry, or both.
To determine whether single-burst LTP is specific to the stimulated synapses, we performed an additional series of experiments using three stimulating electrodes to activate SC synapses in proximal, medial, and distal SR (Fig. 4A). Paired-pulse responses in the same pathway were compared with two-pathway responses at the same interval to determine pathway independence (see Materials and Methods). In six such experiments where three independent pathways were stimulated, LTP was observed only in the pathway activated by using a single-burst conditioning stimulus (always medial SR; potentiation ratio: 1.55 ± 0.06, n = 6; Fig. 4 B and C). Neither LTP nor heterosynaptic long-term depression (LTD) was observed in either of the unconditioned pathways (proximal and distal SR; potentiation ratio: 1.06 ± 0.08 and 0.95 ± 0.07, n = 6, respectively).
Fig. 4.

Single-burst LTP is specific to stimulated synapses. (A) Recording configuration: three stimulation electrodes were placed in distal, medial, and proximal stratum radiatum (dSR, mSR, and pSR, respectively). Pathway independence was confirmed with a cross-facilitation paradigm (see Materials and Methods). In all six experiments the medial pathway (mSR) was used for single-burst LTP induction. (B) EPSP amplitudes recorded with stimulation electrodes in dSR (open circles), mSR (filled circles), and pSR (gray circles) before and after single-burst stimulation of mSR. LTP was only observed in the pathway activated by using the single-burst conditioning stimulus (mSR). Neither heterosynaptic LTP nor LTD was observed in either of the unconditioned pathways (pSR and dSR). (C) Potentiation ratios of pSR, mSR, and dSR.
We also tested whether single-burst LTP of SC synapses induced heterosynaptic LTP or LTD in PP synapses from the entorhinal cortex by placing a stimulating electrode in stratum lacunosum-moleculare (SLM; SI Fig. 7A). Again, no heterosynaptic plasticity was observed in the unstimulated pathway (unstimulated SLM:potentiation ratio 0.98 ± 0.04, n = 8; SI Fig. 7B).
To determine whether single-burst LTP can also be induced at PP synapses, we performed similar experiments using PP as the pathway activated with the single-burst conditioning stimulus and SC as the unstimulated pathway. In these experiments, a single burst induced only modest, yet significant LTP in the PP (SI Fig. 7C; potentiation ratio: 1.17 ± 0.03, n = 8; statistically significant potentiation was observed in five of eight neurons). No heterosynaptic plasticity between SC and SLM synapses was observed (unstimulated medial SR: potentiation ratio 0.98 ± 0.04, n = 8). The larger amount of single-burst LTP observed in SC compared with PP (SI Fig. 7D) suggests that the ability of single bursts to induce LTP in CA1 pyramidal neurons is more robust in SC synapses from CA3 pyramidal neurons than in PP synapses from pyramidal neurons in layer III of entorhinal cortex.
Although bursts of action potentials clearly occur in both CA3 and CA1 pyramidal neurons of behaving animals (see Discussion), reproducing this situation by stimulating CA3 axons while blocking inhibition in the slice could alter the dynamics of dendritic integration and plasticity. Therefore, to determine whether single SC bursts could induce LTP with inhibition intact, we performed a similar series of experiments in the absence of GABA receptor blockers. With appropriate positioning of the stimulating electrode (Fig. 5A; see Materials and Methods) we could elicit 5–8 mV somatic EPSPs (mean: 6.67 ± 0.26 mV, n = 9), which summated during a burst to trigger 1–3 action potentials (Fig. 5B; compare with 2–5 action potentials in GABA blockers). We monitored EPSP amplitude for 5 min before and at least 20 min after the single-burst conditioning stimulus, and we observed significant LTP in 9 of 12 experiments (Fig. 5C). In 3 of the 9 experiments where LTP was induced, 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 50 μM 2-amino-5-phosphonovaleric acid (APV) were bath-applied to block excitatory drive of pyramidal neurons and inhibitory interneurons. In these experiments, CNQX and APV reduced the inhibitory postsynaptic potential (IPSP) by almost 90% (88.3 ± 5.6%), suggesting that most of the IPSP was caused by feedforward inhibition. By contrast, the same test was performed in two of the three experiments not exhibiting LTP, and the IPSP was blocked by <35% (34.6% and 30.2%), suggesting that most of the IPSP was caused by direct stimulation of inhibitory axons. These observations suggest that single-burst LTP can be induced in the presence of feedforward inhibition, but not when inhibitory interneurons are activated directly and synchronously with excitatory inputs. Not surprisingly, the magnitude of LTP with inhibition intact was smaller than in the presence of GABA receptor blockers, but significant LTP was nevertheless induced by the single burst (potentiation ratio: 1.38 ± 0.03; Fig. 5D).
