

Abstract
The c-myb protooncogene is predominantly expressed in hematopoietic cells and plays a vital role in hematopoiesis. Retinoic acid (RA) is able to induce differentiation of several hematopoietic cells. This differentiation is linked to decreased c-myb expression, suggesting that retinoid receptors (RAR/RXR) may down-regulate c-myb gene expression. Furthermore, recent data indicate that RAR inhibits the function of the Myb protein itself. In addition, the Myb-Ets oncogenic fusion protein has been shown to inhibit transcriptional activation by RAR and thyroid hormone receptor. Myb-Ets also antagonizes the biological response of erythrocytic progenitor cells to RA and thyroid hormone. This prompted us to investigate a possible cross talk between RAR and Myb. Here, we demonstrate that RA inhibits the expression of the endogenous Myb target gene tom-1. Conversely, Myb functions as a potent inhibitor of RA-induced biological responses. Functional analysis of Myb mutants in transfection studies revealed that the Myb DNA-binding domain (DBD) is necessary for repression whereas the transactivation domain is dispensable. Furthermore, we show that v-Myb and RAR interact in vitro and in vivo. This interaction requires the DBD of RAR. In contrast, glutathione S-transferase-pulldown assays with v-Myb mutants indicate that the DBD and the C terminus of Myb directly interact with RAR. Our results suggest that the physical interaction between Myb and RAR may play a role in the regulation of hematopoietic gene expression.
Keywords: transcriptional regulation, nuclear receptors, transrepression, myb protooncogene
Retinoids play important roles in development and differentiation and are well-known inhibitors of cell growth (1). Retinoic acid (RA), a vitamin A derivative, exerts biological effects by serving as a ligand for at least two groups of nuclear receptors referred to as RA receptors (RARs) and retinoid X receptors (RXRs). Both belong to the nuclear receptor superfamily of ligand-dependent transcription factors (1). Experimental and clinical observations demonstrate that RARs are involved in the regulation of hematopoiesis. For example, RA induces the HL-60 human leukemia cell line to differentiate into neutrophils (2). This process is mediated through RARs and can be prevented by a dominant-negative RARα mutant (3). Furthermore, the expression of a dominant-negative RARα in normal mouse bone marrow cells can arrest myeloid development (4). Patients with acute promyelocytic leukemia treated with all-trans-RA can achieve temporary remission of the leukemia, presumably by inducing differentiation of the leukemic cells (5).
The c-myb protooncogene is the cellular homolog of the v-myb gene, which initially was identified in the genome of the acutely oncogenic avian myeloblastosis virus (reviewed in ref. 6). v-myb is responsible for the transformation of myelomonocytic hematopoietic cells in vivo and in vitro (reviewed in ref. 7). Considerable evidence suggests that c-myb is involved in proliferation as well as in differentiation of hematopoietic cells. c-myb expression is high in immature cells of all hematopoietic lineages and appears to be essential for their proliferation (7, 8). Induction of terminal differentiation of immature blood cells results in the down-regulation of c-myb mRNA (7, 8). In addition, sustained expression of c-myb blocks terminal differentiation of various hematopoietic cell lines (7, 8). These results suggest that both oncogenic and cellular forms of the Myb protein play important roles in maintaining the immature state of hematopoietic cells by blocking differentiation signals while simultaneously stimulating proliferation. Early attempts to assign a function to Myb were based on its subcellular localization, DNA-binding properties, expression pattern, and transactivation properties. This work led to the conclusion that Myb functions as a transcription factor essential for the proliferation of immature hematopoietic cells (8).
Previous studies showed that c-myb expression decreases during RA-induced differentiation of hematopoietic cells (8). These results suggest that RAR may down-regulate expression of the c-myb protooncogene. In addition, recent data indicated that the transcriptional activation of Myb-responsive reporter genes can be inhibited by RA, thus suggesting that RAR also might inhibit the transcriptional activity of Myb. Furthermore, the introduction of RARα into v-myb-transformed monoblasts suppressed transformation and permitted RA-dependent differentiation into macrophage-like cells (9). Investigation of the Myb-Ets fusion oncoprotein of the E26 avian leukemia retrovirus showed that Myb-Ets inhibits transactivation of both RAR and thyroid hormone receptor (TR). Moreover, Myb-Ets antagonizes the biological response of erythrocytic progenitor cells to RA and thyroid hormone (T3) (10). The hypothesis that RAR and Myb may function as antagonistic regulators of proliferation and differentiation led us to investigate whether Myb alone can inhibit the transcriptional activity of RAR.
