

Abstract
Recent studies indicate that the expression of ERβ in breast cancer is lower than in normal breast, suggesting that ERβ could play an important role in carcinogenesis. To investigate this hypothesis, we engineered estrogen-receptor negative MDA-MB-231 breast cancer cells to reintroduce either ERα or ERβ protein with an adenoviral vector. In these cells, ERβ (as ERα) expression was monitored using RT-PCR and Western blot. ERβ protein was localized in the nucleus (immunocytochemistry) and able to transactivate estrogen-responsive reporter constructs in the presence of estradiol. ERβ and ERα induced the expression of several endogenous genes such as pS2, TGFα or the cyclin kinase inhibitor p21, but in contrast to ERα, ERβ was unable to regulate c-myc proto-oncogene expression. The pure antiestrogen ICI 164, 384 completely blocked ERα and ERβ estrogen-induced activities. ERβ inhibited MDA-MB-231 cell proliferation in a ligand-independent manner, whereas ERα inhibition of proliferation is hormone-dependent. Moreover, ERβ and ERα, decreased cell motility and invasion. Our data bring the first evidence that ERβ is an important modulator of proliferation and invasion of breast cancer cells and support the hypothesis that the loss of ERβ expression could be one of the events leading to the development of breast cancer.
Keywords: Adenoviridae; genetics; Breast Neoplasms; pathology; physiopathology; Cell Division; physiology; Cell Line; Cell Movement; physiology; Estrogen Receptor alpha; Estrogen Receptor beta; Estrogens; physiology; Gene Expression Regulation; physiology; Gene Transfer Techniques; Genes, Reporter; physiology; Genetic Vectors; Humans; Neoplasm Invasiveness; pathology; physiopathology; Receptors, Estrogen; physiology
INTRODUCTION
Estrogens modulate sexual gland development and reproductive functions but have also beneficial effects on cardiovascular, nervous systems or bone integrity (1). Besides, estrogens are potent mitogens in the mammary gland, where they regulate the growth, development and functioning of normal as well as cancerous breast (2, 3). Epidemiological evidences and numerous animal studies have indicated that estrogens play a role in the proliferation and progression of breast cancer; the removal of the ovaries or the treatment with antiestrogens oppose their deleterious effects (4, 5). Though there are growing evidences that estrogens can operate through non genomic pathways (6–8), estrogen receptors, ERα (NR3A1) and ERβ (NR3A2), which belong to a large family of nuclear receptors, are mediating the genomic action of estrogens by acting as ligand dependent transcription factors (9, 10). Most human breast cancers, at least initially, express ERα and the presence of ERα is generally considered as an indication of hormone dependence, eventhough only 60% of ER-positive tumors will respond to adjuvant therapy with tamoxifen (11).
Although ERα has been cloned more than 10 years ago (12), the presence of ERβ has been unrecognized till recently (13, 14). ERα and ERβ have diverged early during evolution (15) and differ mostly in the N-terminal A/B domain, and to a lesser extent in the ligand binding domain (E domain). These differences suggest that the two receptors could serve distinct actions. Indeed, the activation functions AF-1 and AF-2 located respectively in the A/B and LBD domains display activities that are promoter and cell-specific (16–18). Cowley et al. (17) have shown that the AF-1 activity of ERβ is weak compared with that of ERα on estrogen-responsive reporters, whereas their AF-2 activities are similar. In turn, when both AF-1 and AF-2 functions are active in a particular cell and/or on a particular promoter, the activity of ERα greatly exceeds that of ERβ, whereas ERα and ERβ activities are similar when only AF-2 is required. The weaker activity of ERβ in many promoter and cell contexts has also been reported by several groups (18–20).
Moreover, ERα and ERβ knock-out mice have been generated and demonstrated striking different patterns (21, 22). ERβ knock-out mice show significantly reduced fertility in female, with ovaries exhibiting follicular arrest and anovulation. However, these mutant mice have a normal mammary gland development and lactate (21). On the contrary, ERα knock-out mice have an impaired fertility for both sexes and exhibit an estrogen-insensitive mammary gland and genital tract (22), suggesting possible overlapping and distinct action on the expression of genes regulating the important biological functions. Concerning the rodent mammary gland, both estrogen receptors are expressed in the rat mammary gland but the presence and cellular distribution of the two receptors are distinct (23). In prepubertal rats, ERα is detected in 40% of the epithelial cell nuclei. During puberty and pregnancy, ERα expression is strongly decreased, whereas ERα is present in 70% of epithelial cells during lactation. About 60–70% of epithelial cells express ERβ at all stages of breast development. Cells coexpressing both receptors represent up to 60% of the epithelial cells during lactation but are rare during pregnancy. Moreover, more than 90% of ERβ expressing cells cells do not proliferate (23).
In agreement with these observations, recent studies in humans indicate that the ERβ expression is decreased between normal and neoplastic breast, as well as colon and ovarian cancer (24–30), suggesting that ERβ could be an inhibitor of tumorigenesis. In order to test this hypothesis, we have engineered a receptor negative breast cancer cell line to express functional ERβ. In this cell line, ERβ was able to activate the transcription of synthetic promoters in transient transfection experiments as well as natural endogenous promoters. Interestingly ERβ had major effects both on the proliferation, motility and morphology of the cells, suggesting that ERβ could effectively act as an inhibitor of breast cancer development.
MATERIALS AND METHODS
Plasmids
The reporter plasmid ERE2-TK-CAT contains two copies of the consensus ERE cloned upstream of the minimal herpes simplex virus thymidine kinase promoter. CMV-hERα and CMV-hERβ correspond to the wild-type ERα and ERβ cDNAs cloned into CMV5. CMV-GAL reporter was used as an internal control and corresponds to the β-galactosidase gene under the control of the CMV promoter.
Recombinant adenovirus construction and propagation
The complete coding sequence of wild-type hERβ or hERα cDNAs were subcloned in BamHI site of the pACsk12CMV5 shuttle vector. To obtain recombinant viruses, permissive HEK-293 cells (human embryonic kidney cells) were cotransfected with the recombinant pACsk12CMV5-hER plasmid and with pJM17, which contains the remainder of the adenoviral genome as previously described (31–33). In vivo recombination of the plasmids generates infectious viral particles (Ad-hERα or Ad-hERβ). DNA from these viruses were screened for the presence of the hER cDNA by PCR with hER primers, and titered virus stocks were used to infect MDA-MB-231 cells.
Cell Culture and Transient Transfection
HEK-293 cells were cultured in DMEM-F12 supplemented with 10% FCS (fetal calf serum) in the presence of 5% CO2. MDA-MB-231 cells (human breast cancer cells) were cultured in Leibovitz L-15 medium containing 10% FCS. 3.105 cells were plated in 6-well plates in phenol red -free DMEM-F12 supplemented with 10% CDFCS (charcoal-dextran treated FCS) 24h before transfection. Transfections were performed by lipofection (lipofectamine, Life Technologies, Rockville, MA) using 4 μg of CAT reporter construct, 1 μg of the internal reference β-galactosidase reporter plasmid (CMV-GAL) and CMV-hER expression vectors or recombinant viruses per well. Transactivation ability was determined by CAT activity on the whole cell extract as previously described (34).
Whole cell extract preparation and western blot
MDA-MB-231 cells were lysed in TEG (10 mM Tris-HCl, pH 7.4, 1.5 mM EDTA, and 10% glycerol) containing protease inhibitors (5 μg/ml aprotinin, leupeptin and pepstatin A and 0.1 mM phenylmethylsulfonyl fluoride). Then, cells were sonicated and the cellular debris were pelleted by centrifugation at 13000g for 20 minutes in microfuge tubes. 45 μg of whole cell extract proteins were subjected to SDS-PAGE followed by electrotransfer onto a nitrocellulose membrane. The blot was probed with anti hERα (SRA-1000) or hERβ antibody (1:1000) (CWK-F12, (35)) and then incubated with goat anti-mouse IgG horseradish peroxidase conjugated antibody (1 μg/ml). ECL kit from Amersham (Arlington, IL, USA) was used for detection.
Gel Mobility Shift Assays
Briefly, 30000 cpm of the [32P]-labeled (AGCTCTTTGATCAGGTCACTGTGACCTGACTTT) ERE double strand oligonucleotide was combined with 1 μg poly (dI-dC) and 5 μg of MDA-MB-231 whole cell extract (WCE). When indicated, anti-hERα (Stressgen, SRA-1000) or anti-hERβ antibodies (CWK-F12, a kind gift of Pr B.S. Katzenellenbogen, for more details, see (35)) were added. The reaction buffer contained 20 mM Hepes, pH 7.9; 1 mM DTT; 50 mM KCl; 10% glycerol; 2.5 mM MgCl2. Protein-DNA complexes were separated from the free probe by non-denaturating gel electrophoresis with 4% polyacrylamide gels in 0.5 X TBE.
Detection of ERα and ERβprotein by immunocytochemistry
MDA-MB-231 cells were seeded in 10% CDFCS DMEM-F12 on sterile coverslips in six-well plates and infected with the different adenoviruses. 48h after infection, the cells were fixed (formaldehyde 3.7% 12 min/methanol 5 min/acetone 2min) and washed with PBS. The coverslips were incubated for 30 min with PBS containing non-immune rabbit serum (1:40). Then the cells were incubated with the primary antibody (ERα: SRA-1000 1:2000 (Stressgen); ERβ: CWK-F12 1:3000) in PBS for 60 min at room temperature. The cells were then incubated with the secondary antibody (rabbit anti-mouse peroxidase conjugate, Sigma,13000) in PBS-BGG (bovine gamma globulin) for 30 min at room temperature. Finally, the cells were incubated with a DAB (diaminobenzidine) chromogen solution (0.66mg/ml in PBS+0.08% H2O2 (30 vol)) for 10 min at room temperature. The cells were counterstained with hematoxylin.
