

Abstract
Objectives
The reducing capacity of erythrocytes has been used clinically as to estimate resistance to oxidant stress. In this work we targeted the antioxidant capacity of pyridine nucleotide disulfide reductases of these cells by measuring their ability to reduce the disufide α-lipoic acid.
Methods
Erythrocyte reduction of α-lipoic acid and related disulfides was measured as reduction of 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) outside the cells.
Results
Lipoic acid-dependent DTNB reduction by human erythrocytes required D-glucose and consumed NADPH, but not NADH. This activity was inhibited by carmustine and phenylarsine oxide, as expected if α-lipoic acid is reduced by the glutathione and thioredoxin reductase systems. Reduction of hydroxyethyl disulfide, which provides an estimate of total erythrocyte disulfide reduction capacity, was similar to that of α-lipoic acid. Erythrocytes incubated with α-lipoic acid also reduced extracellular ferricyanide, although rates of dehydroascorbate reduction were several-fold greater, probably because intracellular GSH can recycle ascorbate but not α-lipoic acid in erythrocytes.
Conclusion
These results show that α-lipoic acid-dependent DTNB reduction provides a simple method to selectively assess the capacity of pyridine nucleotide disulfide reductases of human erythrocytes. When coupled with other non-destructive assays, such as reduction of hydroxyethyl disulfide and ferricyanide, this assay provides a comprehensive approach to assessing erythrocyte reducing capacity in a variety of clinical conditions associated with oxidant stress.
Keywords: Lipoic acid, dehydroascorbic acid, phenylarsine oxide, ferricyanide, oxidant stress, pyridine nucleotide disulfide reductase, human erythrocytes
Introduction
α-Lipoic acid (LA) is an 8-carbon fatty acid containing a thiolane ring with a disulfide joining carbons 6 and 8. Although the disulfide form can chelate transition metals [1,2], LA is generally considered an antioxidant primarily in its reduced form, dihydrolipoic acid (DHLA) [3,4]. Since the two thiol groups of DHLA are quite susceptible to oxidation, LA is the form used clinically or in animal or culture studies. The disulfide LA is rapidly taken up and reduced by cells to DHLA, most of which exits the cells [5,6]. This extracellular DHLA can be detected by its ability to reduce 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), yielding a bright yellow thiobenzoate anion that can be quantified spectrophotometrically [7]. Within the cell, LA is specifically reduced by NADH- and NADPH-dependent oxidoreductases [5,6,8], so we previously suggested that this escaped DHLA can be used to quantify the pyridine nucleotide-dependent disulfide reductive capacity of cells over a short time period [9].
LA is reduced inside most cells by three enzymes: α-lipoamide dehydrogenase (E.C. 1.8.1.4), thioredoxin reductase (E.C. 1.6.4.5), and glutathione reductase (E.C. 1.6.4.2). The former is NADH-dependent, and the latter two derive their reducing capacity from NADPH. In contrast to other disulfides, LA is not directly reduced by cellular thiols or by GSH [10], and thus its reduction reflects pyridine nucleotide reserves and not those of cellular thiols [9]. Results from cultured cells suggest that both NADH and NADPH are used for LA reduction [5,6,8]. Most studies have used mixed R- and S- isomers of LA, and this may affect which enzymes are used for its reduction. For example, lipoamide dehydrogenase reduces R-LA 18-fold faster than S-LA, whereas glutathione reductase reduces S-LA about 2-fold faster than R-LA [11]. Isomeric preference of thioredoxin reductase has not been described.
If uptake and reduction of LA reflects the pyridine nucleotide-dependent reductive capacity of cells, it is possible that LA-dependent DTNB reduction might be used as an indicator of this aspect of the redox “reserve” of cells. In this work we explored the possibility of using this assay in human erythrocytes. These cells are readily accessible and have been used in the past as an indicator of redox capacity in various clinical conditions [12-17]. Further, erythrocytes lack mitochondria and thus lipoamide dehydrogenase, which allows for focus on cellular NADPH-dependent disulfide reducing capacity.
Materials and methods
Materials
Analytical reagents, including, ascorbic acid, dehydroascorbic acid, DTNB, carmustine (1,3-bis(chloroethyl)-1-nitrosourea, BCNU), racemic α-LA, racemic α-lipoamide, N-ethylmaleimide (NEM), phenylarsine oxide (PAO), thorin, and tetrapentylammonium bromide were supplied by Sigma/Aldrich (St. Louis, MO). The (R) enantiomer of LA was supplied by Charles Bowman and Company (Holland, MI); other derivatives were synthesized at BioLink Life Sciences. PAO was initially dissolved in dimethyl sulfoxide, and diluted so that the highest concentration of the latter was 0.04% (v/v) in cells incubated with 10 μM PAO.
Erythrocyte preparation
Human erythrocytes were obtained by venipuncture from normal human donors typically taking supplemental vitamin C at doses of 50−100 mg daily. At this level of intake, we observed no consistent changes in erythrocyte ascorbate contents (results not shown). Erythrocytes were either prepared immediately or stored less than 24 h as whole blood at 3 °C before use. Erythrocytes were rinsed three times by centrifugation before use in phosphate-buffered saline (PBS, consisting of 140 mM NaCl and 12.5 mM sodium phosphate, pH 7.4). With each rinse, the “buffy” coat of white cells was removed and discarded. Packed cells were diluted with PBS and the packed cell volume calculated as a percent of the total volume occupied by erythrocytes. Rinses in the absence of D-glucose did not affect intracellular ascorbate (results not shown).
Erythrocyte assays
Erythrocytes were rinsed twice by centrifugation in PBS prior to assay of intracellular ascorbate, GSH, and pyridine nucleotides. Ascorbate was measured by HPLC as described [18], except that tetrapentylammonium bromide was used as the ion pair reagent and ascorbate was detected by UV absorbance at 250 nm. GSH was measured by the method of Hissin and Hilf [19], also as previously described [20]. Erythrocyte contents of NADPH and NADH were measured with a spectrophotometric recycling assay [21] with minor modifications as noted [22]. Intracellular concentrations were calculated based on a cytosolic water space of 70% of the packed cell volume [23]. Ferricyanide reduction was measured as the appearance of extracellular ferrocyanide according to the ortho-phenanthroline-based assay of Avron and Shavit [24] and expressed relative to the packed cell volume.
Assay of extracellular DTNB reduction
Erythrocytes at a 1% packed cell volume were suspended in PBS that typically contained 5 mM D-glucose, 0.2 mM DTNB, and the indicated concentrations of LA or its derivatives. After incubations as noted for 30 min at 37 EC with gentle mixing, erythrocytes were pelleted by centrifugation, 1 ml of the supernatant was transferred to a plastic cuvette, and the absorbance at 412 nm was determined in a Beckman DU-640 spectrophotometer. The concentration of the 2-nitro-5-thiobenzoic acid anion of DTNB was calculated based on a molar extinction coefficient of 13,600 [25]. Results were corrected for absorbance of a reagent blank that contained the amounts of LA and DTNB used in the experiment without cells. This blank was usually near zero, indicating that the LA preparations used do not contain contaminants that can reduce DTNB. Haemolysis was not observed under the conditions of these experiments with normal erythrocytes, but it should be noted that haemolysis of fragile erythrocytes will interfere with the assay. Results are expressed relative to the packed erythrocyte volume.