In this report we show that Myb functions as an inhibitor of RA-induced transactivation and that RA antagonizes the induction of the v-Myb target gene tom-1 in vivo. Myb interacts with RAR but not with RXR in vitro and in vivo. We demonstrate that the DNA-binding domain (DBD) of RAR is both necessary and sufficient for interaction. Conversely, we provide evidence that the DBD and the C terminus of Myb physically interact with RAR. The inhibition of RAR-mediated activation by direct protein–protein interactions may, at least in part, explain the ability of Myb to block the differentiation pathway of myeloblast cells.
MATERIALS AND METHODS
Cell Culture and Transfection.
NIH 3T3, CV1, and COS-7 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum. 10.4 cells (expressing v-Myb-ER fusion proteins) were cultured as described (11). Transient transfection assays were carried out as described in (12). Activity of the (TREp)2-thymidine kinase (TK)-luciferase (LUC) reporter plasmid was analyzed by cotransfection of 25 ng each of hRARα and hRXRα expression plasmids. LUC activity was assayed as recommended by the manufacturer (Promega). All experiments were repeated at least three times. Protein analysis of transfected cells were performed by classical Western blotting and immunoprecipitation techniques with whole-cell extracts.
Recombinant Plasmids and Constructs.
The LUC reporter plasmids (TREp)2-TKLUC, βRE2-TKLUC, Galp3TKLUC, and TKLUC were described (12). Expression vectors pCM100, pCM101, pVM116, DIN1, pVM130, and pVM111 were described (13–15). For all experiments hRARα and hRXRα sequences or derivatives were used. The plasmids CMX-hRARα, CMX-hRXRα, CMX-RAR DBD, CMX-RAR ΔDBD, CMX-RAR Nterm, CMX-RAR Cterm, glutathione S-transferase (GST)-hRARα, GST-RAR ΔDBD, GST-RAR DBD, GST-RAR Nterm, and GST-RAR Cterm were described (12). To create pCMX-v-Myb, pCMX-v-MybΔ transactivation domain (TA), pGEX-v-Myb, and pGEX-v-MybΔTA the cDNA inserts of pVM116 and DIN1 were amplified by PCR and cloned into pCMX-NLS and pGEX-1. pCMX-v-MybΔNterm was constructed by inserting the EcoRI–XbaI fragment of pVM116 into pCMX-NLS. To create pCMX-v-MybΔCterm, pCMX-v-Myb was digested with EcoRI and religated. pGEX-v-MybΔNterm was generated by inserting the EcoRI fragment of pCMX-v-Myb, encoding the C-terminal part of v-Myb into pGEX-3X. To create pGEX-v-MybΔCterm the BamHI–EcoRI fragment of pCMX-v-Myb encoding the DBD of v-Myb was inserted into pGEX-1. The expression plasmids Gal4-v-myb and pCMX-RAR-VP16 were described (12, 13).
Reverse Transcription–PCR Analysis.
Cells (10.4) were treated for 18 hr with EtOH, 1 μM RA, 2 μM β-estradiol (E2), or both ligands. Total RNAs were isolated by using TRIZOL (GIBCO/BRL). Five micrograms of RNA was reverse-transcribed by using the first-strand cDNA synthesis kit (Pharmacia). cDNA synthesis and the PCR amplifications were performed according to the manufacturer’s protocol. The annealing temperature for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and tom-1 were 55°C or 60°C, respectively. The following primers were used: tom-1, sense 5′-GTACGAAGATCCCCAAGCCACCAA-3′, antisense 5′-GCCCCACAGCACACCAGTCAGTT-3′; GAPDH, sense 5′-AGAGGTGCTGCCCAGAACATCATC-3′, antisense 5′-GTGGGGGAGACAGAAGGGAACAGA-3′.
In Vitro Binding Assays.
The GST-fusion proteins were expressed according to ref. 12. The in vitro interaction assays with [35S]methionine-labeled, in vitro-translated proteins were performed as recommended in ref. 12. Note that all steps were performed at 4°C and that the interaction buffer contained 50 mM Tris⋅HCl (pH 8), 100 mM NaCl, 10 mM MgCl2, 10% glycerol, 0.3 mM DTT, 0.1% Nonidet P-40.