RNA Isolation, Northern Blot and RT-PCR
Total RNA was isolated from MDA-MB-231 cells using the TRIzol reagent from Life Technologies (Rockville, MA) as described by the manufacturer. Probes were amplified by RT-PCR using specific primers:
| ERα | AAAAGACCGAAGAGGAGGGAGAAT/ATCCGGAACCGAGATGATGTAG, |
| ERβ: | GCCGCCCCATGTGCTGAT/GGACCCCGTGATGGAGGACTT, |
| c-myc: | TACCCTCTCAACGACAGCAGCTCGCCCAAC/TCTTGACATTCTCCTCGGTGTCCGAGGACC, |
| p21: | CGAGTGGGGGCATCATCAAAAAC/TGTTACAGGAGCTGGAAGGTGTTTG, |
| pS2: | TGACTCGGGGTCGCCTTTGGAG/GTGAGCCGAGGCACAGCTGCAG, |
| TGFα: | CCTGTTCGCTCTGGGTATTGTGTTG/CGTGGTCCGCTGATTTCTTCTCTAG). |
Reverse transcription was performed using random primers and GenAmp (Roche, Basel, Switzerland) RT-PCR kit. The amplifying primers are described above. The PCR was performed with Platinium Taq polymerase (Life Technologies) and 1:40 of reverse transcription reaction. Cycles of 30s at 94°C, 1 min at 60°C, 1.30 min at 72°C were done 29 times. A tenth of each PCR was electrophoresed on agarose gel. For Northern blot analysis, 20 μg of total RNA were electrophoresed and then hybridized with the different probes.
Cell proliferation studies
Cells were maintained for 24h in 10% CDFCS DMEM-F12 and then seeded at 30000 cells/well in 24-well dishes in 10% CDFCS DMEM-F12. Cells were infected overnight with the different viruses. The next morning, the medium was removed and replaced with fresh 10% CDFCS DMEM-F12 medium. Treatment with E2 or ICI 164,384 began at the same time. After 2, 4 or 6 days of E2, ICI 164,384 or both compounds treatments, the cells were trypsinized and counted using a Coultronics Coulter counter.
Wound healing assay
Cells were plated in 6-well dishes in DMEM-F12 containing 5% CDFCS. 24h after plating, the cells were infected with the different viruses overnight. The next morning, ethanol or E2 treatment started. After 20h of treatment, wound induced migration was triggered by scraping the cells at day 1 with a blue tip and the wound was pictured immediately. 18h after the wound (day 2), the cells were pictured again. The % of wound filling was calculated by measuring on the pictures the remaining gap space. The ratio of the gap space at day 2 over the gap space at day 1 gives the percentage of wound filling.
Invasion assay
MDA-MB-231 cells infected with Ad5, Ad-hERα or Ad-hERβ (MOI 100) were plated 24h after infection in the upper compartment of a 24-well Transwell (Costar) on a polycarbonate filter (8μm pore size) which was first coated with 30μg of matrigel (Becton Dickinson). The lower compartment of the well was filled with DMEM-F12 supplemented with 10% CDFCS and 30μg/ml fibronectin (Sigma). As a control, the same cells were layered on 24-well plates. Cells were treated with ethanol vehicle or E2 (10−8 M). After 36h of migration, cells which have migrated to the lower side of the filter and cells present in the control plates were trypsinized and counted using a Coulter counter.
Morphology analysis
MDA-MB-231 cells were cultured in DMEM-F12 supplemented with 10% CDFCS. After infection with Ad5, Ad-hERα or Ad-hERβ (MOI 100), cells were tretaed with control vehicle ethanol or E2 (10−8M) for 48h and then pictured using a Zeiss phase contrast microscope.
RESULTS
Adenoviruses elicit a high infection of MDA-MB-231 cells
Replication-deficient adenoviruses encoding hERα or hERβ cDNA sequences were constructed and used to infect ERα-negative MDA-MB-231 cells (Fig. 1A). To control the efficiency of infection of MDA-MB-231 cells, we first treated the cells with an adenovirus coding for the β-galactosidase protein (Fig. 1B). We observed a very efficient infection of the cells when increasing the MOI (multiplicity of infection) from 1 to 100, leading to an infection of about 80% of the cells at the highest MOI.
Fig 1. Schematic representation of adenovirus construction and high infection efficiency of MDA-MB-231 cells.
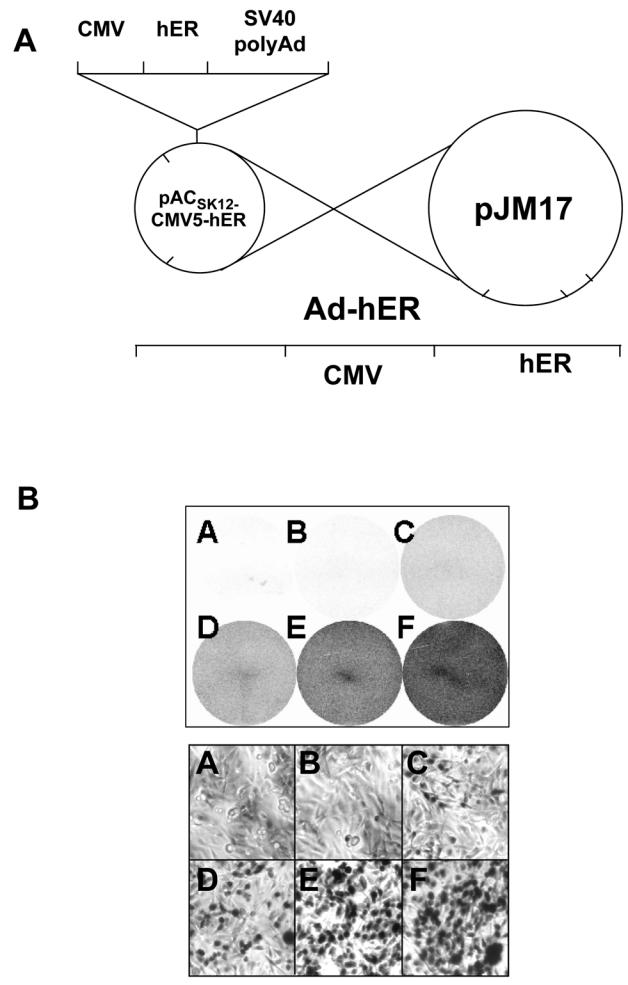
A. Ad-hERα and Ad-hERβ viruses were constructed as described in Materials and Methods using in vivo recombination in HEK-293 cells. The recombination occurs between the shuttle vector pACSK12-CMV5 carrying hERα or hERβ cDNAs and pJM17 adenoviral sequences. B. MDA-MB 231 cells were grown in 6-well plates and infected overnight with no virus (A), or Ad-GAL virus at MOI (multiplicity of infection) 1 (B), 10 (C), or 25 (D), 50 (E), 100 (F). β-galactosidase activity was monitored after 48 h of expression. The upper panel corresponds to a picture of the entire plate and the lower panel to a 200 fold magnification of each well.
Adenoviral mediated expression of ERα and ERβ
ERα and ERβ expression was examined following infection of MDA-MB-231 cells (Fig. 2A, 2B and 2D). We could not detect any expression of ERα or ERβ in MDA-MB-231 cells infected with the non recombinant virus Ad5 as well as in non infected cells (data not shown). On the contrary, after infection with Ad-hERβ virus, a high expression of ERβ could be seen at RNA (Fig. 2A) and protein (Fig. 2B) levels. Similarly, expression of an equivalent amount of ERα was detected in Ad-hERα infected cells (Fig. 2A and 2B). To further check the functionality of the expressed ERβ protein, we analyzed its ability to bind to an ERE (estrogen responsive element) DNA sequence by performing gel shift experiments (Fig. 2C). A specific binding could only be seen when ERα or ERβ extracts were used (lanes 3 and 5). Moreover, the ERβ shifted complex had a faster migration rate than the ERα one. The specificity of the shifted complex could be further demonstrated by using ERα and ERβ specific antibodies (lanes 4 and 6). We then determined the cellular localization of ERα and ERβ expressed proteins by immunocytochemistry (Figure 2D). ERα- and ERβ-infected cells displayed a clear and exclusive nuclear staining when using ERα and ERβ antibodies respectively. These data confirm our previous findings with ERα (31) and suggest that ERβ protein is correctly expressed at RNA and protein levels in MDA-MB-231 cells, is addressed to the nucleus and is able to bind to DNA.
Fig. 2. Adenoviral expression of ERα and ERβ in MDA-MB-231 cells.
