

Abstract
In this issue, Lietha and colleagues (2007) report the structure of focal adhesion kinase (FAK) and reveal how FAK maintains an autoinhibited state. Together with the structure of another tyrosine kinase, ZAP-70 (Deindl et al., 2007), this work highlights the diversity of mechanisms that nature has evolved within the kinase superfamily to regulate their activity through autoinhibition.
The covalent modification of protein hydroxyls by phosphoryl groups is ubiquitous in biology. This simple chemical reaction is so important that the human genome encodes 478 protein kinases, as defined by the presence of a conserved 350 residue catalytic domain (Manning et al., 2002).
“All happy families are alike; each unhappy family is unhappy in its own way.”
—Leo Tolstoy, Anna Karenina
Protein kinases have a common “on” switch, in which they are phosphorylated on one or two serine, threonine, or tyrosine residues of a key segment known as the activation loop. Activation loop phosphorylation stabilizes the assembly of an enzymatic site competent for the Mg2+-dependent transfer of the γ-phosphoryl group of ATP to the substrate protein. Like Tolstoy’s happy families, the active conformations of protein kinases closely resemble one another. A common inactivation mechanism is their dephosphorylation by protein phosphatases. Given the critical role of kinases at the top of signaling cascades and other critical cellular decision points, diverse mechanisms of autoregulation have evolved in addition to dephosphorylation. These mechanisms depend on the composition and order of various domains and posttranslational modification sites in the linker segments that connect the domains. The regulatory domains respond to cellular ligands, mediate protein-protein interactions,and are responsible for the subcellular distribution of the kinases. Some kinases, such as protein kinase A (PKA) (Kim et al., 2005), have evolved such that these regulatory functions are provided by separate chains. Two recent structural studies in Cell, including one in this issue, reveal the autoinhibitory mechanisms for two more kinases, FAK and ZAP-70 (Lietha et al., 2007; Deindl et al., 2007). Every kinase familymay thus be autoinhibited in its own way. Just as the “unhappy family” experience has provided inspiration to dramatists for centuries, the diverse repertoire of kinase autoinhibition mechanisms provides nearly inexhaustible material for the structural biologist in search of drama at the molecular scale.
The prototypical protein tyrosine kinase Src contains a Src-homology 2 (SH2) domain and a Src-homology 3 (SH3) domain in its N-terminal regulatory portion. SH2 domains bind to tyrosine-phosphorylated protein motifs, whereas SH3 domains typically bind to proline-rich sequence motifs. The first structural explanation for the autoinhibitory role of regulatory moieties of protein tyrosine kinases came with the structures of Src and Hck kinases (Figure 1, left) (Sicheri et al., 1997; Xu et al., 1997). The two structures showed how interdomain interactions, stabilized by binding of the SH2 domain to the phosphorylated C terminus, lock the molecule in a closed conformation. This C-terminal phosphorylation is thus inhibitory, in contrast to activation loop phosphorylation. In a remarkable economy of binding surfaces, the SH3 domains of Src, Hck, and another protein tyrosine kinase, Abl (Nagar et al., 2006), dock onto a polyproline type II helix formed by the SH2-kinase linker sequence. Abl possesses all of the regulatory elements of Src but lacks the regulatory C-terminal tail; nevertheless, it has since been shown that a compensatory mechanism exists for the maintenance of the autoinhibited state (Nagar et al., 2006). These structures have been influential in thinking about the allosteric regulation of many other multimodular proteins besides protein kinases.
Figure 1. A Protein Tyrosine Kinase Family Drama.
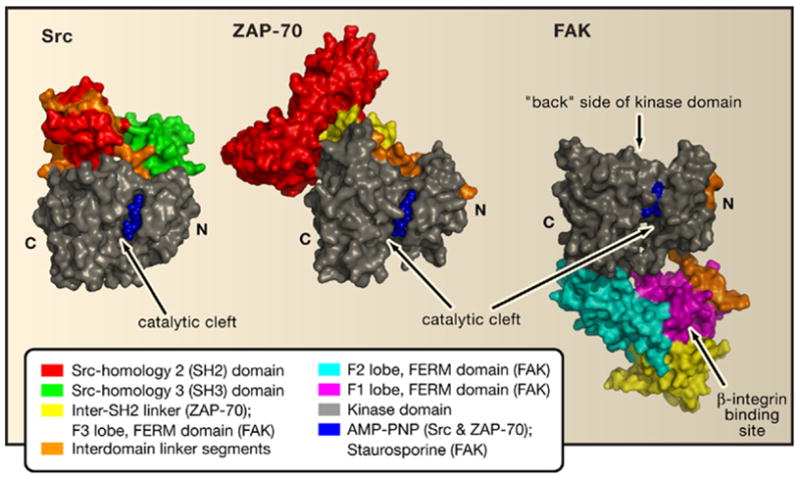
Depicted are autoinhibited assemblies of Src, ZAP-70, and FAK. The structures are aligned based on their kinase domains; important regulatory linker segments are shown in orange. Inhibition is mediated by docking of the regulatory domains to the “back” side of the kinase domain in Src and ZAP-70 but by occlusion of the catalytic cleft on the opposite side by the FERM domain in FAK.