DNA-Binding Studies.
Nuclear extracts of COS-7 cells either untreated or transiently transfected with the v-Myb expression vector pVM116 were used for electrophoretic mobility-shift assays (EMSA). EMSAs were performed in 10 mM Tris⋅HCl (pH 8), 80 mM KCl, 6% glycerol, 0.05% Nonidet P-40, 1 mM DTT, 1 μg of poly(dI-dC) with 32P-labeled βRARE oligonucleotide (5′-GATCTAGGGTTCACCGAAAGTTCACTCGGATC-3′) for 30 min on ice and analyzed on a 5% polyacrylamide gel in 0.5× TBE (90 mM Tris/64.6 mM boric acid/2.5 mM EDTA, pH 8.3) at 4°C. To determine the composition of DNA/protein complexes αRAR (Santa Cruz Biotechnology) or αMyb (2-2-79; ref. 13) antibodies were included in the reaction mixtures.
RESULTS
Conditional Induction of a v-Myb-ER Fusion Protein Blocks RA-Induced Morphological Changes in the Macrophage-Like Cell Line 10.4.
The antagonistic function of Myb and RAR in cell differentiation and proliferation raised the question that Myb might directly interfere with RAR function. To address the physiological significance of this possibility, we investigated whether v-Myb is able to interfere with RA-induced biological responses in hematopoietic cells. The chicken macrophage-like cell line 10.4 contains a conditional v-Myb expression system. The expression vector encoding v-Myb fused to the estrogen receptor (ER) ligand binding domain (v-Myb-ER) was introduced into these cells by stable transfection (11). Treatment with E2 causes reversible changes in the differentiation state and gene expression program of these cells (11). To test whether v-Myb-ER can overcome RA-induced biological responses, 10.4 cells were cultured in the presence or absence of RA and E2. In the absence of both ligands, 10.4 cells grew as adherent cells (Fig. 1A). RA treatment markedly altered the morphology of the 10.4 cells. Cells shaped into a elongated form with a flat and adherent morphology characteristic for more differentiated macrophages (Fig. 1B). However, activation of v-Myb-ER fusion proteins by E2 reversed the RA-induced morphological changes (Fig. 1C). The cells rounded up and grew in suspension as described previously by Burk and Klempnauer (11). These results indicate that in the macrophage-like cell line 10.4 v-Myb is able to overcome differentiation-associated morphological changes induced by RA.
Figure 1.

v-Myb-ER fusion proteins block RA-induced morphological changes in 10.4 macrophage-like cells. (A) 10.4 cells were grown with solvent (EtOH) in the absence of RA and E2, (B) treated with 1 μM RA for 18 h, and (C) treated with 1 μM RA plus 2 μM E2 for 18 h. Light field photomicrographs of representative fields are shown. (D) RA antagonizes the induction of tom-1 expression by E2 in cells expressing a v-Myb-ER fusion protein. Cells (10.4) were grown without ligands (lane 1) or treated for 18 h with 1 μM RA (lane 2) 2 μM E2 (lane 3), or incubated simultaneously with both ligands (lane 4). Expression of tom-1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was analyzed by reverse transcription–PCR.
RA Antagonizes the Induction of the v-Myb Target Gene tom-1.
Previous data demonstrated that RA can inhibit the transcriptional activation of Myb responsive reporter genes in transient transfection experiments (9). To investigate a possible mutual transrepression of both transcription factors, we asked whether RA could block transcriptional activation of an endogenous, Myb-responsive gene, like tom-1 (16), in vivo. To investigate the expression levels of tom-1, we performed reverse transcription–PCR analysis from 10.4 cells treated either with RA, E2, or both ligands. Untreated 10.4 cells express low levels of tom-1 mRNA (Fig. 1D, lane 1). Interestingly, RA treatment led to an accumulation of tom-1 mRNA, indicating that tom-1 also might be a RA target gene. (Fig. 1D, lane 2). As reported, activation of the v-Myb-ER fusion protein by E2 strongly induced tom-1 transcription (Fig. 1D, lane 3). Importantly, however, incubation of the 10.4 cells in the presence of both RA and E2 repressed tom-1 mRNA levels significantly (Fig. 1D, lane 4). In summary, these data indicate that v-Myb and RAR repress each other’s transcriptional activity, resulting in the mutual repression of the endogenous tom-1 gene.
c-Myb Inhibits RA-Induced Gene Activation.