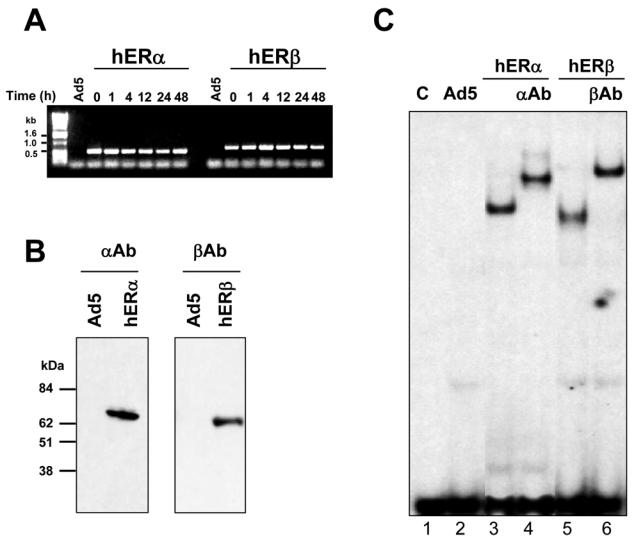
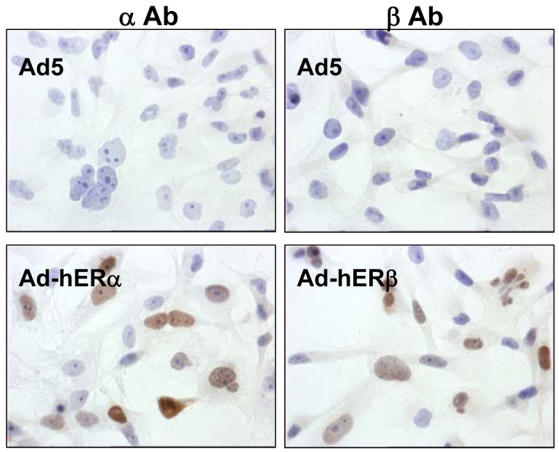
A. MDA-MB-231 cells were infected with the non recombinant (Ad5), Ad-hERα or AdhERβ viruses at MOI 100. After 1 to 48h of treatment with 10−8M E2, hERα and hERβ expression was monitored by RT-PCR using primers located in the ligand binding domain. The PCR products have a size of 542 bp and 703 bp for ERα and ERβ respectively. B. hERα and hERβ protein expression was analysed by Western blot using hERα (αAb) or hERβ (βAb) specific antibodies. C. WCE from non infected cells (C, lane 1), non recombinant viruses (Ad5, lane 2), Ad-hERα (hERα) (lanes 3–4) or Ad-hERβ (hERβ) (lanes 5–6) infected MDA-MB 231 cells were used for gel shift assay using a consensus ERE as a probe. Supershifts were performed using specific anti hERα (αAb) (lane 4) or anti-hERβ (βAb) (lane 6) antibodies. D. MDA-MB-231 cells were infected with Ad5, Ad-hERα or Ad-hERβ (hERβ) at MOI 25 and ERα and ERβ expression was visualized by immunocytochemistry using ERα (αAb) and ERβ (βAb) specific antibodies.
ERβ is able to activate estrogen-sensitive reporter genes
To further assess the functionality of the receptors produced, we analysed their ability to transactivate estrogen-sensitive reporter genes (Fig. 3). As a control, we transfected regular plasmids encoding hERα or hERβ along with the ERE2-TK-CAT reporter (Fig. 3A). We observed a strong activation of the reporter by ERα in the presence of E2. ERβ was also able to activate the transcription in the presence of E2, but the stimulation was half that obtained with ERα (Fig. 3A). We then tested the ability of our recombinant Ad-hERβ virus to activate the ERE2-TK-CAT reporter (Fig. 3B). When increasing MOI of Ad-hERβ were used, a strong ligand-dependent activation of the reporter occurred, demonstrating that the adenovirally produced ERβ exhibits a classical pattern of activation. However, at low MOI, ERβ was less active than ERα, whereas the use of higher MOI of Ad-hERβ virus elicited a good activation of the reporter. We then checked the sensitivity of ERβ to estrogen stimulation (Fig. 3C). We observed a characteristic dose-response curve for ERβ, similar to that obtained for ERα. A slight shift in the sensitivity to E2 was observed for ERβ, which reached its maximal activity at 10−8M, whereas ERα activity was maximal at 10−9M. Similar results have been obtained by others showing that ERβ has a weaker activity than ERα at low concentrations of E2 (36). In order to demonstrate that the expressed receptors were triggering estrogen effect, we analyzed their transactivation ability in the presence of the pure antiestrogen ICI 164, 384 (Fig. 3D). As expected, ICI 164, 384 could not stimulate ERα or ERβ activity but completely shut down both the basal and E2-induced activities of both receptors, suggesting that the basal activity of both receptors was most probably due to remaining traces of E2 in the stripped serum.
Fig. 3. hERα and hERβ can activate the transcription of estrogen-sensitive reporter genes.
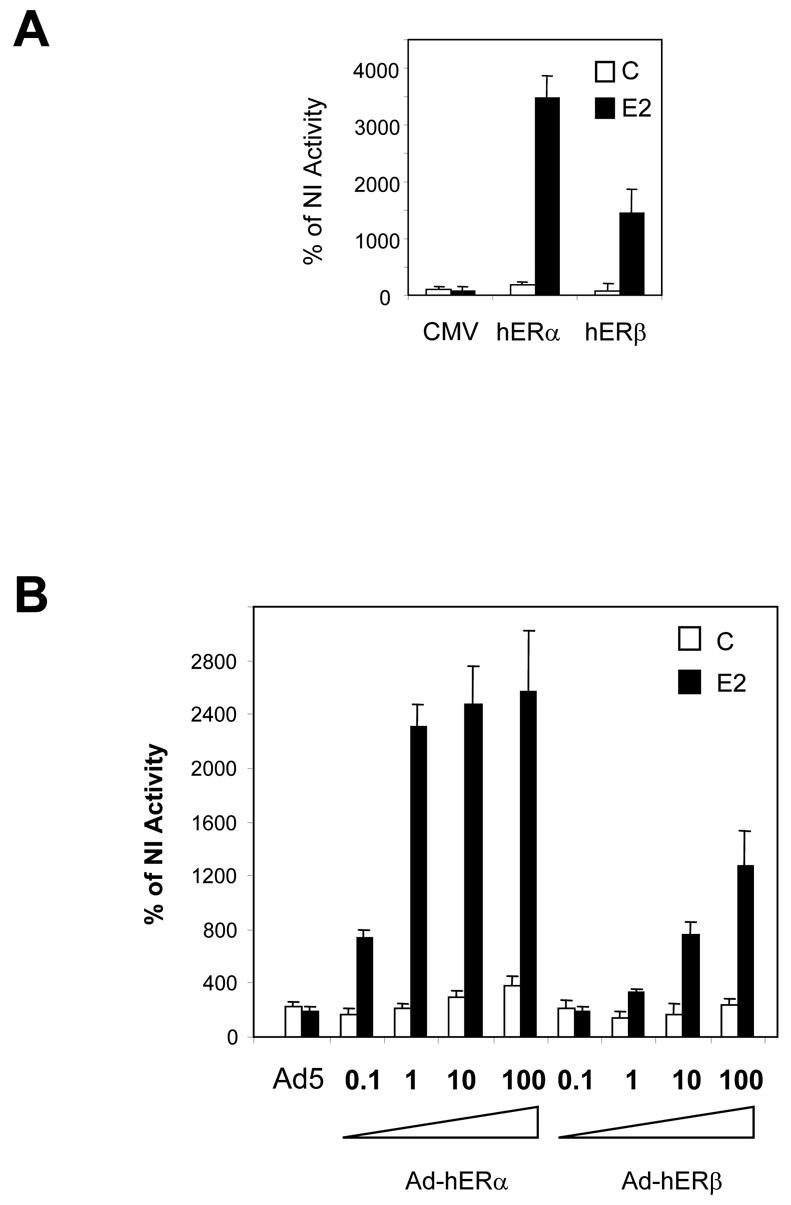
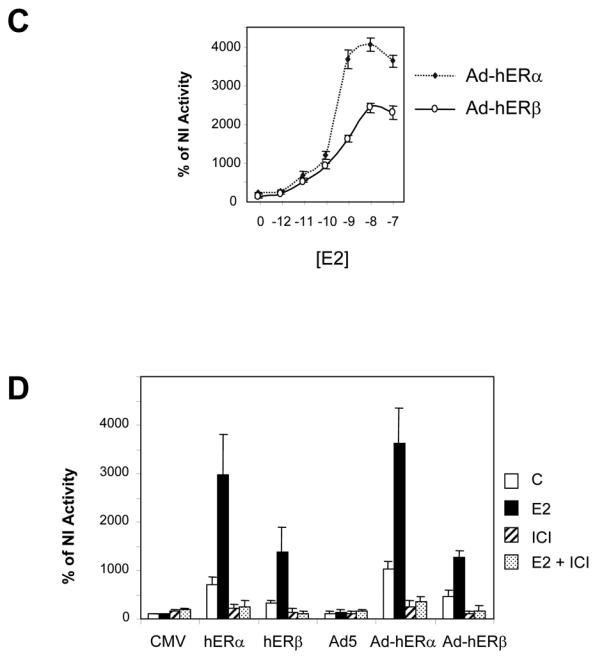
A. Empty CMV5 vector (CMV), CMV-hERα (hERα) or CMV-hERβ (hERβ) vectors were cotransfected in MDA-MB-231 cells with ERE2-TK-CAT reporter constructs and CMV-GAL internal reporter plasmid. Cells were grown for 36 h in the presence of control vehicle ethanol (C) or 10−8M E2. Results are expressed as the percentage of CAT activity in non infected cells (NI) and represent the mean ± SD (n = 5) of CAT activity after normalization for β-galactosidase activity. B. Non infected (NI) or Ad5, Ad-hERα, or Ad-hERβ infected MDA-MB-231 cells were transfected with ERE2-TK-CAT and CMV-GAL reporter constructs. Increasing MOI of Ad-hERα and Ad-hERβ viruses (0.1/1/10/100) were used. Cells were grown for 36 h in the presence of control vehicle ethanol (C) or 10−8M E2. Results are expressed as the percentage of CAT activity in non infected cells (NI) and represent the mean ± SD (n = 6) of CAT activity after normalization for β-galactosidase activity. C. MDA-MB-231 cells were infected with Ad-hERα and Ad-hERβ at MOI 100 and treated with increasing concentrations of E2. Results are expressed as the percentage of CAT activity in non infected cells (NI) and represent the mean ± SD (n = 6) of CAT activity after normalization for β-galactosidase activity. D. MDA-MB-231 cells were either transfected with empty CMV5 vector (CMV), CMV-hERα (hERα) or CMV-hERβ (hERβ) vectors or infected with Ad5, Ad-hERα or Ad-hERβ viruses along with ERE2-TK-CAT and CMV-GAL reporter constructs. Cells were grown for 36 h in the presence of control vehicle ethanol (C), 10−8M E2, ICI 164,384 (10−6M) or the combination of E2 and ICI 164, 384 (10−8 M and 10−6 M respectively). Results are expressed as the percentage of CAT activity in non infected cells (NI) and represent the mean ± SD (n = 6) of CAT activity after normalization for β-galactosidase activity.