Fig. 5.

Single-burst LTP can be induced with intact inhibitory synaptic transmission. (A) Recording configuration: the stimulation electrode was positioned at proximal SR in the CA3 region. At this site monosynaptic inhibitory responses (direct stimulation of inhibitory axons) was negligible. (B) Examples of whole-cell current-clamp responses to single-burst stimulation recorded with inhibitory transmission intact. Each trace represents a recording obtained from a different cell. (C) EPSP amplitude recorded before and after single-burst stimulation of SR. (D) Potentiation ratio after single-burst stimulation (average EPSP 15–20 min after stimulation divided by the average of the 5-min baseline). The magnitude of potentiation with inhibition intact was significantly smaller than in the presence of GABA receptor blockers (ANOVA P < 0.05) but displayed significant LTP compared with baseline (P < 0.01). The number of experiments in each condition is indicated at bottom of the respective bar. Data are presented as mean ± SEM.
Discussion
These experiments suggest that robust and stable LTP can be induced by a single burst of synchronous inputs from CA3 to CA1, provided that the input is strong enough to elicit a dendritic spike (and large enough to be detected as a spikelet in the soma during perisomatic TTX application). As is the case for some other forms of LTP requiring strong synaptic activation, single-burst LTP did not require somatic action potentials (6, 9, 11, 12). The ability of dendritically initiated spikes to provide the necessary depolarization to induce LTP, even during single presynaptic bursts, indicates that this may be an important function of the voltage-gated Na+ and Ca2+ channels expressed in pyramidal neuron dendrites.
One important question to consider is whether the strong, synchronous synaptic activation we used reasonably mimics in vivo activity. Assuming a mean unitary EPSP amplitude of 0.2 mV (13), EPSPs of 5–8 mV represent successful release and activation of 25–40 synapses, ≈0.1% of the Schaffer collateral synapses on a CA1 neuron (14). The presence of inhibition further increases the number of synapses that must be synchronously activated to produce the 5- to 8-mV EPSPs we used to induce single-burst LTP with inhibition intact. Considering that ≈10% of all CA3 pyramidal neurons may be synchronously activated during sharp-wave oscillations (15, 16), which occur not only during sleep but also during exploratory behavior (17), a similarly large percentage of CA3–CA1 synapses are likely to be activated synchronously in vivo. Indeed, given that resting potentials measured in vivo are around −60 mV, synchronous activation of several tens of excitatory synapses is seemingly necessary for CA1 to reach action potential firing threshold.
With stimuli just above threshold, CA1 pyramidal neurons have only weak intrinsic burst-firing properties (18, 19). Therefore, even larger numbers of synapses are likely necessary to drive the bursting normally observed both in CA1 pyramidal neurons in awake, behaving animals (20–24) and in response to the stimuli we used to evoke dendritic spikes. Bursting of the presynaptic CA3 pyramidal neurons also occurs in behaving animals (21–24), with the number and frequency of spikes per burst well within the range we used for our stimulus (five spikes at 100 Hz). Finally, dendritic spikes have been observed in apical dendritic recordings from hippocampal CA1 pyramidal neurons in vivo, particularly during sharp waves (25). Together, the available data in the literature therefore suggest that burst-like activation of large numbers of CA3 pyramidal neurons occurs in vivo, leading to postsynaptic dendritic spikes and bursts in CA1 pyramidal neurons, conditions that would appear consistent with single-burst LTP occurring in the hippocampus of awake animals.
As in other studies, we found that the magnitude of LTP observed in the presence of inhibition was less than when inhibition was blocked (5, 9, 26). Previous studies have demonstrated that LTP can be induced in vitro and in vivo by single bursts preceded by a heterosynaptic or homosynaptic priming stimulus (6, 7, 27). The function of the priming stimulus is not known, but it may facilitate LTP by reducing synaptic inhibition (27–31).
Another study showed that LTP and depotentiation could be induced by a single burst at the peak or trough of cholinergic θ-rhythms, respectively (8). Cholinergic reduction of inhibition (32–34) as well as the timing of the burst at the peak of θ (least inhibition) may also contribute to this form of LTP. Our study extends previous work by demonstrating that single-burst LTP does not require a priming stimulus or a bath-applied modulator, provided that the burst is strong enough to activate dendritic spikes, NMDA receptors, and L-type Ca2+ channels.