Assay of thioredoxin reductase
Reduction of LA derivatives by purified rat liver thioredoxin reductase (catalogue number T9698, Sigma/Aldrich Chemical Co, St. Louis, MO) was assessed at 37 EC by following disappearance of 0.2 mM NADPH spectrophotometrically at 340 nm under in Tris buffer as described. Substrate concentrations of the LA derivatives were 1 mM. Where indicated, thioredoxin prepared from E. coli (catalogue number T0910, Sigma/Aldrich Chemical Co., St. Louis, MO) was added to a concentration of 1.9 μM.
Data analysis
Data are shown as mean ± standard error. Statistical significance was assessed by analysis of variance with appropriate post-hoc testing using the statistical software package Sigmastat 2.0 (Jandel Scientific, St. Louis, MO).
Results
When incubated with D-glucose as an energy source, human erythrocytes reduced R,S-LA in a concentration-dependent and saturable manner (Fig. 1, circles). In studies not shown, reduction of 0.1 mM R,S-LA was linear for at least 30 min with increasing cell numbers (up to 4% packed cell volume). The apparent saturation with increasing R,S-LA in Fig. 1A was not due to depletion of D-glucose, since reduction of DTNB by 1 mM R,S-LA was similar at 1, 2 and 5 mM D-glucose (results not shown). However, the saturation may be due to DTNB depletion at high LA concentrations, since only about 30% of the DTNB remained after a 30 min reaction with 1 mM R,S-LA, assuming generation of 2 moles of the thionitrobenzoate anion with each mole of DHLA. For comparative purposes, a range of LA-derivative concentrations were used in this work, but to accurately assess and compare rates of LA-dependent DTNB reduction in erythrocytes, R,S-LA concentrations 0.1 mM or less or times of 30 min or less should be used. In the absence of D-glucose, there was little DTNB reduction by the cells (Fig. 1A, squares), showing that the reducing equivalents for LA reduction derive from glucose in this cell type. To determine if LA reduction caused a generalized oxidant stress in human erythrocytes, endogenous ascorbate (Fig. 1B) and GSH (Fig. 1C) concentrations were measured. Neither ascorbate nor GSH were affected by reduction of R,S-LA concentrations as high as 1 mM. Omission of D-glucose also had no effect on steady-state intracellular concentrations of ascorbate (Fig. 1B, squares) or GSH (Fig. 1C, squares) during incubations with R,S-LA. This shows that the cells were unable to use reducing equivalents from either endogenous antioxidant for LA reduction. In cells that were incubated with 100 μM DHA for 15 min, intracellular ascorbate concentrations increased from 70−100 μM to 1.5 mM. Subsequent treatment of cells with R,S-LA concentrations as high as 1 mM again had no effect on intracellular concentrations of either ascorbate or GSH (results not shown). This indicates that even a redox stress from forcing the cells to reduce DHA to ascorbate did not affect pyridine nucleotide reserve.
Fig. 1.
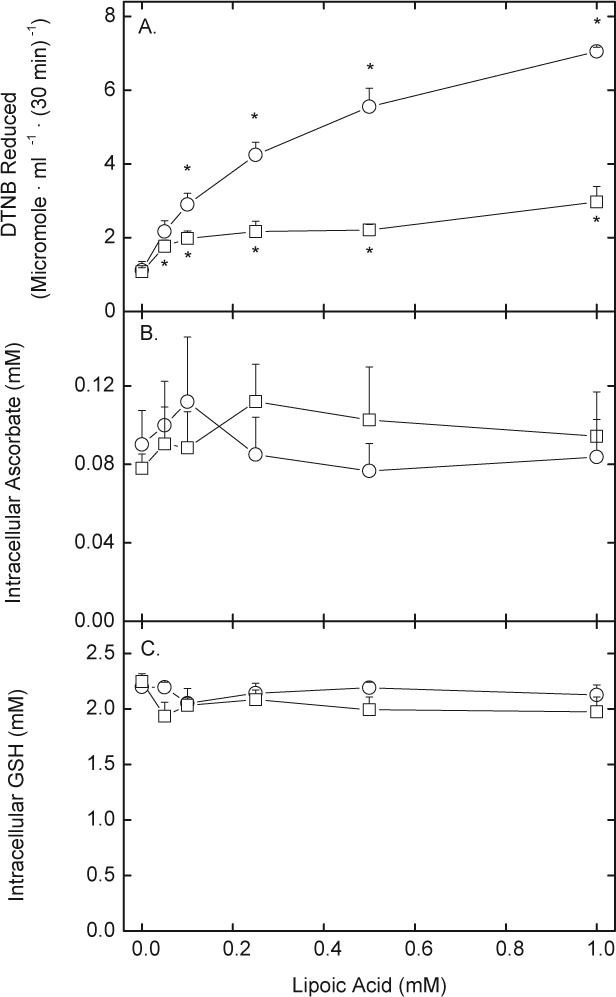
LA-dependent DTNB reduction and oxidant stress. Erythrocytes at a 1% packed cell volume were incubated at 37 °C in PBS with 0.2 mM DTNB and increasing concentrations of R,S-LA in the presence (circles) or absence (squares) of 5 mM d-glucose. After 30 min, the cells were pelleted by centrifugation and medium aliquots were taken for assay of DTNB reduction (A). After two rinses by centrifugation, the cells were taken for assay of intracellular ascorbate (B), GSH, (C) NADPH (D), and NADH. Results are from at least 4 experiments, with an “*” indicating p < 0.05 compared to the sample not treated with LA.
NADPH appeared to be a major source of reducing equivalents for LA in these cells. As shown in Fig. 2A, erythrocyte concentrations of NADPH decreased about 50% in D-glucose-treated cells incubated with increasing concentrations of R,S-LA for 30 min (circles). On the other hand, cells prepared and incubated in the absence of D-glucose contained less than 30% of the NADPH found in D-glucose-treated cells, and this was severely depleted by subsequent incubation with R,S-LA (Fig. 2A, squares). In contrast, NADH concentrations were unaffected by LA reduction when D-glucose was present (Fig. 2B, circles), although NADH (squares) was also severely decreased by lack of glucose.
Fig. 2.