To further investigate the effect of Myb on RAR function we cotransfected c-Myb and RAR expression vectors together with (TREp)2-TKLUC reporter plasmids in CV1 cells. As shown in Fig. 2A, increasing amounts of c-Myb expression vectors inhibited the RA-induced reporter activity in a concentration-dependent manner (Fig. 2A, lanes 2 and 3). Transfection of a control vector, containing the c-Myb sequence in antisense orientation, did not affect RA-induced activity of the reporter construct (Fig. 2A, lane 4). The activity of the TKLUC reporter plasmid was not altered (Fig. 2A, lanes 5 and 6). These results demonstrate that c-Myb-mediated repression is not caused by unspecific squelching, but is rather specific for RA-dependent transactivation. To exclude the possibility that c-Myb-mediated repression is cell-type-specific or -dependent on a particular RA response element, we analyzed whether c-Myb is able to repress the RA-induced activity of the two different reporter constructs (TREp)2-TKLUC and βRE2-TKLUC in NIH 3T3 cells. Expression of c-Myb potently inhibited RA-dependent activation of both reporter constructs (Fig. 2B, lanes 2 and 5). The control vector failed to influence the activity of the reporter constructs (Fig. 2B, lanes 3 and 6). Taken together, these results indicate that c-Myb-mediated repression is neither dependent on a particular RA response element nor on a specific cell line. Interestingly, immunoprecipitation assays from cells transiently transfected with the same amounts of either RAR or c-Myb expression plasmids demonstrated that the amount of RAR protein was much higher compared with that of c-Myb (data not shown).
Figure 2.

Myb inhibits RA-mediated transcriptional activation in a dose-dependent manner in different cell lines. (A) (TREp)2-TKLUC reporter plasmids or the control plasmids TKLUC were cotransfected in CV1 cells together with constant amounts of plasmids expressing hRARα and hRXRα and various amounts of expression vectors encoding full-length c-Myb (pCM100) or with control plasmids containing the c-Myb sequence in antisense orientation (pCM101). The cells were either untreated (filled bars) or treated with 10−6 M RA (striped bars). Numbers indicate the amount of cotransfected plasmid DNA in μg. (B) Transcriptional activity of reporter plasmids (TREp)2-TKLUC and βRE2-TKLUC in NIH 3T3 cells, cotransfected with 2.5 μg of expression vectors pCM100 or pCM101. Cells were treated with 10−6 M RA. (C) CV1 cells were transfected with 5 μg of expression vectors encoding hRARα (lane 1) or hRARα and full-length c-Myb. Whole-cell extracts were prepared 48 h after transfection and used in immunoprecipitation experiments with αRAR antibodies. (D) The TA of Myb is dispensable for repression of RA-mediated gene activation. Reporter plasmids βRE2-TKLUC were cotransfected in NIH 3T3 cells with 2.5 μg of various expression vectors coding for c-Myb (pCM100), v-Myb (pVM116), or the indicated mutants of v-Myb (pVM130 and DIN1). As controls, the expression vectors pCM101or pVM111 containing a frame-shift mutation in the v-Myb coding region close to its 5′ end were transfected. Reporter activity is shown as fold activation. Cells were treated with 10−6 M RA. The structure of the c-Myb, v-Myb, and v-Myb mutant proteins are illustrated schematically. L, leucine zipper. (E) Whole-cell extracts from cells transiently transfected with expression vectors coding for v-Myb (pVM116) (lanes 1 and 4) or the v-Myb mutants pVM130 (lanes 2 and 5) and DIN1 (lanes 3 and 6) were analyzed by Western blotting with the αMyb antibodies 5E11 that recognize the DBD of Myb (lanes 1–3) and 2-2-79, recognizing the C terminus of Myb (lanes 4–6).
These results indicate that the RAR protein is either more stable or much better expressed than the Myb protein. Consequently, we cotransfected reduced amounts of RAR expression plasmids or used endogenously expressed RAR. Expression of c-Myb did not alter RAR protein levels in transfected cells, demonstrating that c-Myb-mediated repression is not simply caused by blocking the transcription rate of the CMX-driven RAR expression plasmids (Fig. 2C, lanes 1–2).
The TA of v-Myb Is Dispensable for Repression.