ERα and ERβ have common but also distinct target genes
Very little is known about the specific target genes of ERβ. Therefore, we examined in these ERβ-positive cells the expression of 4 genes, TGFα (Transforming growth factor alpha), p21, c-myc proto-oncogene and pS2 genes, which are also regulated in ERα-infected cells in the presence of E2 (Fig. 4A, B). ERα and ERβ were able to activate the expression of pS2, p21 and TGFα genes in an estrogen-dependent manner. ERβ was 2 to 3 fold less potent than ERα to stimulate pS2, p21 and TGFα expression than ERα. pS2 activation was maximum at 48h of E2 treatment for both receptors. For TGFα and p21 genes the maximal activation was reached at 24h for both receptors, suggesting that these genes exhibit an earlier response than pS2. Very interestingly, ERα almost completely abolished the expression of c-myc in the presence of E2, whereas ERβ had no significant effect. These data suggest that ERα and ERβ effects on target genes differ both in the amplitude of regulation and in the nature of the genes regulated. To evaluate whether antiestrogens could also modulate the expression of these genes, we performed the same experiments in the presence of ICI 164,384, alone or in combination with E2 (Fig. 4C). ICI 164,384 was able to decrease the basal level of expression of pS2, p21 and TGFα and most interestingly completely reverse the induction of pS2, p21 and TGFα genes by E2 in ERα and ERβ infected cells.
Fig. 4. Modulation of endogenous gene expression by hERα and hERβ.
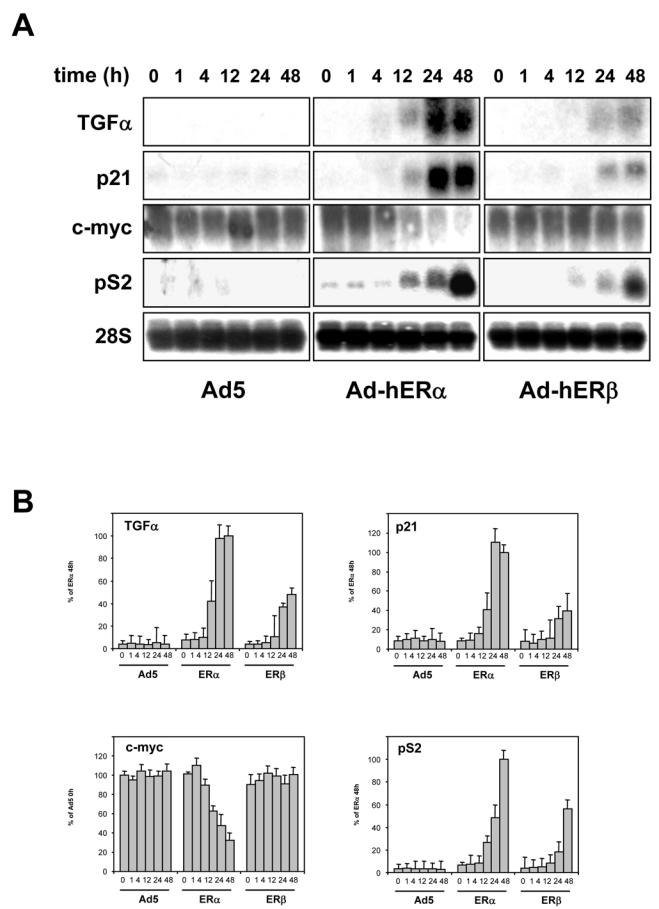
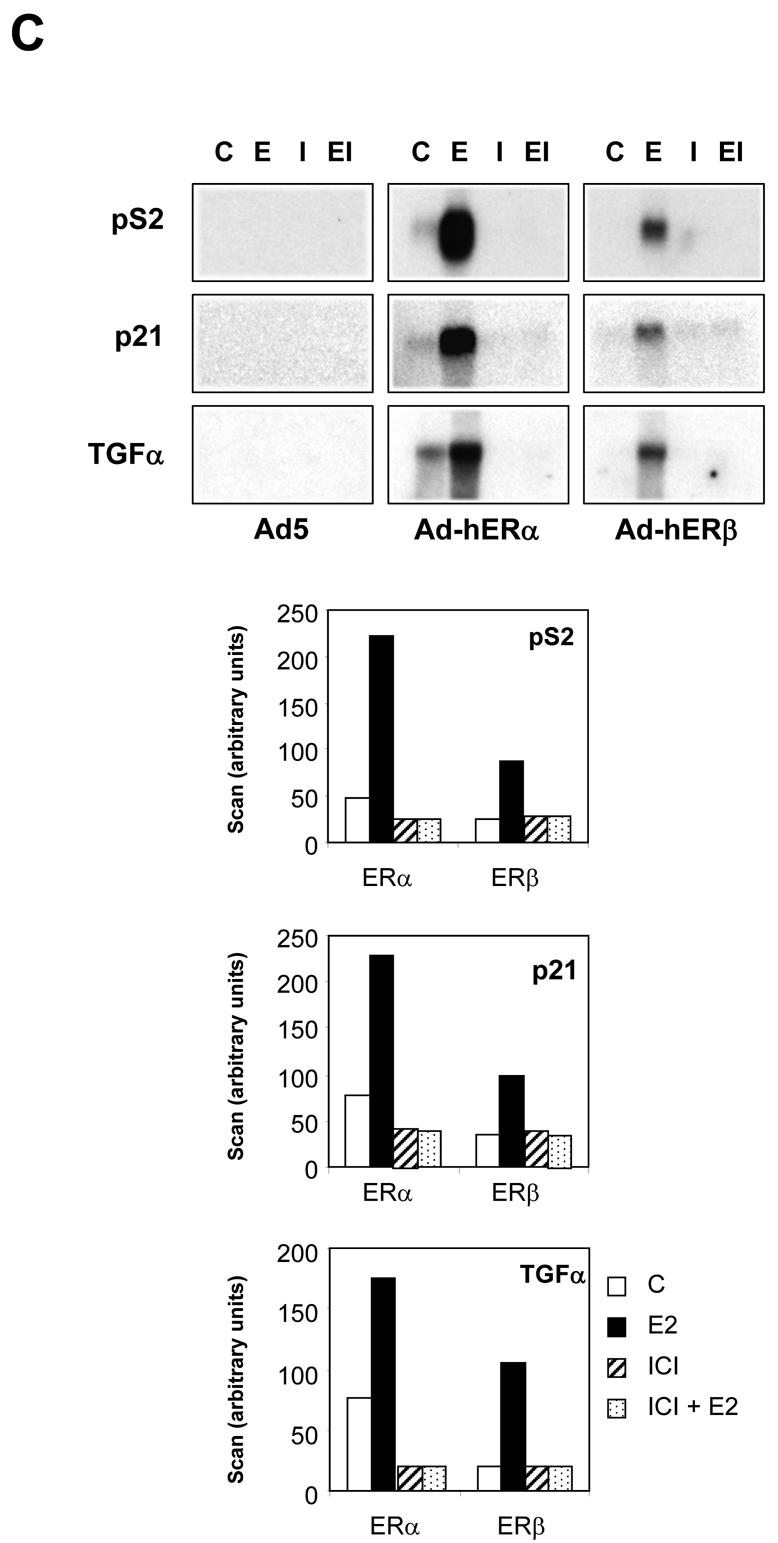
MDA-MB-231 cells were infected at MOI 100 with the different viruses. 24 h after infection, the E2 treatment began sequentially. All cells were harvested at the same moment following different times of E2 exposure and RNA extracted. 20 μg of total RNA were used for Northern blot and hybridized with TGFα, p21, c-myc or pS2 probes. Equal loading was checked with an RNA 28S probe. Data of a representative experiment are shown here. B. Quantification of Northern experiments after normalization by 28S RNA levels. Results are the mean ± SD (n = 3) of 3 experiments. C. The same experiments were performed in the presence of control vehicle ethanol (C), E2 (10−8M) (E), ICI 164,384 (10−6M) alone (I) or in combination (EI). Data of a representative experiment are shown here and the quantification after normalization with 28S RNA is indicated below. Results are expressed in arbitrary units of scan.
ERα and ERβ are potent inhibitors of the proliferation
The main question was to determine if ERβ expression could modulate the proliferation rate of MDA-MB-231 cells. Control cells (non-infected or Ad5 infected) had a similar growth pattern in the absence or in the presence of estrogens (Fig. 5A, left panel). When MDA-MB-231 cells were infected with Ad-hERα virus (Fig. 5A, middle panel), they proliferated at the same rate as naive cells in the absence of estrogens. But when E2 was added, a strong inhibition (50%) of the proliferation occurred, which is in agreement with our previous work (31). Very interestingly, ERβ was also able to inhibit the proliferation of MDA-MB-231 cells, but this effect was totally ligand-independent: a 40% inhibition occurred whether or not estrogens were present (Fig. 5A, right panel). This is to our knowledge, the first direct evidence that ERβ can be involved in the proliferation control of breast cancer cells. To determine the effects of pure antiestrogens on cell proliferation, we performed experiments in the presence of ICI 164,384 (Fig. 5B). ICI 164,384 had no effect by itself on the proliferation of naive, ERα or ERβ expressing cells. However, ICI 164,384 completely reversed E2-triggered inhibition in Ad-hERα infected cells. Moreover, ICI 164,384 or in combination with E2 could not modulate ERβ expressing cells proliferation rate. These data confirm that inhibition of the proliferation by ERα is ligand-dependent, whereas the inhibition by ERβ was ligand-independent.