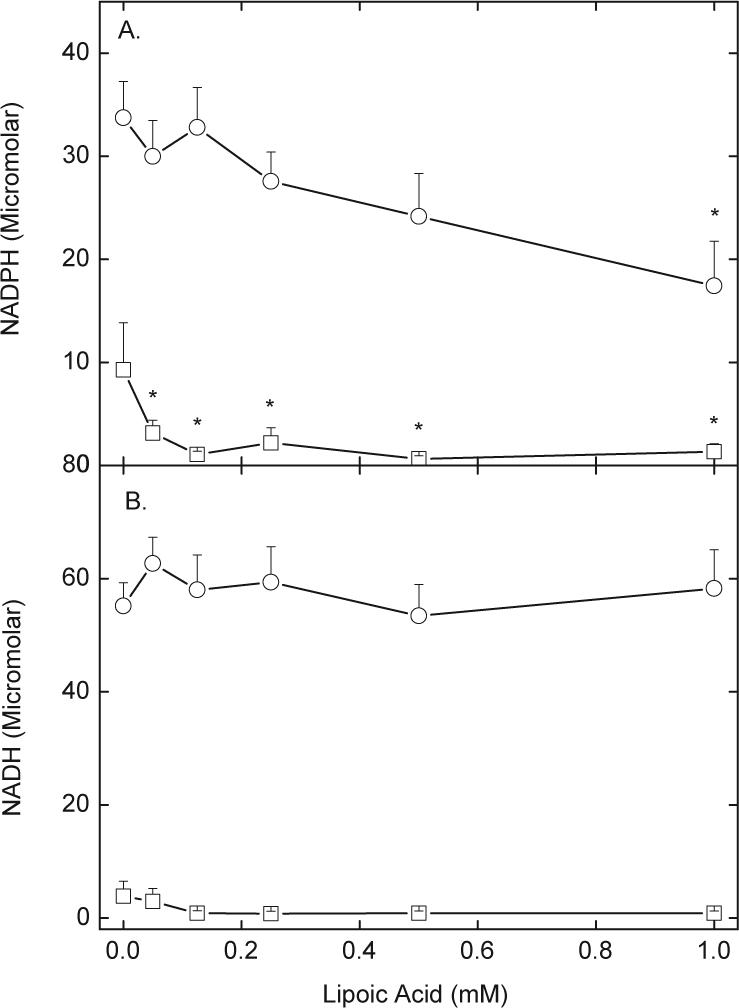
LA and glucose effects on erythrocyte pyridine nucleotide contents. Erythrocytes were incubated at 37 °C in PBS with increasing concentrations of R,S-LA in the presence (circles) or absence (squares) of 5 mM D-glucose. After 30 min, the cells were rinsed twice by centrifugation in PBS and taken for assay of intracellular NADPH (A, N = 6 experiments) or NADH (B, N = 4 experiments). An “*” indicates p < 0.05 compared to the sample not treated with LA.
To examine the mechanisms by which LA is reduced, erythrocytes were treated with thiol reagents known to affect GSH and NADPH-dependent enzymes capable of reducing LA. Exposure of erythrocytes to increasing concentrations of BCNU for 5 min at 37 EC, followed by addition of 0.2 mM DTNB and 0.1 mM R,S-LA for an additional 30 min decreased rates of DTNB reduction to about 30% of control (circles, Fig. 3A). GSH concentrations were also reduced to 25% of control in a manner slightly less sensitive than seen with DTNB reduction (squares, Fig. 3A). The non-specific alkylating agent NEM also decreased the capacity of the cells to reduce DTNB and decreased intracellular GSH in parallel (Fig. 3B). PAO, which is reactive with vicinal thiols, was much more potent in inhibiting LA-dependent DTNB reduction than the other two agents, again with a corresponding decrease in intracellular GSH (Fig. 3C). Neither NEM nor PAO decreased rates of DTNB reduction below basal rates in the absence of R,S-LA, however (results not shown). Together, these results indicate that LA reduction by erythrocytes was very sensitive to thiol reagents, but no more sensitive than the redox status of the cells as reflected by intracellular GSH concentrations. In results not shown, the arsine oxide reagent thorin, which does not enter cells because it contains two negative charges, at concentrations up to 0.2 mM, neither decreased DTNB reduction due to R,S-LA nor lowered intracellular GSH.
Fig. 3.
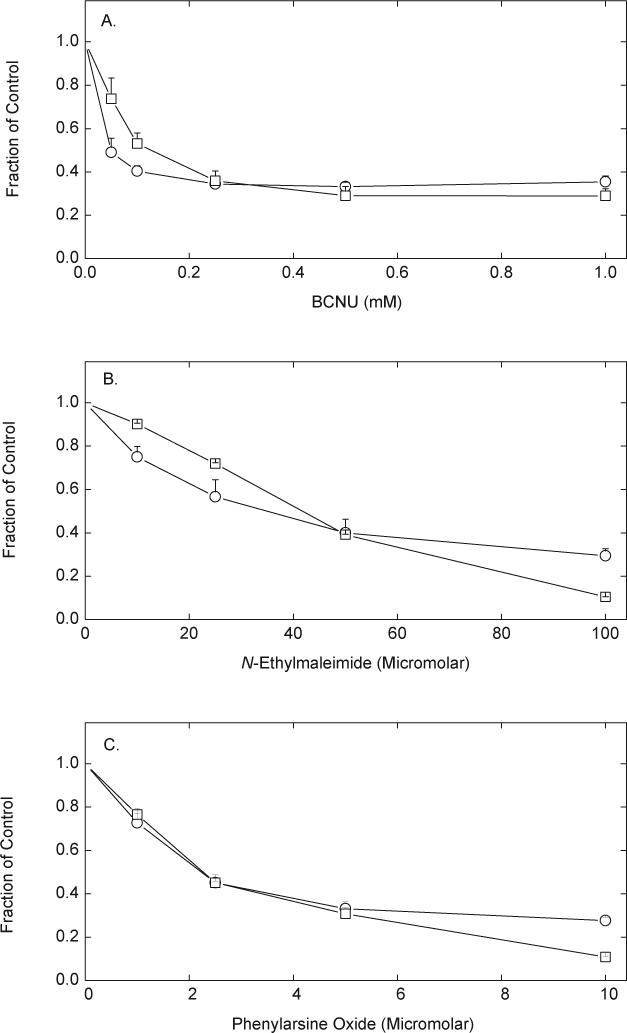
Inhibition of LA-dependent DTNB reduction by thiol reagents. Erythrocytes (1%) were incubated at 37 °C in PBS containing 5 mM D-glucose and the indicated agent at the concentrations noted in the 3 panels for 5 min, followed by addition of 0.2 mM DTNB and 0.1 mM R,S-LA. After 30 min, the cells were pelleted by centrifugation and aliquots of the medium were taken for assay of DTNB reduction (circles) and GSH content (squares). Results from 4 experiments with each agent are expressed as a fraction of the rate of LA-stimulated DTNB reduction or GSH content in cells not treated with the agent. Control rates of LA-stimulated DTNB reduction were 2.2 to 3.5 μmol mL−1 (30 min)−1, and GSH contents were 2−2.2 mM. Decreases due to the agents were significant at all agent concentrations for both DTNB reduction and GSH.