In the cortex, strong single shocks have been shown to induce LTD (35), suggesting that the ability of brief stimuli to induce synaptic plasticity may generalize to other brain regions. We did not observe LTD, however, nor did we observe heterosynaptic LTD of unstimulated synapses. Combined with the modest single-burst LTP at PP synapses in CA1, these comparisons suggest that the type of plasticity induced by single bursts may vary between different classes of synapses.
Although LTP is widely acknowledged as the primary candidate mechanism for the cellular basis of learning and memory, the connection is difficult to prove and certainly not universally accepted (36–39). One common criticism of this link is that many stimuli are required to induce most forms of LTP or STDP, whereas many forms of learning require only a single trial. For example, in rodents, fear conditioning, conditioned taste aversion, and inhibitory avoidance can all be learned in a single trial (40–44). In humans, the hippocampus is required for episodic memories, which must by definition be formed on a single trial (45, 46). Rodents also perform a number of hippocampus-dependent tasks in ways that indicate the capacity for episodic memory (46, 47). It has therefore been suggested that stimuli briefer than those normally used to induce LTP are necessary for the hippocampus to encode experience (48). Consistent with this idea, a recent report demonstrated that LTP is induced during single-trial learning (44).
How single-burst LTP relates to synaptic plasticity that occurs in vivo, in response to naturally occurring firing patterns (such as multiple bursts) and during different behavioral and neuromodulatory states, clearly requires further study. For example, it is not clear what neural activity occurs during a single-trial task to probe learning or during a single episode of experience. Nevertheless, our findings indicate that very brief electrophysiological events can lead to LTP. Together with previous reports (7, 8, 35), this finding enhances the notion that natural neural activity might lead to memory storage at hippocampal synapses, even during brief, one-time events, as required for single-trial learning and episodic memory.
Materials and Methods
Hippocampal Slice Preparation and Electrophysiology.
Transverse hippocampal slices (300 μm) were made from 3- to 5-week-old Wistar rats by using standard artificial cerebrospinal fluid (ACSF) and intracellular solutions as described previously (9). Recording temperature was 35 ± 2°C. In some recordings, the GABAA and GABAB receptor antagonists SR95531 (4 μM) and CGP52432 (1 μM) were added to the ACSF. Somatic and dendritic whole-cell current-clamp recordings were made in conjunction with one or two bridge amplifiers (BVC-700; Dagan, Minneapolis, MN). Electrode resistance in the bath for somatic recordings ranged from 2 to 4 MΩ, and series resistance ranged from 8 to 20 MΩ. The dendritic electrode resistance was 6–9 MΩ, and series resistance ranged from 14 to 35 MΩ. The average membrane resting potential was −65.6 ± 0.2 mV (n = 66 neurons). Field potentials were recorded with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) by using ACSF-filled patch-clamp electrodes with a tip resistance of ≈5 MΩ. Electrophysiological traces were digitized by an ITC-16 board (Instrutech, Port Washington, NY) under control of macros custom programmed in IGOR Pro (WaveMetrics, Lake Oswego, OR). EPSP amplitude was monitored by using 0.05-Hz synaptic stimulation. When noted, 2.5 μM TTX was pressure-applied (PMI-100; Dagan) through a patch pipette positioned near the soma and axon initial segment under visual guidance. The pressure injection preceded the stimulation by 2 s and lasted 0.5 s. The flow of solution was monitored by using 0.1% Fast Green in the TTX pipette and optimized to avoid TTX diffusion into SR. TTX application prevented axonal action potential generation in response to large (1.5–3 nA, 5 ms) somatic current injections. In all neurons, the EPSP amplitude during TTX application at the time of LTP induction remained unchanged. All other drugs were bath-applied and present throughout the entire experiment.
Synaptic Stimulation.
Bipolar stainless-steel electrodes (FHC, Bowdoin, ME) were used in conjunction with Dagan BSI-950 biphasic stimulators. Stimulating electrodes were positioned in distal stratum radiatum, closer to SLM than stratum pyramidale. Electrodes were always positioned at least 100 μm away from the recorded neuron and toward CA3. Thus, the activated synapses are likely distributed across several dendritic branches.
In some experiments, up to three separate stimulation electrodes were placed in proximal, medial, and distal SR and in SLM (see Fig. 4 and SI Fig. 7). To confirm pathway independence, the paired-pulse ratio (150-ms interpulse interval) was determined separately for each of the stimulated pathways; then adjacent pathways were paired at the same interval. Only those recordings that exhibited paired-pulse facilitation in all stimulated pathways and no significant cross-facilitation after 10 repetitions were included.
Data Analysis.