To delineate the domains in Myb that are responsible for the repression of RAR transactivation, we asked whether v-Myb, the oncogenic counterpart of c-Myb, is able to inhibit RA-induced transactivation. We used v-Myb because the viral protein already contains several naturally occurring point mutations in addition to amino- and carboxy-terminal deletions (7, 8). As shown in Fig. 2D, v-Myb blocked RA-dependent activation of the reporter construct βRE2-TKLUC as potently as c-Myb (Fig. 2D, compare lanes 2 and 4). The control vectors did not alter RA-induced activation of the reporter gene (Fig. 2D, lanes 3 and 5). Deletion of the v-Myb DBD resulted in a mutant (pVM130) that failed to repress (Fig. 2D, lane 6). In contrast, a mutant in which the TA was deleted (DIN1) repressed nearly as efficiently as the wild type (Fig. 2D, lane 7). Proper expression of v-Myb and the v-Myb mutants were determined by Western blotting (Fig. 2E). Our results indicate that the TA of v-Myb is not necessary for repression, whereas the DBD is absolutely required.
v-Myb Inhibits RAR DNA Binding.
Possible mechanisms by which Myb might inhibit RAR-mediated transcription are the inhibition of RAR DNA binding or the occupation of the RARE itself, hence displacing RAR from the target DNA. To investigate the significance of these possibilities we performed EMSAs with a radiolabeled oligonucleotide containing the βRARE as a probe and nuclear extracts from COS-7 cells. Endogenous RAR/RXR formed a DNA/protein complex (Fig. 3, lane 1). The complex was specifically up-shifted by an αRAR mAb (Fig. 3, lane 2), whereas an αMyb-specific antibody had no influence on the mobility of the DNA/protein complex (Fig. 3, lane 3). Importantly, when we performed the EMSA with the same amount of protein from COS-7 cells transiently transfected with v-Myb expression plasmids, no specific RAR-containing complex could be detected. As expected, no direct binding of v-Myb to the βRARE was observed (Fig. 3, compare lanes 1 and 4). Taken together, these data demonstrate that the expression of v-Myb inhibits the ability of RAR/RXR to bind target DNA.
Figure 3.
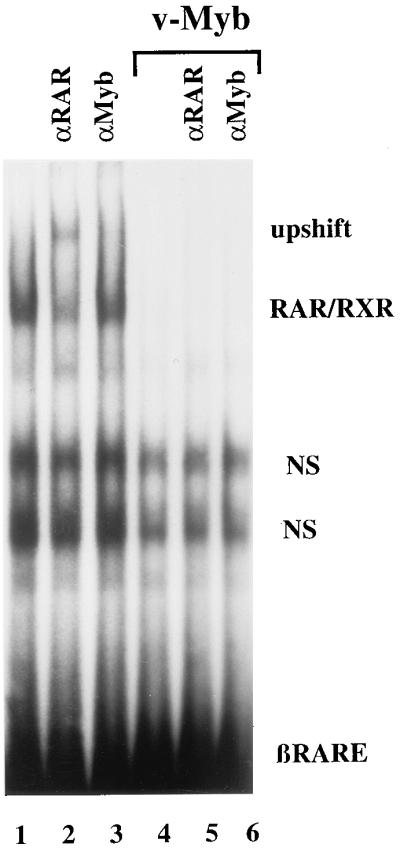
v-Myb inhibits RAR/RXR-DNA complex formation. EMSAs were performed with nuclear extracts from COS-7 cells either untreated (lanes 1–3) or transiently transfected with v-Myb expression vectors (pVM116) (lanes 4–6). A 32P-labeled oligonucleotide containing the βRARE was used as a probe. The composition of DNA/protein complexes were determined by adding the following antibodies to the reaction mixtures: αRAR (lanes 2 and 5) and αMyb (lanes 3 and 6). In addition, two nonspecific complexes (NS) were observed.
RAR and v-Myb Physically Interact in Vitro.
One possible mechanism by which v-Myb and RAR might repress each other’s function is through direct protein–protein interactions. To test this hypothesis, we performed GST-pulldown experiments with GST-RAR and GST-RXR fusion proteins and in vitro-translated, [35S]methionine-labeled v-Myb. GST-RAR interacted very strongly with v-Myb protein (Fig. 4A, lane 2) and c-Myb (data not shown). This interaction was not dependent on RA (data not shown). In contrast, equal amounts of GST-RXR failed to interact with v-Myb (Fig. 4A, lane 3). Likewise, GST protein alone exhibited no specific binding (Fig. 4A, lane 4). Performing the pulldown experiments with recombinant GST-v-Myb proteins further supported our results. In vitro-translated [35S]methionine-labeled RAR but not RXR was retained by immobilized GST-v-Myb (Fig. 4A, lanes 6 and 9). These results suggest that specific interactions of v-Myb with RAR but not with RXR might be responsible for inhibition of RA-dependent transactivation.