To attempt to separate effects of the thiol reagents on different types of intracellular thiols, cells that had been exposed for 10 min to either NEM or PAO were then treated with or without 2 mM dithiothreitol for 20 min. The cells were then rinsed to remove intra- and extracellular dithiothreitol before exposure to R,S-LA. The rationale was to determine whether dithiothreitol could reverse the effects of either thiol reagent on rates of LA-dependent DTNB reduction or on GSH concentrations. The ability of NEM to inhibit either LA-dependent DTNB reduction or to decrease GSH was not affected by dithiothreitol (compare third and sixth pair of bars, Fig. 4A). Although dithiothreitol did not reverse the ability of PAO to lower intracellular GSH concentrations, it completely reversed the inhibition of LA-dependent DTNB reduction due to PAO (compare third and sixth pair of bars, Fig 4B). The latter result suggests that GSH was not involved in the inhibition of LA-dependent DTNB reduction by PAO. This result supports the notion that either glutathione reductase or thioredoxin reductase, or both, are responsible for LA reduction by erythrocytes.
Fig. 4.
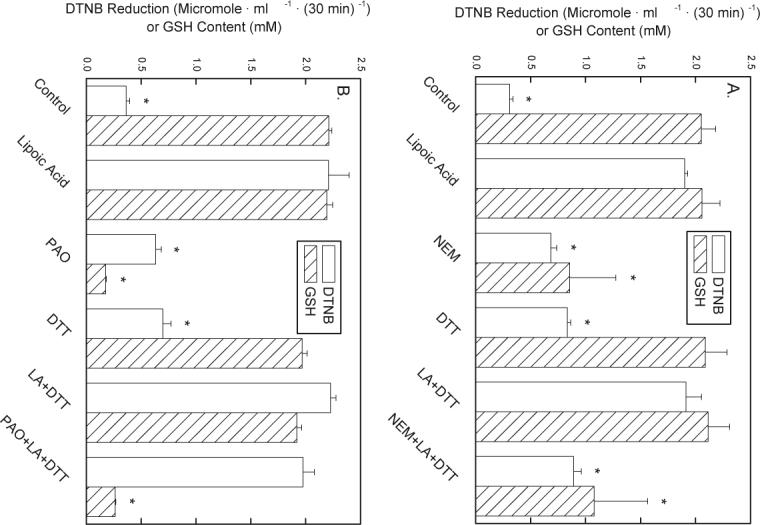
Reversibility of NEM- and PAO-induced decreases in LA-dependent DTNB reduction and GSH. Erythrocytes (1%) were incubated at 37 °C in PBS containing 5 mM d-glucose and either 50 μM NEM (A, N = 4 experiments) or 5 μM PAO (B, N = 6 experiments), where indicated. After 10 min, 2 mM dithiothreitol (DTT) was added where indicated and incubations were continued for another 20 min. The cells were then rinsed three times by centrifugation in PBS, and suspended in PBS that contained 5 mM d-glucose, 0.2 mM DTNB, and 0.1 mM R,S-LA, where indicated. After another 30 min of incubation at 37 °C, the cells were pelleted by centrifugation. Medium aliquots were assayed for DTNB reduction (open bars) and the cell contents of GSH (hatched bars) were determined. An “*” indicates p < 0.05 compared to the sample treated with LA alone.
Since different enzymes may reduce different isomers of LA with varying efficiencies, reduction of R- and S- LA was compared. Erythrocytes reduced the S-isomer of LA (circles) about 40−50% more efficiently than the R-isomer (squares) when both were present at low concentrations (Fig. 5A). We also compared uptake and reduction of the two forms of LA with that of hydroxyethyl disulfide. This small uncharged disulfide would not be expected to use a transporter to cross the cell membrane, and should be reduced by GSH- as well as NADPH-dependent mechanisms to β-mercaptoethanol. The latter will then readily diffuse out of the cells and react with extracellular DTNB. Rates of reduction of hydroxyethyl disulfide were in the range observed for R- and S-LA over the same concentration range (triangles, Fig. 5A).
Fig. 5.
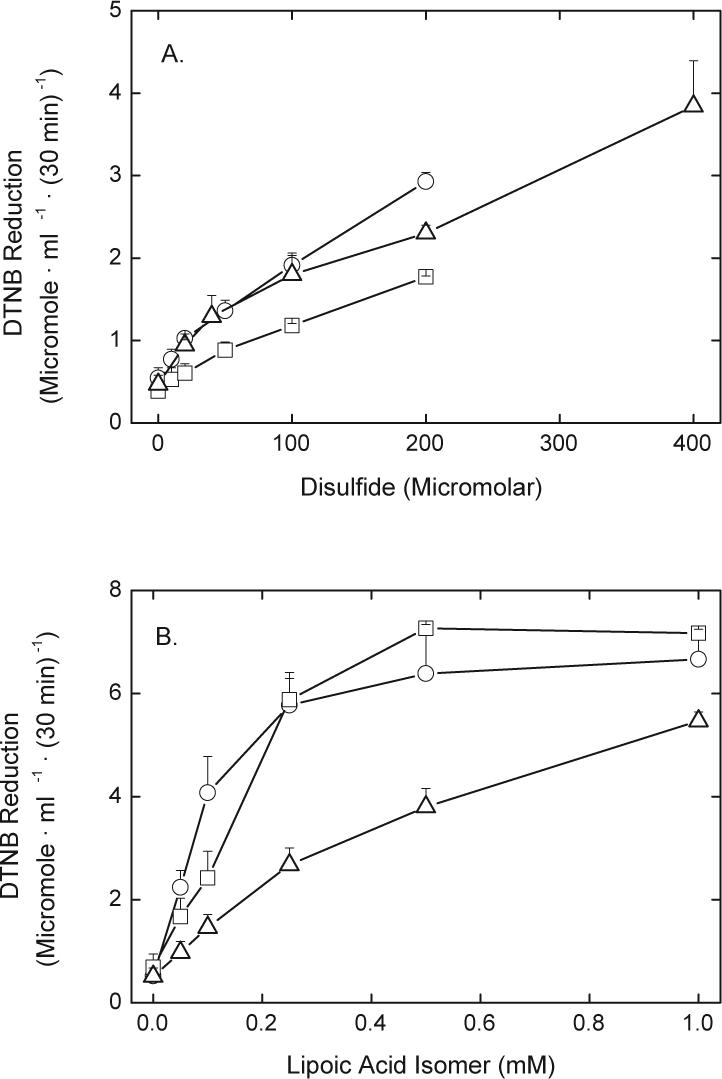
DTNB reduction by lipoic acid derivatives. Erythrocytes were incubated under the conditions described in the legend to Fig. 1 in the presence of 5 mM d-glucose for 30 min before assay of DTNB reduction. Panel A: S-LA (circles) or R-LA (squares), or hydroxyethyl disulfide (triangles). Panel B: R,S-lipoamide (circles), R-lipoamide (squares), or N-benzyl-R-lipoamide (triangles). Analysis of the results from 4 experiments in panel A showed that the two curves were significantly different (p < 0.05), and in panel B the analysis of the data from 4 experiments showed that the N-benzyl-R-lipoamide experimental data was significantly different than that from the other two experiments (p < 0.05).