The technical challenges to determining structures of full-length or almost full-length multimodular kinases are still considerable. Conformational plasticity is required for regulation but is an enemy of crystallization. Furthermore, heterogeneity in the phosphorylation state of the kinase must be avoided. These and other barriers have limited the number of such structures, in spite of their importance. FAK and ZAP-70 are essential nonreceptor tyrosine kinases in adherent cells and T cells, respectively. In responding to extracellular signals transduced by receptors localized in the cell membrane, FAK and ZAP-70 mediate the tyrosine phosphorylation of downstream effectors, thereby promoting diverse signaling events. In the case of FAK, these signals modulate cellular growth, survival, and migration, whereas in the case of ZAP-70, they regulate T cell proliferation and the secretion of cytokines. The two new structures of autoinhibited ZAP-70 and FAK represent a significant accomplishment in the field. ZAP-70 and FAK contain a tandem pair of SH2 domains and a FERM (band 4.1, ezrin, radixin, moesin) domain, respectively, in their regulatory regions. The FERM domain is a trilobed structure that is known to bind to phosphorylated protein sequence motifs and, in some cases, to lipids. The new structures show how these domains regulate the activity of the kinase catalytic domains.
The tandem SH2 domains of ZAP-70 are special because they bind to a cognate doubly phosphorylated sequence known as an ITAM (immunoreceptor tyrosine-based activation motif) peptide in the cytoplasmic tails of the TCR ζ and CD3 chains. The ZAP-70 structure highlights the role of interdomain linkers and regulatory interactions with the “back” side of the catalytic domain (Figure 1, middle), as in Src and Hck. The active conformation of the ITAM-bound SH2 domains (Hatada et al., 1995) requires the breakdown of the autoinhibitory interactions seen in the new ZAP-70 interactions, offering a rationale for kinase activation. Upon activation, two linker tyrosine residues are exposed for phosphorylation, which may prevent reversion to the inactive conformation even after ITAM disengagement.
FAK illustrates an entirely different mode of inhibition that not only involves distinct binding surfaces but achieves inhibition by blocking access to the active site. The F2 lobe of the FERM domain docks onto the kinase domain C lobe, covering the substratebinding site (Figure 1, right). Lietha et al. (2007) convincingly demonstrate that activation of FAK involves a sequential displacement of the FERM domain from the C lobe of the kinase and concomitant tyrosine phosphorylation of the activation loop. Furthermore, in contrast to the Src family, it is apparent that its autoinhibition does not involve displacement of the αC helix of the kinase N lobe. The binding site for one major class of known ligands, the cytoplasmic tails of β-integrins, is distal to the site of contact with the catalytic domain. This means that binding of β-integrin tails does not offer an obvious mechanism for the displacement of the FERM domain from its autoinhibitory conformation. Thus, the mechanism by which the FERM domain is displaced, leading to the activation of FAK, is still an open question.
There is now a growing list of kinase families in which autoinhibition has been characterized structurally. In addition to the Src, Abl, ZAP-70, and FAK families of cytoplasmic protein tyrosine kinases, autoinhibition has also been structurally demonstrated in the AGC serine/threonine kinase family (for example, PKA), the receptor tyrosine kinase family, and the calcium/calmodulin-dependent protein kinase (CaMK) family. All three kinase families mediate inhibition in unique ways, from a pseudosubstrate sequence in the case of PKA (Kim et al., 2005) to a juxtamembrane segment in the receptor tyrosine kinases that occupies the catalytic cleft (Wybenga-Groot et al., 2001) to a C-terminal autoinhibitory helix-loop-helix segment in the case of CaMKI (Goldberg et al., 1996). How many unique mechanisms of kinase autoinhibition are there? A quick glance at the human protein kinase family tree should leave kinase family dramatists feeling reassured that they are unlikely to run out of material any time soon.
Acknowledgments
This work was supported by the intramural program of the NIH through the National Institute of Diabetes and Digestive and Kidney Diseases (J.H.H.). T.A.L. is a recipient of an EMBO Long-Term Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Deindl S, Kadlecek TA, Brdicka T, Cao X, Weiss A, Kuriyan J. Cell. 2007;129:735–746. doi: 10.1016/j.cell.2007.03.039. [DOI] [PubMed] [Google Scholar]
- Goldberg J, Nairn AC, Kuriyan J. Cell. 1996;84:875–887. doi: 10.1016/s0092-8674(00)81066-1. [DOI] [PubMed] [Google Scholar]
- Hatada MH, Lu X, Laird ER, Green J, Morgenstern JP, Lou M, Marr CS, Phillips TB, Ram MK, Theriault K, et al. Nature. 1995;377:32–38. doi: 10.1038/377032a0. [DOI] [PubMed] [Google Scholar]
- Kim C, Xuong NH, Taylor SS. Science. 2005;307:690–696. doi: 10.1126/science.1104607. [DOI] [PubMed] [Google Scholar]
- Lietha D, Cai X, Ceccarelli DFJ, Li Y, Schaller MD, Eck MJ. Cell. 2007 doi: 10.1016/j.cell.2007.05.041. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Nagar B, Hantschel O, Seeliger M, Davies JM, Weis WI, Superti-Furga G, Kuriyan J. Mol. Cell. 2006;21:787–798. doi: 10.1016/j.molcel.2006.01.035. [DOI] [PubMed] [Google Scholar]
- Sicheri F, Moarefi I, Kuriyan J. Nature. 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- Wybenga-Groot LE, Baskin B, Ong SH, Tong J, Pawson T, Sicheri F. Cell. 2001;106:745–757. doi: 10.1016/s0092-8674(01)00496-2. [DOI] [PubMed] [Google Scholar]
- Xu W, Harrison SC, Eck MJ. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]