Fig. 5. hERα and hERβ are able to repress the proliferation of MDA-MB-231 cells.
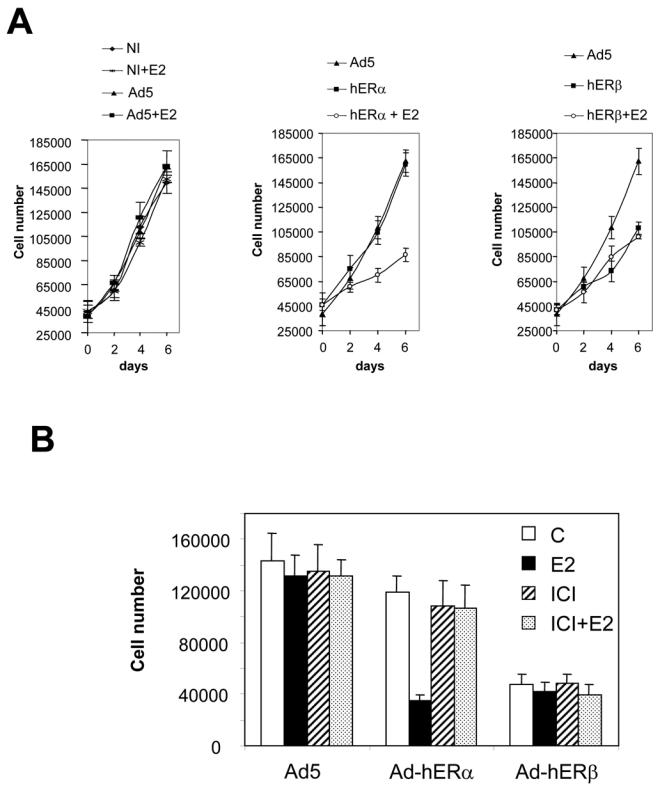
MDA-MB-231 cells were either non infected (NI) or infected with Ad5, Ad-hERα or AdhERβ viruses at MOI 100. A. The cells were treated with ethanol vehicle or E2 (10−8M) 24 h after the beginning of the infection. Proliferation rate was determined by counting the cells at day 2, 4 and 6. Results represent the mean ± SD of 4 determinations. B. The effect of the pure antiestrogen ICI 164,384 was evaluated by treating the cells either with ICI 164,384 (10−6M) alone or in combination with E2 (10−8M). On day 4, cells were counted and results represent the mean ± SD of 3 determinations.
ER expression has profound effects on invasion, motility and morphology of the cells
It was of interest to determine whether ERβ expression might affect cell motility and therefore modulate the invasiveness of the cells. To address this issue, we performed wound healing-induced migration experiments (Figure 6A, B). Infected cells were forced to migrate through the space created by scraping the monolayer with a tip. After 18h of migration, Ad5-infected MDA-MB-231 cells had filled 85% of the wound (Figure 6B). On the contrary, ERα-infected cells had only partially (50%) filled the wound. This lack of migration was not significantly affected when E2 was added. ERβ was also able to inhibit the cell motility. This inhibition occurred in the absence or in the presence of E2 (filling of only 30–40% of the space), suggesting that ERβ was a more potent inhibitor of motility than ERα. We then evaluated the migration ability of these cells using the classic Transwell in vitro assay. In this assay, cells are encouraged to migrate from the upper compartment coated with matrigel to the lower compartment coated with fibronectin, which serves as a chemoattractant. After 36h of migration, we observed that Ad-hERα and Ad-hERβ migrating cells represent respectively 70% and 50% of the control migrating cells (Figure 6C). Addition of E2 did not change the migration rate of any kind of cells. In conclusion, motility and invasion assays are in close agreement suggesting that ERβ is a more potent inhibitor of cell migration than ERα.
Fig. 6. hERβ is a strong inhibitor of motility and invasion.
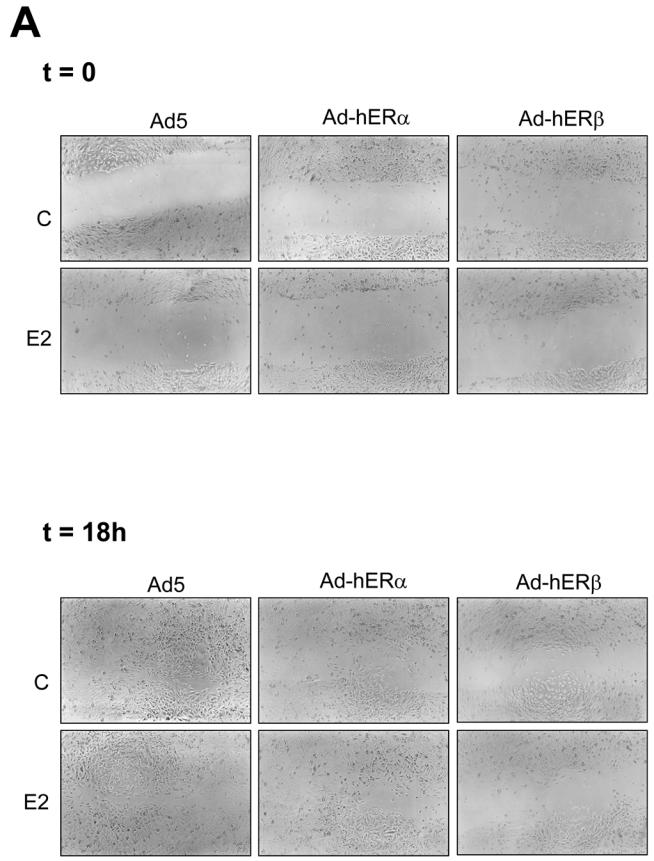
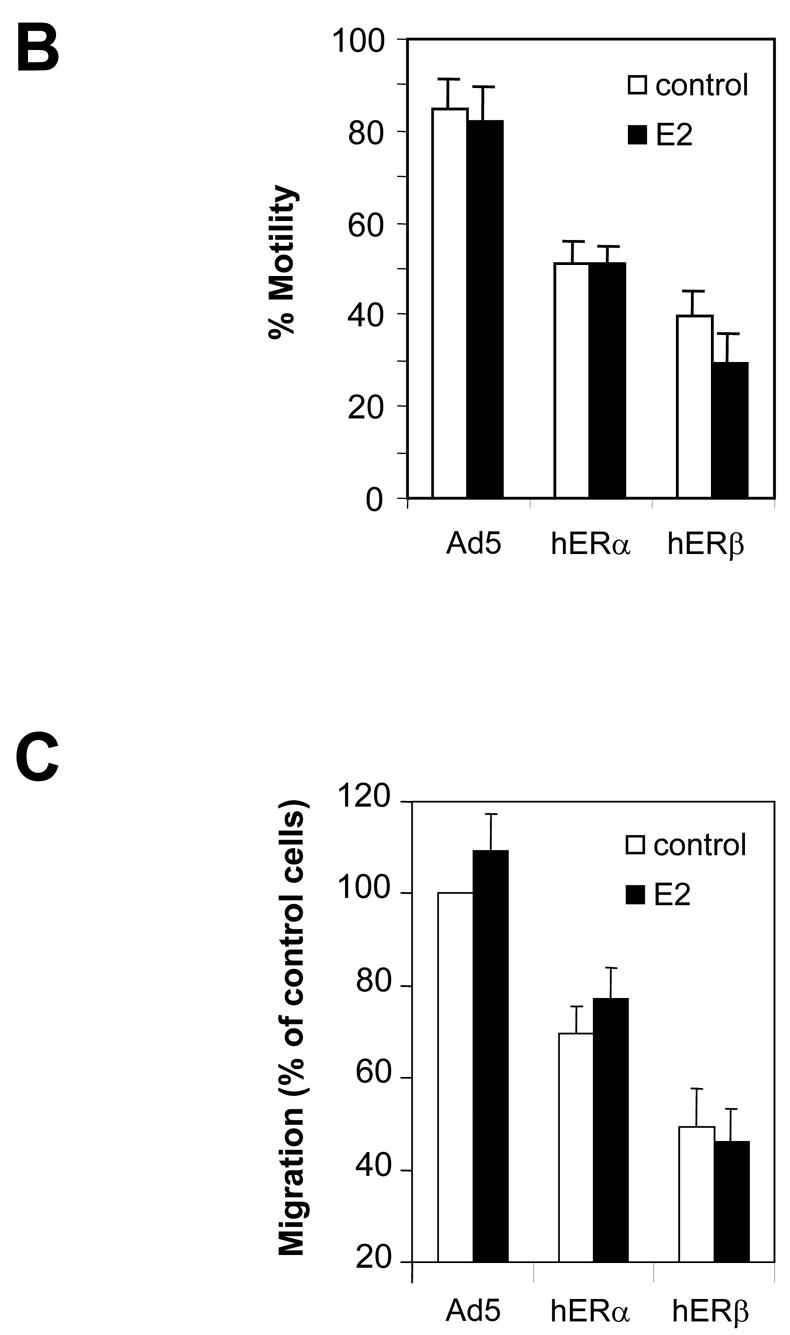
A. MDA-MB-231 cells were infected with Ad5, Ad-hERα or Ad-hERβ viruses at MOI 100. 24 h after the beginning of the infection, the cells were then treated with ethanol (control) or E2 (10−8M). After 48h of ligand treatment, cells were scratched with a blue tip and pictured (t=0). The wound was pictured again 18h after the scratch (t=18h). Pictures of a representative assay are shown here. B. Results are shown as the % of wound filling after 18h of migration and represent the mean ± SD of 3 experiments. C. MDA-MB-231 cells were infected with Ad5, Ad-hERα or Ad-hERβ (MOI 100). Cells were plated on transwell or on control plates and treated with ethanol vehicle or E2 (10−8 M) 24h after infection. Cells which have migrated to the lower side of the filter and cells present in the control plates were counted after 36h of migration. The percentage of control migrating cells was set up to 100. Results are expressed as the percentage of control migrating cells and represent the mean ± SD of four experiments.