Since movement of LA and its derivatives across the cell membrane might be affected by charge and lipophilicity of the derivative, we tested DTNB reduction generated by R-lipoamide, R,S-lipoamide, and N-benzyl-R-lipoamide. As shown in Fig. 5B, both R,S- (circles) and R-lipoamide (squares) caused higher rates of DTNB reduction at concentrations of 0.2 mM and less than those found for the different isomers of LA shown in Figs. 1 and 5A. In contrast, rates of DTNB reduction induced by N-benzyl-R-lipoamide (triangles, Fig. 5B) were about 40% less than those of R-lipoamide.
Inhibitors of likely transport mechanisms for LA were also studied. As shown in Fig. 6, increasing concentrations of octanoate inhibited DTNB reduction induced by both R- and S-LA, although the maximal inhibition was only about 16%. Biotin and pantothenic acid are known to share a transporter with LA in other cells, but neither inhibited LA-dependent DTNB reduction at concentrations up to 0.5 mM (results not shown). These results suggest that neither the entry of LA nor the exit of DHLA in erythrocytes is significantly dependent on transporters known to be used in other cells.
Fig. 6.
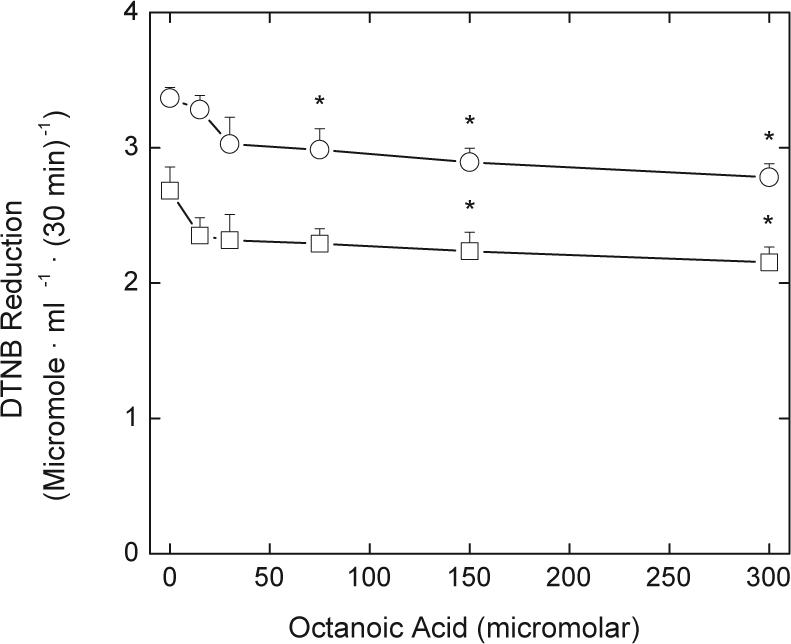
Inhibition of LA-dependent DTNB reduction by octanoic acid. Erythrocytes at a 1% packed cell volume were incubated at 37 °C in PBS containing 5 mM d-glucose and the indicated concentrations of octanoic acid. After 5 min, DTNB was added to a final concentration of 0.2 mM, followed by addition of either S-LA (circles) or R-LA (squares) to an initial concentration of 0.1 mM. After 30 min the cells were pelleted by centrifugation and DTNB reduction measured in aliquots of the supernatant. Analysis of results from four experiments showed that the two curves were significantly different (p < 0.05). An “*” indicates p < 0.05 compared to the sample not treated with octanoate.
Since the results with PAO suggest that thioredoxin reductase is the major enzyme responsible for LA reduction in erythrocytes, we tested rates of reduction of purified mammalian thioredoxin reductase with LA isomers. None of the derivatives were reduced appreciably by thioredoxin reductase alone. However, when thioredoxin was added to a concentration of 1.9 μM, reduction was readily detected. Reduction was linear for 3 min at 37 EC, with similar rates for the different derivatives as follows (units are nmol NADPH • (nmol thioredoxin reductase)−1 • (min)−1 ± SD from 3 experiments): R,S-LA, 216 ± 58; R-LA, 244 ± 75; S-LA, 254 ± 71; and R,S-lipoamide, 263 ± 34.
DHA is also taken up by erythrocytes and reduced to ascorbate. Ascorbate in turn donates electrons to a trans-plasma membrane oxidoreductase that transfers the electrons to extracellular ferricyanide. Since DHLA will also reduce ferricyanide, to compare the ability of erythrocytes to reduce R,S-LA and DHA, we measured rates of ferricyanide reduction in cells treated with each antioxidant precursor. As shown in Fig. 7, the rate of DHA-dependent ferricyanide reduction was several-fold higher than that of LA-dependent ferricyanide reduction across the same concentration range of the agents.
Fig. 7.
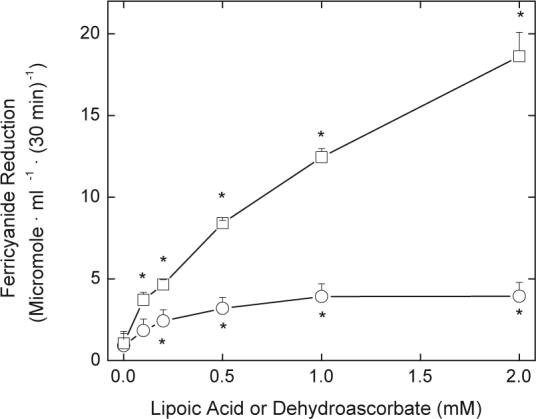
Ferricyanide reduction of DHA and LA. Erythrocytes (1%) were incubated at 37 °C in PBS containing 5 mM d-glucose, 1 mM ferricyanide, and the indicated concentration of either R,S-LA (circles) or DHA (squares). After 30 min the cells were pelleted by centrifugation and aliquots of the supernatant were taken for assay of ferrocyanide. Results are shown from 3 experiments with an “* indicating p < 0.05 compared to a sample not treated with LA or DHA. These two curves were different from one another at p < 0.05.