Analysis of electrophysiology data and statistical tests were performed by using IGOR Pro and Excel (Microsoft, Redmond, WA). Statistical analysis of multigroup data were performed by using a single-factor analysis of variance (Prism 4; GraphPad Software, San Diego, CA). When a significant difference between the groups was observed, Tukey's multiple comparison tests were performed to determine the level of significance for each pairwise comparison. For integral calculation of single-burst responses all action potentials were truncated. All measurements are presented as mean ± SEM.
Supplementary Material
Acknowledgments
We thank Heinz Beck, Jason Hardie, Michael Häusser, Tim Jarsky, Mayank Mehta, and Shannon Moore for comments on the manuscript. This work was supported by National Institutes of Health Grants NS-35180, MH-074866, and RR-01549 and the Alexander von Humboldt Foundation.
Abbreviations
- EPSP
excitatory postsynaptic potential
- LTD
long-term depression
- LTP
long-term potentiation
- PP
perforant path
- SC
Schaffer collateral
- SLM
stratum lacunosum-moleculare
- SR
stratum radiatum
- STDP
spike-timing dependent plasticity
- TTX
tetrodotoxin.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0707919104/DC1.
References
- 1.Martin SJ, Grimwood PD, Morris RG. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- 2.Bliss TV, Collingridge GL. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 3.Dan Y, Poo MM. Neuron. 2004;44:23–30. doi: 10.1016/j.neuron.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 4.Sjöström PJ, Turrigiano GG, Nelson SB. Neuron. 2001;32:1149–1164. doi: 10.1016/s0896-6273(01)00542-6. [DOI] [PubMed] [Google Scholar]
- 5.Abraham WC, Gustafsson B, Wigstrom H. Neurosci Lett. 1986;70:217–222. doi: 10.1016/0304-3940(86)90466-0. [DOI] [PubMed] [Google Scholar]
- 6.Diamond DM, Dunwiddie TV, Rose GM. J Neurosci. 1988;8:4079–4088. doi: 10.1523/JNEUROSCI.08-11-04079.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rose GM, Dunwiddie TV. Neurosci Lett. 1986;69:244–248. doi: 10.1016/0304-3940(86)90487-8. [DOI] [PubMed] [Google Scholar]
- 8.Huerta PT, Lisman JE. Neuron. 1995;15:1053–1063. doi: 10.1016/0896-6273(95)90094-2. [DOI] [PubMed] [Google Scholar]
- 9.Golding NL, Staff NP, Spruston N. Nature. 2002;418:326–331. doi: 10.1038/nature00854. [DOI] [PubMed] [Google Scholar]
- 10.Golding NL, Spruston N. Neuron. 1998;21:1189–1200. doi: 10.1016/s0896-6273(00)80635-2. [DOI] [PubMed] [Google Scholar]
- 11.Gustafsson B, Wigstrom H, Abraham WC, Huang YY. J Neurosci. 1987;7:774–780. doi: 10.1523/JNEUROSCI.07-03-00774.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McNaughton BL, Douglas RM, Goddard GV. Brain Res. 1978;157:277–293. doi: 10.1016/0006-8993(78)90030-6. [DOI] [PubMed] [Google Scholar]
- 13.Magee JC, Cook EP. Nat Neurosci. 2000;3:895–903. doi: 10.1038/78800. [DOI] [PubMed] [Google Scholar]
- 14.Megias M, Emri Z, Freund TF, Gulyas AI. Neuroscience. 2001;102:527–540. doi: 10.1016/s0306-4522(00)00496-6. [DOI] [PubMed] [Google Scholar]
- 15.Ylinen A, Bragin A, Nadasdy Z, Jando G, Szabo I, Sik A, Buzsaki G. J Neurosci. 1995;15:30–46. doi: 10.1523/JNEUROSCI.15-01-00030.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Csicsvari J, Hirase H, Mamiya A, Buzsaki G. Neuron. 2000;28:585–594. doi: 10.1016/s0896-6273(00)00135-5. [DOI] [PubMed] [Google Scholar]
- 17.O'Neill J, Senior T, Csicsvari J. Neuron. 2006;49:143–155. doi: 10.1016/j.neuron.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 18.Metz AE, Jarsky T, Martina M, Spruston N. J Neurosci. 2005;25:5763–5773. doi: 10.1523/JNEUROSCI.0624-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yue C, Remy S, Su H, Beck H, Yaari Y. J Neurosci. 2005;25:9704–9720. doi: 10.1523/JNEUROSCI.1621-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harris KD, Hirase H, Leinekugel X, Henze DA, Buzsaki G. Neuron. 2001;32:141–149. doi: 10.1016/s0896-6273(01)00447-0. [DOI] [PubMed] [Google Scholar]