In correlation with these observations on the reduction of cell motility and invasion following ER expression, we have tested whether cell morphology was affected. Strikingly, ERβ led to a change in the morphology of the cells (Fig. 7). Infected cells lose their fibroblastic appearance and acquired an “epithelioid-like” shape. The cells were enlarged and more rounded. ERα expression also modified the morphology of the cells and led to a more flattened shape of the cells. This change was even more pronounced in the presence of estradiol creating a characteristic structure of “branching” cells.
Fig. 7. hERα and hERβ alter the morphology of MDA-MB-231 cells.
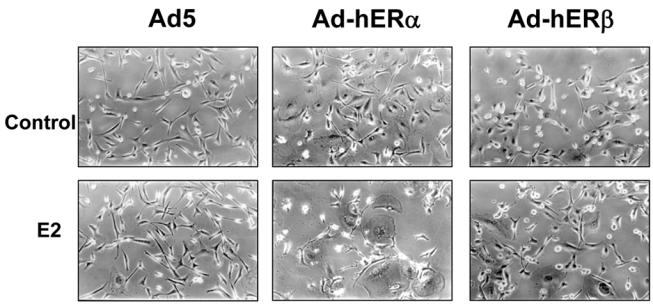
MDA-MB-231 cells were infected with Ad5, Ad-hERα Ad-hERβ viruses at MOI 100. 24 h after the beginning of the infection, the cells were then treated with ethanol (control) or E2 (10−8M). After 48h of ligand treatment, cells were pictured under a phase contrast microscope.
DISCUSSION
The use of recombinant adenoviruses has enabled us to express ERβ in breast cancer cells devoid of detectable endogenous ERs. ERβ protein appears to be fully functional as shown by DNA binding, cellular localization, transient transfection experiments and regulation of estrogen-regulated endogenous genes. Thus, our data suggest that this novel model exhibits all the interesting features required for the study of ERβ action in breast cancer cells and could thus be predictive of its role in human tumors.
In MDA-MB-231 cells, expressed ERβ regulated the activity of reporter constructs and endogenous genes. The weaker activity of ERβ on reporter genes compared to ERα is likely due to a lack of ERβ AF-1 activity as suggested by several studies (17, 19, 20, 36). Indeed, depending on the cellular and promoter context, AF-1 function has a negligeable or high activity, which in turn leads to a greatly enhanced activity of ERα when gene regulation requires both AF-1 and AF-2. On the contrary, when only AF-2 is active, both receptors exhibit similar activities.
Interestingly, we show here that ERβ can induce the expression of pS2, p21 and TGFα, whereas it has no effect c-myc expression. We and others have previously shown that in cellular models in which ERα was exogenously expressed, ERα could induce cathepsin D, pS2, p21 and TGFα expression in the presence of E2 (31, 37–39), whereas it was able to down regulate c-myc, TGFβ2, BRCA-1, BRCA-2 and c-fos/c-jun expression (31, 37, 40). This suggests that ERα and ERβ target genes are partially overlapping but that there are also target genes only regulated by one type of receptor. To date, only a limited number of promoters regulated specifically by one E2 liganded-ER isotype have been identified. This is the case of the osteopontin (41) and hTERT (catalytic subunit of human telomerase) (42) promoters which are up-regulated by ERα and not by ERβ. There is only one demonstration of a gene exclusively regulated by ERβ and not by ERα in the presence of estrogens. This is the case of methallothionein II gene which is specifically up-regulated by ERβ in SAOS-2 cells but is regulated by ERα and ERβ in LNCaPLN3 cells (43). Interestingly, c-myc RNA levels (whose expression is generally correlated with the proliferation rate) were not affected by ERβ in the presence of E2 in contrast to what is observed in ERα-MDA-MB-231 cells. p21 RNA levels were increased by ERβ in the presence of E2. p21 expression is definitely induced in numerous growth arrested cells (44), even if there are no growth abnormalities in p21-null mice (45). Moreover, p21 is also involved in some cases in the differentiation process, without affecting proliferation (46). Therefore, p21 up-regulation observed in E2-treated ERβ expressing cells might be related to differentiation, as suggested by the morphological changes observed. The change in morphology from a “fibroblastic” to an “epithelioid-like” shape of MDA-MB-231 cells infected with ERβ has been reported for other engineered cell lines, such as MDA-MB-231 cells stably expressing PR (47). Interestingly, the proliferation of these cells was inhibited by the addition of progesterone, the corresponding receptor ligand. It will be of interest to evaluate whether ERβ expression leads to changes in adhesion properties of the cells and in particular to determine whether adhesion molecule expression is altered.
Our work represents the first direct evidence that ERβ is involved in the control of the proliferation of breast cancer cells. Surprisingly, ERβ inhibition of the proliferation was ligand-independent, whereas ERβ was able to regulate reporter genes and endogenous gene expression in a ligand-dependent manner. Exogenous ERα expression using stable or retroviral infected cell lines has already been reported (31, 48–51). All these studies have shown an E2-dependent decreased proliferation of ERα expressing cells, ranging from a modest to a high level of inhibition. Therefore, our data suggest that ERα and ERβ inhibit the proliferation through distinct mechanisms. To date, only one study has reported the stable expression of ERβ (52). These authors used rat-1 cells and compared ERα and ERβ transfectants. ERβ did not affect the proliferation, but in this model, in disagreement with all other studies, ERα had no ability either to repress proliferation in the presence of estradiol. More interestingly, in contrast to ERα, the effect of exogenous expression of ERβ on proliferation seems to be relevant to the clinical situation. Indeed, numerous studies have shown that the ERβ/ERα ratio was decreased between normal to cancerous tissues, as in breast, colon and ovarian cancers (24–30), suggesting that ERβ could play a negative role on tumorigenesis. Roger et al. (27) have shown that ERβ protein was expressed in 85% of epithelial cells of normal mammary gland and this expression was not significantly altered in non-proliferative breast benign disease (BBD). On the contrary, ERβ expression was decreased in proliferative BBD and was nearly completely shut down in high grade carcinoma in situ (DCIS), suggesting that the presence of ERβ is associated with non proliferative states of the disease. What is still under question is whether ERβ expression in breast cancers could be considered as a good prognostic indicator. In invasive breast cancer, other studies have shown that ERβ protein expression was associated with less invasive and proliferative tumors (negative axillary node status, low grade, low S-phase fraction) suggesting that ERβ might be a good prognostic indicator (53). This conclusion was also supported by Omoto et al. (54), eventhough they could not see a significant correlation between ERβ expression and other known clinical parameters. Finally, in terms of adjuvant hormonal therapy (AHT), the conclusions are rather contradictory at present as some studies suggest that ERβ expressing tumors are associated to a better survival of patients under AHT (55) whereas other results suggest that ERβ is up-regulated in tamoxifen resistant tumors and could be involved in tamoxifen resistance (56, 57).
In agreement with previous work (58), our data show that ERα and ERβ activities on an ERE-containing reporter and on estrogen regulated genes can be inhibited by the pure antiestrogen ICI 164,384. Moreover, several studies have underlined the differences between ERα and ERβ in terms of response to estrogens or anti-estrogens on AP-1 sites. Indeed, ERβ is able to potentiate AP-1 containing reporters in the presence of antiestrogens but not in the presence of estrogens. ERα stimulates AP-1 activity in the presence of estrogens and anti-estrogens in endometrial cells (58–60), but antiestrogens have no effect on AP-1 activity in breast cancer cells (60, 61). Of particular note, ERβ is overall more potent than ERα on AP-1 sites, whereas the contrary occurs on EREs (17, 19, 20, 36, 58).
We also show that ERα and ERβ inhibit migration and invasion in a ligand-independent manner. These effects of ERβ are in close agreement with a previous report showing that ERα inhibits the migration of ERα-negative breast cancer cells (48, 62). In the context of breast cancer, such a reduction of invasion and motility would certainly lead to less aggressive cancers with a lower rate of metastasis. These results fit also well with numerous reports describing that ER-positive breast cancer cells are generally less invasive than ER-negative breast cancer cells (63–66) and that ERβ expressing tumors are less metastatic (53). Moreover, reintroduction of ERα in ER-negative breast cancer cells decreases their invasion and metastaic potential (48). Thus, both ERα and ERβ are able to reverse the invasive phenotype of MDA-MB-231 cells into less invasive cells, mimicking the situation of ER-positive breast cancer cells.
In conclusion, our results strongly support the idea that ERβ could be a potent proliferation gatekeeper as well as an inhibitor of cell motility and invasion. The decreased expression of ERβ observed between normal and cancerous breast could be one of the events leading to an uncontrolled proliferation of the cells. Our data suggest that the use of ERβ itself or of some of its target genes could be of interest to design a gene therapy approach against hormone-unresponsive breast cancer.
Acknowledgments
We thank Pr B.S. Katzenellenbogen for the gift of ERβ antibody, ERα and ERβ cDNAs and ERE2-TK-CAT construct. We are also grateful to Dr P. Moullier and AFM (Association Française contre les Myopathies) for their support to produce the viruses. We acknowledge the participation of A. Licznar and M. Lacroix to some experiments during their graduate courses. We thank Dr P. Roger for critically reviewing this manuscript and J.Y. Cance for the photographic work. This work was supported by grants from ARC (Association pour la Recherche contre le Cancer, grant n°5405), la Ligue Nationale contre le Cancer (Comité du Gard), Inserm and CNRS.