Discussion
As in cultured tumour and endothelial cells [7,9], human erythrocytes took up and reduced LA to DHLA, which was released and then detected outside cells by its reduction of DTNB. This reduction was dependent on the concentration of LA and required cellular glucose metabolism. Similar glucose dependence was previously documented in cultured cell lines [7,9]. Since there was no LA-dependent DTNB reduction in cells deficient in glucose 6-phosphate dehydrogenase-deficient cells [7], activity of the pentose phosphate cycle is also likely to be required, even in cells containing mitochondria and thus lipoamide dehydrogenase. Further evidence that NADPH derived from the pentose phosphate cycle is required for LA reduction in erythrocytes was the finding in the present work that LA depleted erythrocyte NADPH without affecting NADH. Additionally, reduction of LA concentrations as high as 1 mM did not cause a significant generalized oxidant stress in these short-term incubations, since neither GSH nor ascorbate was affected. On the other hand, it has been recently shown that incubation of human erythrocytes for 4 h or more at 37 °C with 0.1 to 1 mM LA decreased total cellular thiols [26]. Since DHLA caused only minimal thiol depletion [26], initial reduction of LA and its continued “redox” cycling with time can cause an oxidant stress in erythrocytes.
The selective depletion of NADPH as a result of LA reduction favors the notion that NADPH-dependent disulfide oxidoreductases were responsible for erythrocyte LA reduction. This was further supported by testing thiol reagents with differential selectivity for GSH and the enzymes presumed to be involved. Of the agents tested, NEM is relatively specific for thiols, although it is not specific for any type of thiol and will alkylate both protein and low molecular weight thiols. Whereas BCNU is typically used as an inhibitor of glutathione reductase [27], it also can carbamoylate active site cysteines of other enzymes, probably including the active selenol of thioredoxin reductase [28,29]. PAO reacts with nearby or vicinal thiols and probably selenols [30]. It inhibits thioredoxin reductase at concentrations of 1 μM or less [30,31], which agrees well with the observed half-maximal effect on LA-dependent DTNB reduction of about 2 μM in erythrocytes. On the other hand, all three of the agents decreased erythrocyte GSH in concert with their effects on LA-dependent DTNB reduction. For NEM and BCNU, this likely reflects direct reaction with GSH or inhibition of glutathione reductase. PAO does not directly react with GSH [30] or inhibit glutathione reductase ([30] and May, J.M., unpublished observations). GSSG is not a substrate for thioredoxin reductase [32], and only a poor substrate for the thioredoxin reductase/thioredoxin system [33]. Nonetheless, inhibition of TR by PAO could indirectly lower GSH by causing cellular oxidant stress. In this regard, we previously showed that PAO caused a more severe depletion of ascorbate than GSH in ascorbate-loaded erythrocytes [31]. Although this was considered to reflect inhibition of thioredoxin reductase-dependent ascorbate recycling, at least part of the decrease in ascorbate could have been due to non-specific cellular oxidant stress. Inhibition of the recently discovered enzyme selenoprotein thioredoxin-glutathione reductase-1 could also have played a role in the decrease in GSH with the agents. This enzyme can reduce GSSG to GSH [34], although its sensitivity to thiol inhibitors is not known. However, the enzyme is likely present in very small amounts in tissues outside the testes [35].
The effect of PAO to inhibit LA-dependent DTNB reduction was completely reversed by treating the cells with dithiothreitol without reversing its effect on GSH depletion. Given the selectivity of PAO for thioredoxin reductase, this result strongly implicates the thioredoxin reductase system in erythrocyte LA-dependent DTNB reduction. This conclusion requires that the PAO-thiol or PAO–selenol adducts on thioredoxin reductase or PAO-thiol adducts on thioredoxin are more sensitive to release by dithiothreitol than are those on the GSH-regenerating enzymes.
The mechanism of LA uptake and DHLA efflux from erythrocytes is unclear. Strong inhibition of R,S-LA uptake and reduction by octanoic acid implicated a medium chain fatty acid transporter in endothelial cells [9]. However, octanoic acid had little effect on LA-dependent DTNB reduction in erythrocytes, irrespective of whether R- or S-LA was used. This suggests that a medium change fatty acid transporter plays little role in this activity in erythrocytes. As in cultured endothelial cells [9], neither biotin nor pantothenic acid inhibited LA-dependent DTNB reduction, making it unlikely that a multivitamin transporter [36] mediates LA uptake in either cell type. Lipoamide induced DTNB reduction only slightly more effectively than LA in erythrocytes, and N-benzyl-R-lipoamide was less effective. It might be expected that these uncharged and more hydrophobic LA derivatives would cross the plasma membrane better than LA, but this was not a major factor. LA has been reported to readily cross erythrocyte ghost membranes, but not to be taken up by phospholipid liposomes [8]. This suggests that there may be an as yet unknown transporter that facilitates LA entry or DHLA exit from erythrocytes.
Erythrocytes were modestly more effective in reducing S-LA than R-LA. Mammalian glutathione reductase reduced S-LA about 2-fold faster than R-LA, which could contribute to the differences seen here. We found little difference in specificity of the thioredoxin reductase system for R- versus S-LA, or for R,S-lipoamide. In contrast to published results [37], we observed little activity of thioredoxin reductase with LA derivatives alone. However, when thioredoxin, the natural substrate for the enzyme, was included in the reaction mixture, there was reduction at rates comparable to those measured previously with the enzyme alone [37]. This suggests that thioredoxin could mediate LA and lipoamide reduction in cells.
Erythrocyte ferricyanide reduction was up to 4-fold greater when erythrocytes were pre-incubated with DHA than with the same concentrations of LA. Ferricyanide remains outside cells because of its size and negative charge [38]. It is directly reduced by thiols and by ascorbic acid. However, it is also an electron acceptor for a trans-plasma membrane oxidoreductase [23] that uses intracellular ascorbate as its primary electron donor [39], generating the ascorbate free radical within erythrocytes [20]. LA-dependent ferricyanide reduction will occur only with DHLA that escapes the cells, just as with LA-dependent DTNB reduction. Indeed, rates of LA-dependent DTNB reduction (Fig. 1A, circles) were comparable to those of LA-dependent ferricyanide reduction (Fig. 8) under identical incubation conditions. This suggests that both extracellular agents capture most of the DHLA released from the cells. The markedly greater ferricyanide reduction due to loading the cells with DHA rather than with LA could be due to several factors. First, whereas reduction of both DHA and LA occurs within cells, trans-plasma membrane electron transfer from ascorbate to ferricyanide may be faster than efflux of DHLA. Second, the cells in dilute suspension may not be able to recover all the extracellular LA generated by reaction of DHLA with DTNB. Third, the cells may be more efficient at reducing DHA than LA. This might relate to the observations that both DHA [40] and LA [37] are reduced by thioredoxin reductase, but only DHA is reduced by GSH and GSH-dependent enzymes [41]. Whereas ascorbate-dependent ferricyanide reduction has been used as an integrated measure of the global reduction capacity of erythrocytes [42-44], LA-dependent DTNB reduction is likely to reflect more specifically the activity and capacity of pyridine nucleotide disulfide oxidoreductases, and thioredoxin reductase in particular.