Figure 4.

v-Myb and RAR physically interact in vitro. (A) v-Myb physically interacts with RAR but not with RXR. The ability of RAR and RXR proteins to interact with v-Myb was evaluated by GST-pulldown assays. RAR and RXR were expressed as GST-fusion proteins, immobilized to glutathione-linked Sepharose beads and tested for interaction with in vitro-translated, [35S]methionine-labeled v-Myb. In the reciprocal experiment GST-v-Myb was tested with in vitro-translated, [35S]methionine-labeled RAR and RXR. The same amounts of either GST or GST fusion proteins were linked to glutathione Sepharose beads. The input control in lanes 1, 5, and 8 reflects 10% of the total amount of [35S]methionine-labeled proteins used for the pulldown experiments. (B) The DBD of RAR is necessary and sufficient for interaction with v-Myb. To localize the domain within RAR necessary for interaction with v-Myb either full-length RAR or various mutants thereof were expressed as GST-fusion proteins and tested for interaction with in vitro-translated, [35S]methionine-labeled v-Myb. In the reciprocal set of experiments GST-v-Myb was tested with in vitro-translated, [35S]methionine-labeled RAR or mutants thereof. The input control reflects 10% of the total amount of [35S]methionine-labeled proteins used for the pulldown assay. The full-length hRAR protein is composed of the N-terminal (A/B) domain, the DBD, and the C-terminal ligand binding domain (Ligand). (C) Several domains of v-Myb are involved in interaction with RAR. v-Myb and various mutants of v-Myb were either expressed as GST-fusion proteins or in vitro translated in the presence of [35S]methionine and tested in GST-pulldown experiments. v-Myb and v-Myb mutant proteins are illustrated schematically. L, leucine zipper.
The DBD of RAR Is Both Necessary and Sufficient for Interaction with v-Myb.
Next, we asked which region of RAR is responsible for the interaction with v-Myb. A mutant RAR in which the DBD is deleted (GST-RAR ΔDBD) was unable to interact with in vitro-translated v-Myb (Fig. 4B, lane 3), whereas the isolated RAR DBD (GST-RAR DBD) interacted even better than full-length RAR (Fig. 4B, compare lanes 2 and 4). The mutant GST-RAR Nterm, which contains amino acids 1–87 of RAR, or GST-RAR Cterm, which contains amino acids 174–462, failed to interact with v-Myb (Fig. 4B, lanes 5 and 6). Experiments performed with immobilized GST-v-Myb showed that the in vitro-translated RAR DBD bound very strongly to v-Myb (Fig. 4B, lane 15). As expected, the RAR mutant in which the DBD is deleted failed to bind to GST-v-Myb (Fig. 4B, lane 12). These results further support our conclusion that the RAR DBD is necessary and sufficient to mediate direct protein–protein interactions with v-Myb.
Several Domains of v-Myb Are Involved in the Interaction with RAR.
To determine which domains in v-Myb are necessary for interaction with RAR we assayed various v-Myb deletion mutants in pulldown experiments. As shown above in vitro-translated RAR bound to GST-v-Myb but not to GST (Fig. 4C, compare lanes 2 and 6). A mutant in which the TA of v-Myb is deleted (GST-v-MybΔTA) bound very efficiently to RAR (Fig. 4C, lane 3). In addition, the DBD deletion mutant GST-v-MybΔN and a mutant containing the DBD alone (GST-v-MybΔC) also interacted strongly with in vitro-translated RAR (Fig. 4C, lanes 4 and 5). These results suggest that at least two domains of v-Myb can interact with RAR. To confirm these results we performed the reciprocal set of experiments by using immobilized GST-RAR. Accordingly, GST-RAR retained labeled full-length v-Myb, v-MybΔTA, the DBD deletion mutant v-MybΔN, and the mutant containing the DBD alone (v-MybΔC) (Fig. 4C, lanes 8, 11, 14, and 17).
v-Myb Interacts with RAR in Vivo.