ABBREVIATIONS
- Ad
adenovirus
- Ad5
non recombinant adenovirus
- Ad hERα or β
recombinant adenovirus with hERα or β
- AF
activation function
- BGG
bovine gamma globulin
- DAB
diaminobenzidine
- ERE
estrogen responsive element
- hERα or β
human estrogen receptor α or β
- WCE
whole cell extract
References
- 1.Gustafsson JA. Estrogen receptor beta--a new dimension in estrogen mechanism of action. J Endocrinol. 1999;163:379–83. doi: 10.1677/joe.0.1630379. [DOI] [PubMed] [Google Scholar]
- 2.Russo J, Russo IH. Differentiation and breast cancer. Medicina (B Aires) 1997;57:81–91. [PubMed] [Google Scholar]
- 3.Haslam SZ, Counterman LJ, Nummy KA. Effects of epidermal growth factor, estrogen, and progestin on DNA synthesis in mammary cells in vivo are determined by the developmental state of the gland. J Cell Physiol. 1993;155:72–8. doi: 10.1002/jcp.1041550110. [DOI] [PubMed] [Google Scholar]
- 4.Hilakivi-Clarke L, Clarke R, Lippman M. The influence of maternal diet on breast cancer risk among female offspring. Nutrition. 1999;15:392–401. doi: 10.1016/s0899-9007(99)00029-5. [DOI] [PubMed] [Google Scholar]
- 5.Liao DZ, Pantazis CG, Hou X, Li SA. Promotion of estrogen-induced mammary gland carcinogenesis by androgen in the male Noble rat: probable mediation by steroid receptors. Carcinogenesis. 1998;19:2173–80. doi: 10.1093/carcin/19.12.2173. [DOI] [PubMed] [Google Scholar]
- 6.Pietras RJ, Szego CM. Cell membrane estrogen receptors resurface. Nat Med. 1999;5:1330. doi: 10.1038/70877. [DOI] [PubMed] [Google Scholar]
- 7.Collins P, Webb C. Estrogen hits the surface. Nat Med. 1999;5:1130–1. doi: 10.1038/13453. [DOI] [PubMed] [Google Scholar]
- 8.Ruehlmann DO, Mann GE. Rapid non-genomic vasodilator actions of oestrogens and sex steroids. Curr Med Chem. 2000;7:533–41. doi: 10.2174/0929867003375038. [DOI] [PubMed] [Google Scholar]
- 9.Nuclear Receptors Nomenclature C. A unified nomenclature system for the nuclear receptor superfamily [letter] Cell. 1999;97:161–3. doi: 10.1016/s0092-8674(00)80726-6. [DOI] [PubMed] [Google Scholar]
- 10.Beato M, Herrlich P, Schutz G. Steroid hormone receptors: many actors in search of a plot. Cell. 1995;83:851–7. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- 11.McGuire WL. Endocrine therapy of breast cancer. Annu Rev Med. 1975;26:353–63. doi: 10.1146/annurev.me.26.020175.002033. [DOI] [PubMed] [Google Scholar]
- 12.Green S, Walter P, Kumar V, Krust A, Bornert JM, Argos P, Chambon P. Human oestrogen receptor cDNA: sequence, expression and homology to v-erbA. Nature. 1986;320:134–139. doi: 10.1038/320134a0. [DOI] [PubMed] [Google Scholar]
- 13.Kuiper G, Enmark E, Peltohuikko M, Nilsson S, Gustafsson JA. Cloning of a novel estrogen receptor expressed in rat prostate and ovary. PNAS. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mosselman S, Polman J, Dijkema R. ER-beta - identification and characterization of a novel human estrogen receptor. Febs Lett. 1996;392:49–53. doi: 10.1016/0014-5793(96)00782-x. [DOI] [PubMed] [Google Scholar]
- 15.Kelley ST, Thackray VG. Phylogenetic analyses reveal ancient duplication of estrogen receptor isoforms. J Mol Evol. 1999;49:609–14. doi: 10.1007/pl00006582. [DOI] [PubMed] [Google Scholar]
- 16.Tora L, White J, Brou C, Tasset D, Webster N, Scheer E, Chambon P. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell. 1989;59:477–87. doi: 10.1016/0092-8674(89)90031-7. [DOI] [PubMed] [Google Scholar]
- 17.Cowley SM, Parker MG. A comparison of transcriptional activation by ER alpha and ER beta. J Steroid Biochem Mol Biol. 1999;69:165–75. doi: 10.1016/s0960-0760(99)00055-2. [DOI] [PubMed] [Google Scholar]
- 18.McInerney EM, Weis KE, Sun J, Mosselman S, Katzenellenbogen BS. Transcription activation by the human estrogen receptor subtype beta (ER-beta) studied with ER-beta and ER-alpha receptor chimeras. Endocrinology. 1998;139:4513–4522. doi: 10.1210/endo.139.11.6298. [DOI] [PubMed] [Google Scholar]
- 19.Hall JM, McDonnell DP. The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology. 1999;140:5566–78. doi: 10.1210/endo.140.12.7179. [DOI] [PubMed] [Google Scholar]
- 20.Delaunay F, Pettersson K, Tujague M, Gustafsson JA. Functional differences between the amino-terminal domains of estrogen receptors alpha and beta. Mol Pharmacol. 2000;58:584–90. doi: 10.1124/mol.58.3.584. [DOI] [PubMed] [Google Scholar]
- 21.Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, Sar M, Korach KS, Gustafsson JA, Smithies O. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. PNAS. 1998;95:15677–82. doi: 10.1073/pnas.95.26.15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 23.Saji S, Jensen EV, Nilsson S, Rylander T, Warner M, Gustafsson J. Estrogen receptors alpha and beta in the rodent mammary gland. PNAS. 2000;97:337–342. doi: 10.1073/pnas.97.1.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foley EF, Jazaeri AA, Shupnik MA, Jazaeri O, Rice LW. Selective loss of estrogen receptor beta in malignant human colon. Cancer Res. 2000;60:245–8. [PubMed] [Google Scholar]
- 25.Campbell-Thompson M, Lynch IJ, Bhardwaj B. Expression of estrogen receptor (ER) subtypes and ERbeta isoforms in colon cancer. Cancer Res. 2001;61:632–40. [PubMed] [Google Scholar]
- 26.Iwao K, Miyoshi Y, Egawa C, Ikeda N, Noguchi S. Quantitative analysis of estrogen receptor-beta mRNA and its variants in human breast cancers. Int J Cancer. 2000;88:733–6. doi: 10.1002/1097-0215(20001201)88:5<733::aid-ijc8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 27.Roger P, Sahla ME, Makela S, Gustafsson JA, Baldet P, Rochefort H. Decreased Expression of Estrogen Receptor {beta} Protein in Proliferative Preinvasive Mammary Tumors. Cancer Res. 2001;61:2537–2541. [PubMed] [Google Scholar]
- 28.Rutherford T, Brown WD, Sapi E, Aschkenazi S, Munoz A, Mor G. Absence of estrogen receptor-beta expression in metastatic ovarian cancer. Obstet Gynecol. 2000;96:417–21. doi: 10.1016/s0029-7844(00)00917-0. [DOI] [PubMed] [Google Scholar]
- 29.Leygue E, Dotzlaw H, Watson PH, Murphy LC. Altered estrogen receptor alpha and beta messenger RNA expression during human breast tumorigenesis. Cancer Res. 1998;58:3197–3201. [PubMed] [Google Scholar]
- 30.Pujol P, Rey JM, Nirde P, Roger P, Gastaldi M, Laffargue F, Rochefort H, Maudelonde T. Differential expression of estrogen receptor-alpha and -beta messenger RNAs as a potential marker of ovarian carcinogenesis. Cancer Res. 1998;58:5367–5373. [PubMed] [Google Scholar]
- 31.Lazennec G, Katzenellenbogen BS. Expression of human estrogen receptor using an efficient adenoviral gene delivery system is able to restore hormone-dependent features to estrogen receptor-negative breast carcinoma cells. Mol Cell Endocrinol. 1999;149:93–105. doi: 10.1016/s0303-7207(98)00254-8. [DOI] [PubMed] [Google Scholar]
- 32.Graham FL, Smilet J, Russel WC, Nairn R. Characteristics of a human cell line transformed by DNA from adenovirus type 5. J Gen Virol. 1977;36:59–72. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 33.McGregory WJ, Bautista DS, Graham FL. A simple technique for the rescue of early region 1 mutations into infectious human adenovirus type 5. Virology. 1988;163:614–617. doi: 10.1016/0042-6822(88)90302-9. [DOI] [PubMed] [Google Scholar]
- 34.Lazennec G, Alcorn JL, Katzenellenbogen BS. Adenovirus-mediated delivery of a dominant negative estrogen receptor gene abrogates estrogen-stimulated gene expression and breast cancer cell proliferation. Mol Endocrinol. 1999;13:969–80. doi: 10.1210/mend.13.6.0318. [DOI] [PubMed] [Google Scholar]
- 35.Choi I, Ko C, Park-Sarge O-K, Nie R, Hess R, Graves C, Katzenellenbogen BS. Human estrogen receptor beta-specific monoclonal antibodies: Characterization and use in studies of estrogen receptor beta protein expression in reproductive tissues. Mol Cell Endocrinol. 2001 doi: 10.1016/s0303-7207(01)00492-0. in press. [DOI] [PubMed] [Google Scholar]
- 36.Pettersson K, Delaunay F, Gustafsson JA. Estrogen receptor beta acts as a dominant regulator of estrogen signaling. Oncogene. 2000;19:4970–8. doi: 10.1038/sj.onc.1203828. [DOI] [PubMed] [Google Scholar]