In this work we have evaluated the mechanism of LA-dependent DTNB reduction in human erythrocytes. LA and DHLA readily cross the erythrocyte membrane, but little of this transfer occurs on the medium chain fatty acid transporter, as it does in other cells. LA is reduced exclusively by NADPH-dependent disulfide oxidoreductases in erythrocytes. Since human erythrocytes can be studied ex vivo in different clinical conditions, measurements of LA-dependent DTNB reduction can be used in combination with assay of hydroxyethyl disulfide reduction to assess both NADPH- and GSH-dependent disulfide reduction by the cells. Further, use of these assays in conjunction with measurement of ascorbate-dependent ferricyanide reduction will provide a global approach to assessing erythrocyte reduction capacity or redox reserve.
Acknowledgment
This publication was made possible by grant number R21AT001062 from NCCAM/NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ou P, Tritschler HJ, Wolff SP. Thioctic (lipoic) acid: A therapeutic metal-chelating antioxidant. Biochem Pharmacol. 1995;50:123–126. doi: 10.1016/0006-2952(95)00116-h. [DOI] [PubMed] [Google Scholar]
- 2.Bonomi F, Pagani S. Removal of ferritin-bound iron by DL-dihydrolipoate and DL-dihydrolipoamide. Eur J Biochem. 1986;155:295–300. doi: 10.1111/j.1432-1033.1986.tb09489.x. [DOI] [PubMed] [Google Scholar]
- 3.Packer L, Tritschler HJ. Alpha-lipoic acid: The metabolic antioxidant. Free Radic Biol Med. 1996;20(4):625–626. doi: 10.1016/0891-5849(95)02129-9. [DOI] [PubMed] [Google Scholar]
- 4.Bilska A, Wlodek L. Lipoic acid - the drug of the future? Pharmacol Rep. 2005;57(5):570–577. [PubMed] [Google Scholar]
- 5.Handelman GJ, Han D, Tritschler H, Packer L. α-lipoic acid reduction by mammalian cells to the dithiol form, and release into the culture medium. Biochem Pharmacol. 1994;47:1725–1730. doi: 10.1016/0006-2952(94)90298-4. [DOI] [PubMed] [Google Scholar]
- 6.Jones W, Li X, Perriott LM, Whitesell RR, May JM. Uptake, recycling, and antioxidant functions of α-lipoic acid in endothelial cells. Free Radic Biol Med. 2002;33:83–93. doi: 10.1016/s0891-5849(02)00862-6. [DOI] [PubMed] [Google Scholar]
- 7.Biaglow JE, Donahue J, Tuttle S, Held K, Chrestensen C, Mieyal J. A method for measuring disulfide reduction by cultured mammalian cells: relative contributions of glutathione-dependent and glutathione-independent mechanisms. Anal Biochem. 2000;281(1):77–86. doi: 10.1006/abio.2000.4533. [DOI] [PubMed] [Google Scholar]
- 8.Constantinescu A, Pick U, Handelman GJ, Haramaki N, Han D, Podda M, Tritschler HJ, Packer L. Reduction and transport of lipoic acid by human erythrocytes. Biochem Pharmacol. 1995;50(2):253–261. doi: 10.1016/0006-2952(95)00084-d. [DOI] [PubMed] [Google Scholar]
- 9.May JM, Qu ZC, Nelson DJ. Cellular disulfide-reducing capacity: an integrated measure of cell redox capacity. Biochem Biophys Res Commun. 2006;344(4):1352–1359. doi: 10.1016/j.bbrc.2006.04.065. [DOI] [PubMed] [Google Scholar]
- 10.Jocelyn PC. The standard redox potential of cysteine-cystine from the thiol-disulfide exchange reaction with glutathione and lipoic acid. Eur J Biochem. 1967;2:327–331. doi: 10.1111/j.1432-1033.1967.tb00142.x. [DOI] [PubMed] [Google Scholar]
- 11.Pick U, Haramaki N, Constantinescu A, Handelman GJ, Tritschler HJ, Packer L. Glutathione reductase and lipoamide dehydrogenase have opposite stereospecificities for α-lipoic acid enantiomers. Biochem Biophys Res Commun. 1995;206:724–730. doi: 10.1006/bbrc.1995.1102. [DOI] [PubMed] [Google Scholar]
- 12.Canestrari F, Galli F, Giorgini A, Albertini MC, Galiotta P, Pascucci M, Bossù M. Erythrocyte redox state in uremic anemia: Effects of hemodialysis and relevance of glutathione metabolism. Acta Haematol. 1994;91:187–193. doi: 10.1159/000204332. [DOI] [PubMed] [Google Scholar]
- 13.Henning SM, Zhang JZ, McKee RW, Swendseid ME, Jacob RA. Glutathione blood levels and other oxidant defense indices in men fed diets low in vitamin C. J Nutr. 1991;121:1969–1975. doi: 10.1093/jn/121.12.1969. [DOI] [PubMed] [Google Scholar]
- 14.Hunt NH, Stocker R. Oxidative stress and the redox status of malaria-infected erythrocytes. Blood Cells. 1990;16:499–526. [PubMed] [Google Scholar]
- 15.Yoshida K, Hirokawa J, Tagami S, Kawakami Y, Urata Y, Kondo T. Weakened cellular scavenging activity against oxidative stress in diabetes mellitus: Regulation of glutathione synthesis and efflux. Diabetologia. 1995;38:201–210. doi: 10.1007/BF00400095. [DOI] [PubMed] [Google Scholar]
- 16.Youdim KA, Shukitt-Hale B, MacKinnon S, Kalt W, Joseph JA. Polyphenolics enhance red blood cell resistance to oxidative stress: in vitro and in vivo. Biochim Biophys Acta Gen Subj. 2000;1523(1):117–122. doi: 10.1016/s0304-4165(00)00109-4. [DOI] [PubMed] [Google Scholar]
- 17.Coleman MD. Monitoring diabetic antioxidant status: a role for in vitro methaemoglobin formation. Environ Toxicol Pharmacol. 2001;10(4):207–213. doi: 10.1016/s1382-6689(01)00084-9. [DOI] [PubMed] [Google Scholar]
- 18.May JM, Qu Z-C, Mendiratta S. Protection and recycling of α-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch Biochem Biophys. 1998;349:281–289. doi: 10.1006/abbi.1997.0473. [DOI] [PubMed] [Google Scholar]
- 19.Hissin PJ, Hilf R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal Biochem. 1976;74:214–226. doi: 10.1016/0003-2697(76)90326-2. [DOI] [PubMed] [Google Scholar]
- 20.May JM, Qu ZC, Whitesell RR, Cobb CE. Ascorbate recycling in human erythrocytes: Role of GSH in reducing dehydroascorbate. Free Radic Biol Med. 1996;20(4):543–551. doi: 10.1016/0891-5849(95)02130-2. [DOI] [PubMed] [Google Scholar]