To determine whether the interaction between RAR and v-Myb also occurs in intact cells, we performed a series of mammalian two-hybrid assays. NIH 3T3 cells were transiently transfected with a vector encoding v-Myb fused to the Gal4 DBD (Gal4-v-myb) or in combination with an expression vector encoding RAR tagged with the activation domain of VP16 (RAR-VP16). As shown in Fig. 5, Gal4-v-myb slightly induced the activity of the Galp3TKLUC reporter gene (Fig. 5, lane 2). Cotransfection of RAR-VP16 and Gal4-v-myb expression plasmids significantly increased the activity of the reporter gene (Fig. 5, lane 3). This increase was moderately strengthened by the addition of RA. Transfection of RAR-VP16 in the presence of a control vector encoding the Gal4 DBD only slightly affected the activity of the reporter construct (Fig. 5, lane 4). Direct interaction between v-Myb and RAR was confirmed further by coimmunoprecipitation experiments (data not shown). Taken together, these results indicate that v-Myb is able to interact with the RAR in vivo.
Figure 5.

v-Myb interacts with RAR in vivo. Galp3TKLUC reporter plasmids were cotransfected with 250 ng of plasmids expressing Gal4-v-myb or Gal4 alone or in combination with 50 ng of expression vectors encoding RAR-VP16 in NIH 3T3 cells. The cells were either untreated (filled bars) or treated with 10−6 M RA (striped bars).
DISCUSSION
Several lines of evidence indicate that Myb and RAR can function as mutual antagonists able to prevent or to promote normal hematopoietic differentiation, respectively. This evidence led us to investigate whether a reciprocal repression mechanism, such as the mutual repression of BZLF1 and RAR (12, 17), also may function between Myb and RAR. In this study, we demonstrate that Myb is a potent inhibitor of RA-induced, differentiation-associated morphological changes of the well-studied chicken macrophage-like cell line 10.4. In addition, we demonstrate that functional interaction between endogenous RAR and v-Myb inhibits the induction of the endogenous Myb target gene tom-1.
Furthermore, we provide evidence that Myb is a potent inhibitor of RA-induced gene activity. Myb-mediated repression is not dependent on a particular RA response element or a specific cell line. In contrast to the oncogenic Myb-Ets fusion protein (10), the cellular form of Myb (c-Myb) as well as the truncated viral form (v-Myb) seem to be sufficient for inhibition of RA-induced gene activation. Thus, Myb and RAR represent a further example of cross talk between a proliferation-inducing protooncogene and a member of the nuclear receptor superfamily (17). To elucidate the mechanism by which Myb interferes with RAR activity, we tested the repression function of several different v-Myb deletion mutants. Our studies demonstrate that the DBD of Myb is necessary for repression whereas the TA is dispensable.
Recent data show that CREB-binding protein (CBP) can function as a coactivator for both c-Myb and RAR (18). These data could imply that the inhibition of RAR activity by Myb might be the result of competition for limiting amounts of CBP. However, the fact that the TA of Myb, which interacts with CBP, is dispensable for repression strongly argues against a possible involvement of CBP in Myb-mediated repression. Instead, our results support a mechanism for repression in which RAR and Myb physically associate and sequester each other into an inactive complex. Accordingly, we were able to demonstrate interaction of Myb and RAR in vitro and in vivo. GST-pulldown experiments reveal that Myb interacts with RAR, but not with RXR, indicating that the interaction with RAR might be responsible for inhibition of RA-dependent transactivation. In contrast to the results shown here, the well-characterized cross talk between the viral transactivator BZLF1 and the retinoid receptors involves both RAR and RXR (12).
Further pulldown experiments demonstrate that the RAR DBD is necessary and sufficient for the interaction with v-Myb, reinforcing the idea that the DBDs of nuclear receptors are involved not only in protein–DNA interactions, but also in protein–protein interactions with transcription factors that do not belong to the nuclear receptor superfamily.
Our data show that at least two domains, the DBD and the C-terminal part of v-Myb, are involved in protein–protein interactions with RAR. In this context it is interesting to note that both domains of Myb are already known to be involved in protein–protein interactions (19, 20). In contrast, transfection experiments show that the DBD of Myb is necessary for repression of RA-dependent transactivation.
This finding indicates that although two domains of Myb are able to physically interact with RAR, the DBD is absolutely necessary for the observed repression in vivo.