- 37.Jeng MH, Jiang SY, Jordan VC. Paradoxical regulation of estrogen-dependent growth factor gene expression in estrogen receptor (ER)-negative human breast cancer cells stably expressing ER. Cancer Lett. 1994;82:123–128. doi: 10.1016/0304-3835(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 38.Thomas TJ, Faaland CA, Adhikarakunnathu S, Watkins LF, Thomas T. Induction of p21 (CIP1/WAF1/SID1) by estradiol in a breast epithelial cell line transfected with the recombinant estrogen receptor gene: a possible mechanism for a negative regulatory role of estradiol. Breast Cancer Res Treat. 1998;47:181–93. doi: 10.1023/a:1005925931215. [DOI] [PubMed] [Google Scholar]
- 39.Touitou I, Vignon F, Cavailles V, Rochefort H. Hormonal regulation of cathepsin D following transfection of the estrogen or progesterone receptor into three sex steroid hormone resistant cancer cell lines. J Steroid Biochem Mol Biol. 1991;40:231–7. doi: 10.1016/0960-0760(91)90187-a. [DOI] [PubMed] [Google Scholar]
- 40.Wang W, Smith Rr, Burghardt R, Safe SH. 17 beta-Estradiol-mediated growth inhibition of MDA-MB-468 cells stably transfected with the estrogen receptor: cell cycle effects. Mol Cell Endocrinol. 1997;133:49–62. doi: 10.1016/s0303-7207(97)00142-1. [DOI] [PubMed] [Google Scholar]
- 41.Vanacker JM, Pettersson K, Gustafsson J, Laudet V. Transcriptional targets shared by estrogen receptor- related receptors (ERRs) and estrogen receptor (ER) alpha, but not by ERbeta. Embo J. 1999;18:4270–4279. doi: 10.1093/emboj/18.15.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Misiti S, Nanni S, Fontemaggi G, Cong YS, Wen J, Hirte HW, Piaggio G, Sacchi A, Pontecorvi A, Bacchetti S, Farsetti A. Induction of hTERT expression and telomerase activity by estrogens in human ovary epithelium cells. Mol Cell Biol. 2000;20:3764–71. doi: 10.1128/mcb.20.11.3764-3771.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harris HA, Henderson RA, Bhat RA, Komm BS. Regulation of Metallothionein II Messenger Ribonucleic Acid Measures Exogenous Estrogen Receptor-{beta} Activity in SAOS-2 and LNCaPLN3 Cells. Endocrinology. 2001;142:645–652. doi: 10.1210/endo.142.2.7952. [DOI] [PubMed] [Google Scholar]
- 44.Cariou S, Donovan JC, Flanagan WM, Milic A, Bhattacharya N, Slingerland JM. Down-regulation of p21WAF1/CIP1 or p27Kip1 abrogates antiestrogen-mediated cell cycle arrest in human breast cancer cells. PNAS. 2000;97:9042–6. doi: 10.1073/pnas.160016897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weiss RH, Joo A, Randour C. p21Waf1/Cip1 Is an Assembly Factor Required for Platelet-derived Growth Factor-induced Vascular Smooth Muscle Cell Proliferation. J Biol Chem. 2000;275:10285–10290. doi: 10.1074/jbc.275.14.10285. [DOI] [PubMed] [Google Scholar]
- 46.Di Cunto F, Topley G, Calautti E, Hsiao J, Ong L, Seth PK, Dotto GP. Inhibitory function of p21Cip1/WAF1 in differentiation of primary mouse keratinocytes independent of cell cycle control. Science. 1998;280:1069–72. doi: 10.1126/science.280.5366.1069. [DOI] [PubMed] [Google Scholar]
- 47.Lin VC, Ng EH, Aw SE, Tan MG, Bay BH. Progesterone induces focal adhesion in breast cancer cells MDA-MB-231 transfected with progesterone receptor complementary DNA. Mol Endocrinol. 2000;14:348–58. doi: 10.1210/mend.14.3.0426. [DOI] [PubMed] [Google Scholar]
- 48.Garcia M, Derocq D, Freiss G, Rochefort H. Activation of estrogen receptor transfected into a receptor-negative breast cancer cell line decreases the metastatic and invasive potential of the cells. PNAS. 1992;89:11538–11542. doi: 10.1073/pnas.89.23.11538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang SY, Jordan VC. Growth regulation of estrogen receptor-negative breast cancer cells transfected with complementary DNAs for estrogen receptor. J Natl Cancer Inst. 1992;84:580–91. doi: 10.1093/jnci/84.8.580. [DOI] [PubMed] [Google Scholar]
- 50.Kushner PJ, Hort E, Shine J, Baxter JD, Greene GL. Construction of cell lines that express high levels of the human estrogen receptor and are killed by estrogens. Mol Endocrinol. 1990;4:1465–1473. doi: 10.1210/mend-4-10-1465. [DOI] [PubMed] [Google Scholar]
- 51.Zajchowski DA, Sager R. Induction of estrogen-regulated genes differs in immortal and tumorigenic human mammary epithelial cells expressing a recombinant estrogen receptor. Mol Endocrinol. 1991;5:1613–23. doi: 10.1210/mend-5-11-1613. [DOI] [PubMed] [Google Scholar]
- 52.Cheng J, Malayer JR. Responses to stable ectopic estrogen receptor-beta expression in a rat fibroblast cell line. Mol Cell Endocrinol. 1999;156:95–105. doi: 10.1016/s0303-7207(99)00134-3. [DOI] [PubMed] [Google Scholar]
- 53.Jarvinen TA, Pelto-Huikko M, Holli K, Isola J. Estrogen receptor beta is coexpressed with ERalpha and PR and associated with nodal status, grade, and proliferation rate in breast cancer. Am J Pathol. 2000;156:29–35. doi: 10.1016/s0002-9440(10)64702-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Omoto Y, Inoue S, Ogawa S, Toyama T, Yamashita H, Muramatsu M, Kobayashi S, Iwase H. Clinical value of the wild-type estrogen receptor beta expression in breast cancer. Cancer Lett. 2001;163:207–12. doi: 10.1016/s0304-3835(00)00680-7. [DOI] [PubMed] [Google Scholar]
- 55.Mann S, Laucirica R, Carlson N, Younes PS, Ali N, Younes A, Li Y, Younes M. Estrogen receptor beta expression in invasive breast cancer. Hum Pathol. 2001;32:113–118. doi: 10.1053/hupa.2001.21506. [DOI] [PubMed] [Google Scholar]
- 56.Speirs V, Malone C, Walton DS, Kerin MJ, Atkin SL. Increased expression of estrogen receptor beta mRNA in tamoxifen-resistant breast cancer patients. Cancer Res. 1999;59:5421–4. [PubMed] [Google Scholar]
- 57.Speirs V, Kerin MJ. Prognostic significance of oestrogen receptor beta in breast cancer. Br J Surg. 2000;87:405–9. doi: 10.1046/j.1365-2168.2000.01402.x. [DOI] [PubMed] [Google Scholar]
- 58.Paech K, Webb P, Kuiper G, Nilsson S, Gustafsson JA, Kushner PJ, Scanlan TS. Differential ligand activation of estrogen receptors ER-alpha and ER-beta at AP1 sites. Science. 1997;277:1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- 59.Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, Katzenellenbogen BS, Enmark E, Gustafsson JA, Nilsson S, Kushner PJ. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol Endocrinol. 1999;13:1672–85. doi: 10.1210/mend.13.10.0357. [DOI] [PubMed] [Google Scholar]
- 60.Webb P, Lopez GN, Uht RM, Kushner PJ. Tamoxifen activation of the estrogen receptor/AP-1 pathway: potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol Endocrinol. 1995;9:443–56. doi: 10.1210/mend.9.4.7659088. [DOI] [PubMed] [Google Scholar]
- 61.Philips A, Chalbos D, Rochefort H. Estradiol increases and anti-estrogens antagonize the growth factor- induced activator protein-1 activity in MCF7 breast cancer cells without affecting c-fos and c-jun synthesis. J Biol Chem. 1993;268:14103–8. [PubMed] [Google Scholar]
- 62.Platet N, Cunat S, Chalbos D, Rochefort H, Garcia M. Unliganded and liganded estrogen receptors protect against cancer invasion via different mechanisms [In Process Citation] Mol Endocrinol. 2000;14:999–1009. doi: 10.1210/mend.14.7.0492. [DOI] [PubMed] [Google Scholar]
- 63.Tong D, Czerwenka K, Sedlak J, Schneeberger C, Schiebel I, Concin N, Leodolter S, Zeillinger R. Association of in vitro invasiveness and gene expression of estrogen receptor, progesterone receptor, pS2 and plasminogen activator inhibitor-1 in human breast cancer cell lines. Breast Cancer Res Treat. 1999;56:91–7. doi: 10.1023/a:1006262501062. [DOI] [PubMed] [Google Scholar]
- 64.Thompson EW, Paik S, Brunner N, Sommers CL, Zugmaier G, Clarke R, Shima TB, Torri J, Donahue S, Lippman ME, et al. Association of increased basement membrane invasiveness with absence of estrogen receptor and expression of vimentin in human breast cancer cell lines. J Cell Physiol. 1992;150:534–44. doi: 10.1002/jcp.1041500314. [DOI] [PubMed] [Google Scholar]
- 65.Sommers CL, Byers SW, Thompson EW, Torri JA, Gelmann EP. Differentiation state and invasiveness of human breast cancer cell lines. Breast Cancer Res Treat. 1994;31:325–35. doi: 10.1007/BF00666165. [DOI] [PubMed] [Google Scholar]
- 66.Madsen MW, Briand P. Relationship between tumorigenicity, in vitro invasiveness, and plasminogen activator production of human breast cell lines. Eur J Cancer. 1990;26:793–7. doi: 10.1016/0277-5379(90)90154-l. [DOI] [PubMed] [Google Scholar]