- 21.Zerez CR, Lee SJ, Tanaka KR. Spectrophotometric determination of oxidized and reduced pyridine nucleotides in erythrocytes using a single extraction procedure. Anal Biochem. 1987;164:367–373. doi: 10.1016/0003-2697(87)90506-9. [DOI] [PubMed] [Google Scholar]
- 22.May JM, Qu ZC, Cobb CE. Reduction and uptake of methylene blue by human erythrocytes. Am J Physiol Cell Physiol. 2004;286(6):C1390–C1398. doi: 10.1152/ajpcell.00512.2003. [DOI] [PubMed] [Google Scholar]
- 23.Orringer EP, Roer ME. An ascorbate-mediated transmembrane-reducing system of the human erythrocyte. J Clin Invest. 1979;63:53–58. doi: 10.1172/JCI109277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Avron M, Shavit N. A sensitive and simple method for determination of ferrocyanide. Anal Biochem. 1963;6:549–554. doi: 10.1016/0003-2697(63)90149-0. [DOI] [PubMed] [Google Scholar]
- 25.Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 26.Coleman MD, Rimmer GS, Haenen GR. Effects of Lipoic Acid and Dihydrolipoic Acid on Total Erythrocytic Thiols under Conditions of Restricted Glucose in vitro. Basic Clin Pharmacol Toxicol. 2007;100(2):139–144. doi: 10.1111/j.1742-7843.2006.00025.x. [DOI] [PubMed] [Google Scholar]
- 27.Frischer H, Ahmad T. Severe generalized glutathione reductase deficiency after antitumor chemotherapy with BCNU (1,3-bis(chloroethyl)-1-nitrosourea). J Lab Clin Med. 1977;89:1080–1091. [PubMed] [Google Scholar]
- 28.Gromer S, Schirmer RH, Becker K. The 58 kDa mouse selenoprotein is a BCNU-sensitive thioredoxin reductase. FEBS Lett. 1997;412(2):318–320. doi: 10.1016/s0014-5793(97)00816-8. [DOI] [PubMed] [Google Scholar]
- 29.Schallreuter KU, Gleason FK, Wood JM. The mechanism of action of the nitrosourea anti-tumor drugs on thioredoxin reductase, glutathione reductase and ribonucleotide reductase. Biochim Biophys Acta. 1990;1054(1):14–20. doi: 10.1016/0167-4889(90)90199-n. [DOI] [PubMed] [Google Scholar]
- 30.Biaglow JE, Ayene IS, Tuttle SW, Koch CJ, Donahue J, Mieyal JJ. Role of vicinal protein thiols in radiation and cytotoxic responses. Radiat Res. 2006;165(3):307–317. doi: 10.1667/rr3505.1. [DOI] [PubMed] [Google Scholar]
- 31.Mendiratta S, Qu Z-C, May JM. Enzyme-dependent ascorbate recycling in human erythrocytes: role of thioredoxin reductase. Free Radic Biol Med. 1998;25:221–228. doi: 10.1016/s0891-5849(98)00060-4. [DOI] [PubMed] [Google Scholar]
- 32.Luthman M, Holmgren A. Rat liver thioredoxin and thioredoxin reductase: purification and characterization. Biochemistry. 1982;21(26):6628–6633. doi: 10.1021/bi00269a003. [DOI] [PubMed] [Google Scholar]
- 33.Holmgren A. Reduction of disulfides by thioredoxin. Exceptional reactivity of insulin and suggested functions of thioredoxin in mechanism of hormone action. J Biol Chem. 1979;254:9113–9119. [PubMed] [Google Scholar]
- 34.Sun QA, Kirnarsky L, Sherman S, Gladyshev VN. Selenoprotein oxidoreductase with specificity for thioredoxin and glutathione systems. Proc Natl Acad Sci USA. 2001;98(7):3673–3678. doi: 10.1073/pnas.051454398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su D, Novoselov SV, Sun QA, Moustafa ME, Zhou Y, Oko R, Hatfield DL, Gladyshev VN. Mammalian selenoprotein thioredoxin-glutathione reductase. Roles in disulfide bond formation and sperm maturation. J Biol Chem. 2005;280(28):26491–26498. doi: 10.1074/jbc.M503638200. [DOI] [PubMed] [Google Scholar]
- 36.Prasad PD, Wang H, Kekuda R, Fujita T, Fei YJ, Devoe LD, Leibach FH, Ganapathy V. Cloning and functional expression of a cDNA encoding a mammalian sodium- dependent vitamin transporter mediating the uptake of pantothenate, biotin, and lipoate. J Biol Chem. 1998;273:7501–7506. doi: 10.1074/jbc.273.13.7501. [DOI] [PubMed] [Google Scholar]
- 37.Arnér ESJ, Nordberg J, Holmgren A. Efficient reduction of lipoamide and lipoic acid by mammalian thioredoxin reductase. Biochem Biophys Res Commun. 1996;225(1):268–274. doi: 10.1006/bbrc.1996.1165. [DOI] [PubMed] [Google Scholar]
- 38.Székely M, Mányai S, Straub FB. Über den Mechanismus der osmotischen Hämolyse. Acta Physiol Acad Sci Hung. 1952;3:571–583. [PubMed] [Google Scholar]
- 39.May JM, Qu Z-C, Whitesell RR. Ascorbate is the major electron donor for a transmembrane oxidoreductase of human erythrocytes. Biochim Biophys Acta. 1995;1238:127–136. doi: 10.1016/0005-2736(95)00120-r. [DOI] [PubMed] [Google Scholar]
- 40.May JM, Mendiratta S, Hill KE, Burk RF. Reduction of dehydroascorbate to ascorbate by the selenoenzyme thioredoxin reductase. J Biol Chem. 1997;272:22607–22610. doi: 10.1074/jbc.272.36.22607. [DOI] [PubMed] [Google Scholar]
- 41.Winkler BS, Orselli SM, Rex TS. The redox couple between glutathione and ascorbic acid: A chemical and physiological perspective. Free Radic Biol Med. 1994;17:333–349. doi: 10.1016/0891-5849(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 42.Kadlubowski M, Agutter PS. Changes in the activities of some membrane-associated enzymes in vivo during ageing of the normal human erythrocyte. Br J Haematol. 1977;37:111–125. [PubMed] [Google Scholar]
- 43.Lykkesfeldt J, Moos T. Age-dependent change in Vitamin C status: a phenomenon of maturation rather than of ageing. Mech Ageing Dev. 2005;126(8):892–898. doi: 10.1016/j.mad.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 44.Matteucci E, Giampietro O. Transmembrane electron transfer in diabetic nephropathy. Diabetes Care. 2000;23(7):994–999. doi: 10.2337/diacare.23.7.994. [DOI] [PubMed] [Google Scholar]