- 21.Kandel ER, Spencer WA. J Neurophysiol. 1961;24:243–259. doi: 10.1152/jn.1961.24.3.243. [DOI] [PubMed] [Google Scholar]
- 22.Ranck JB., Jr Exp Neurol. 1973;41:461–531. doi: 10.1016/0014-4886(73)90290-2. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki SS, Smith GK. Exp Neurol. 1985;89:90–95. doi: 10.1016/0014-4886(85)90267-5. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki SS, Smith GK. Exp Neurol. 1985;89:71–89. doi: 10.1016/0014-4886(85)90266-3. [DOI] [PubMed] [Google Scholar]
- 25.Kamondi A, Acsady L, Buzsaki G. J Neurosci. 1998;18:3919–3928. doi: 10.1523/JNEUROSCI.18-10-03919.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wigstrom H, Gustafsson B. J Physiol (Paris) 1986;81:228–236. [PubMed] [Google Scholar]
- 27.Larson J, Lynch G. Science. 1986;232:985–988. doi: 10.1126/science.3704635. [DOI] [PubMed] [Google Scholar]
- 28.Pitler TA, Alger BE. J Neurosci. 1992;12:4122–4132. doi: 10.1523/JNEUROSCI.12-10-04122.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlson G, Wang Y, Alger BE. Nat Neurosci. 2002;5:723–724. doi: 10.1038/nn879. [DOI] [PubMed] [Google Scholar]
- 30.Davies CH, Collingridge GL. J Physiol (London) 1996;496:451–470. doi: 10.1113/jphysiol.1996.sp021698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson RI, Nicoll RA. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- 32.Pitler TA, Alger BE. J Physiol (London) 1992;450:127–142. doi: 10.1113/jphysiol.1992.sp019119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krnjevic K, Reiffenstein RJ, Ropert N. Neuroscience. 1981;6:2465–2474. doi: 10.1016/0306-4522(81)90092-0. [DOI] [PubMed] [Google Scholar]
- 34.Kim J, Isokawa M, Ledent C, Alger BE. J Neurosci. 2002;22:10182–10191. doi: 10.1523/JNEUROSCI.22-23-10182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holthoff K, Kovalchuk Y, Yuste R, Konnerth A. J Physiol (London) 2004;560:27–36. doi: 10.1113/jphysiol.2004.072678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barnes CA. Neuron. 1995;15:751–754. doi: 10.1016/0896-6273(95)90166-3. [DOI] [PubMed] [Google Scholar]
- 37.Lisman J, Lichtman JW, Sanes JR. Nat Rev Neurosci. 2003;4:926–929. doi: 10.1038/nrn1259. [DOI] [PubMed] [Google Scholar]
- 38.Shors TJ, Matzel LD. Behav Brain Sci. 1997;20:597–655. doi: 10.1017/s0140525x97001593. [DOI] [PubMed] [Google Scholar]
- 39.Stevens CF. Neuron. 1998;20:1–2. doi: 10.1016/s0896-6273(00)80426-2. [DOI] [PubMed] [Google Scholar]
- 40.Blank T, Nijholt I, Kye MJ, Radulovic J, Spiess J. Nat Neurosci. 2003;6:911–912. doi: 10.1038/nn1101. [DOI] [PubMed] [Google Scholar]
- 41.Irvine EE, Vernon J, Giese KP. Nat Neurosci. 2005;8:411–412. doi: 10.1038/nn1431. [DOI] [PubMed] [Google Scholar]
- 42.Misane I, Tovote P, Meyer M, Spiess J, Ogren SO, Stiedl O. Hippocampus. 2005;15:418–426. doi: 10.1002/hipo.20067. [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto T, Shimura T, Sako N, Yasoshima Y, Sakai N. Behav Brain Res. 1994;65:123–137. doi: 10.1016/0166-4328(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 44.Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- 45.Burgess N, Maguire EA, O'Keefe J. Neuron. 2002;35:625–641. doi: 10.1016/s0896-6273(02)00830-9. [DOI] [PubMed] [Google Scholar]
- 46.Eichenbaum H, Fortin NJ. J Exp Anal Behav. 2005;84:619–629. doi: 10.1901/jeab.2005.80-04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morris RG. Philos Trans R Soc London Ser B. 2001;356:1453–1465. doi: 10.1098/rstb.2001.0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morris RG, Frey U. Philos Trans R Soc London Ser B. 1997;352:1489–1503. doi: 10.1098/rstb.1997.0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}