In summary, we demonstrate that Myb and RAR interact both in vitro and in vivo. This interaction results in the repression of RAR activity. Our data suggest that this repression mechanism might account for some of the biological effects of Myb and RAR during hematopoiesis.
Acknowledgments
We thank Drs. Karl-Heinz Klempnauer, Oliver Burk, Ronald M. Evans, and Kazuhiko Umesono for materials. We also thank the members of the Schüle laboratory, Sigrun Mink, Heike Pahl, Darren Daniels, and Bernd Groner for helpful discussions and critical reading of the manuscript, and Corina Schüle for providing the artwork and administrative assistance. This work was supported by a grant of the Deutsche Forschungsgemeinschaft to R.S. (Schu 688/2-2).
ABBREVIATIONS
- RA
retinoic acid
- RAR
retinoic acid receptor
- RXR
retinoid X receptor
- TR
thyroid hormone receptor
- T3
thyroid hormone
- E2
β-estradiol
- LUC
luciferase
- TK
thymidine kinase
- GST
glutathione S-transferase
- DBD
DNA-binding domain
- TA
transactivation domain
- EMSA
electrophoretic mobility-shift assay
References
- 1.Gudas L J, Sporn M B, Roberts A B. In: The Retinoids. Sporn M B, Roberts A B, Goodman D S, editors. New York: Raven; 1994. pp. 443–520. [Google Scholar]
- 2.Breitman T R, Selonick T E, Collins S J. Proc Natl Acad Sci USA. 1980;77:2936–2940. doi: 10.1073/pnas.77.5.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robertson K A, Emami B, Collins S J. Blood. 1992;80:1885–1889. [PubMed] [Google Scholar]
- 4.Tsai S, Collins S J. Proc Natl Acad Sci USA. 1993;90:7153–7157. doi: 10.1073/pnas.90.15.7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang M E, Ye Y-C, Chen S-R, Chai J-R, Lu J-X, Zhoa L, Gu H T, Wang Z-Y. Blood. 1988;76:567–572. [PubMed] [Google Scholar]
- 6.Shen-Ong G L. Biochim Biophys Acta. 1990;1032:39–52. doi: 10.1016/0304-419x(90)90011-o. [DOI] [PubMed] [Google Scholar]
- 7.Graf T. Curr Opin Genet Dev. 1992;2:249–255. doi: 10.1016/s0959-437x(05)80281-3. [DOI] [PubMed] [Google Scholar]
- 8.Tompson M A, Ramsay R G. BioEssays. 1995;17:341–350. doi: 10.1002/bies.950170410. [DOI] [PubMed] [Google Scholar]
- 9.Smarda J, Sugarman J, Glass C, Lipsick J. Mol Cell Biol. 1995;15:2474–2481. doi: 10.1128/mcb.15.5.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rascle A, Ferrand N, Gandrillon O, Samarut J. Mol Cell Biol. 1996;16:6338–6351. doi: 10.1128/mcb.16.11.6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burk O, Klempnauer K-H. EMBO J. 1991;10:3713–3719. doi: 10.1002/j.1460-2075.1991.tb04939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pfitzner E, Becker P, Rolke A, Schüle R. Proc Natl Acad Sci USA. 1995;92:12265–12269. doi: 10.1073/pnas.92.26.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foos G, Grimm S, Klempnauer K-H. EMBO J. 1992;11:4619–4629. doi: 10.1002/j.1460-2075.1992.tb05564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foos G, Natour S, Klempnauer K-H. Oncogene. 1993;8:1775–1782. [PubMed] [Google Scholar]
- 15.Klempnauer K-H, Arnold H, Biedenkapp H. Genes Dev. 1989;3:1582–1589. doi: 10.1101/gad.3.10.1582. [DOI] [PubMed] [Google Scholar]
- 16.Burk O, Worpenberg S, Haenig B, Klempnauer K-H. EMBO J. 1997;16:1371–1380. doi: 10.1093/emboj/16.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schüle R, Evans R M. Trends Genet. 1991;56:119–127. [Google Scholar]
- 18.Janknecht R, Hunter T. Nature (London) 1996;383:22–23. doi: 10.1038/383022a0. [DOI] [PubMed] [Google Scholar]
- 19.Mink S, Kerber U, Klempnauer K-H. Mol Cell Biol. 1996;16:1316–1325. doi: 10.1128/mcb.16.4.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Favier D, Gonda T J. Oncogene. 1994;9:305–311. [PubMed] [Google Scholar]