

Abstract
The double Holliday junction (dHJ) is a central intermediate to homologous recombination, but biochemical analysis of the metabolism of this structure has been hindered by the lack of a substrate that adequately replicates the endogenous structure. We have synthesized a novel dHJ substrate that consists of two small, double stranded DNA circles conjoined by two Holliday junctions (HJs). Its biochemical synthesis is based on the production of two pairs of single stranded circles from phagemids, followed by their sequential annealing with reverse gyrase. The sequence between the two HJs is identical on both strands, allowing the HJs to migrate without the generation of unpaired regions of DNA, whereas the distance between the HJs is on the order of gene conversion tracts thus far measured in Drosophila and mouse model systems. The structure of this substrate also provides similar topological constraint as would occur in an endogenous dHJ. Digestion of the dHJ substrate by T7 endonuclease I resolves the substrate into crossover and non-crossover products, as predicted by the Szostak model of double strand break repair. This substrate will greatly facilitate the examination of the mechanism of resolution of double Holliday junctions.
Homologous recombination is an important pathway for the repair of double stranded DNA breaks, the restart of stalled replication forks, and the generation of genetic diversity during meiosis (1). One of the predicted intermediates of homologous recombination is the reciprocal exchange of single strands of DNA between two homologous sequences across the region of the original break (shown diagrammatically in the column of Fig. 1), a structure termed a double Holliday junction (dHJ)2 (2, 3). It was postulated that the digestion of this structure by endonucleases could give rise to gene conversion events either with (crossovers, CO) or without (non-crossovers, NCO) the exchange of sequences flanking the original break site (see Fig. 1, left branch).
FIGURE 1. A modified version of the Szostak model of double strand break repair.
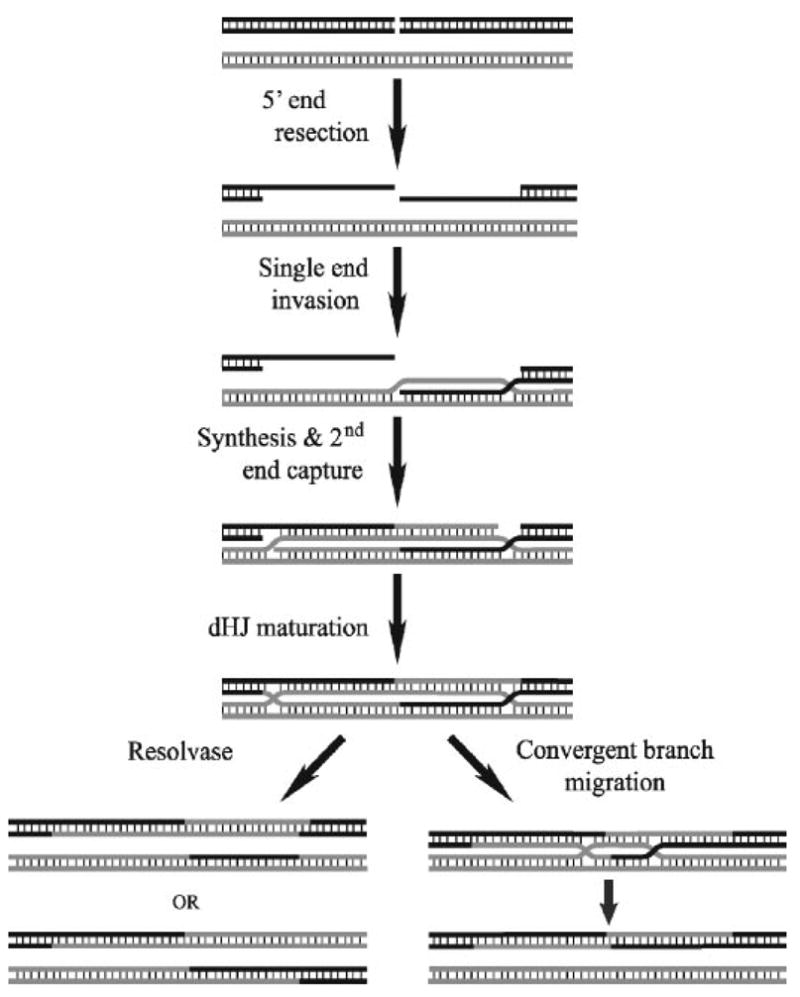
After a double strand break occurs, the 5′-ends of the break are resected to generate 3′-ssDNA. One of these single strands invades homologous sequence to form a D-loop. DNA synthesis extends this D-loop, and the second ssDNA is captured and also extended. This structure is then matured into a dHJ that may be resolved by two different pathways. In the left pathway, a structure-specific endonuclease (resolvase) digests the structure with the orientation of the cuts relative to one another determining if the product of the reaction will be a crossover (bottom) or a non-crossover (top). It has been proposed that the dHJ can be resolved by another pathway, represented on the right, in which the two HJs are convergently branch-migrated until all the linkages between the two strands are removed. This pathway would result exclusively in non-crossover product formation.
Since that time, many of the components of the model have been discovered. In yeast, it has been demonstrated that exonucleases resect the 5′-strands of the DNA at a double stranded break to reveal ssDNA with a 3′-end which can invade a homolog or sister chromatid to initiate DNA synthesis (4–6). Work by Newlon, Kleckner, and their respective co-workers (7–9) demonstrated that double Holliday junctions exist in vivo, and subsequently Hunter and Kleckner (10) also demonstrated the existence of a stable single-end invasion intermediate that precedes dHJ formation. Numerous endonucleases (or their activities) have been discovered that specifically bind to and cleave synthetic HJs from organisms ranging from bacteria and viruses to yeast and mammalian systems (11–13). When studied in vitro, these endonucleases can cleave a dHJ substrate to yield both CO and NCO products (this work). However, in both yeast and mouse meiosis the generation of crossover and non-crossover products has been shown to be distinctly different processes, not the differential digestion of a common intermediate (14, 15). One of the proposed mechanisms for the generation of NCO products is the collapse of smaller dHJs by a topoisomerase partnered with a helicase (see Fig. 1, right branch) (16, 17).
Currently, the only dHJ-containing substrates that are available for the biochemical study of these processes are constructed from oligonucleotides (18, 19). Although these substrates certainly replicate the dHJ structure, there are many other characteristics of an endogenous dHJ that these substrates cannot recapitulate. Because of technical constraints in the construction of the oligonucleotide substrates, the structures used for these studies are small, with a very short distance between the two HJs, which are necessarily immobile, unlike an endogenous dHJ. In addition to this, these oligonucleotide substrates cannot recapitulate all of the topological constraints inherent to the endogenous structures.
We have synthesized a substrate that consists of two small, double stranded DNA circles conjoined by two Holliday junctions. The sequence between the two HJs is identical on both strands, allowing the HJs to migrate without the generation of unpaired, single stranded regions of DNA. This substrate is also topologically constrained, as migration of a HJ will generate negative writhe behind the HJ and positive writhe in front. Because these writhes will be generated in two topologically isolated regions, this recapitulates the topological constraints that must be intrinsic to endogenous dHJs. The distance between the two HJs is 165 bp, a distance that is comparable to gene conversion tracts that have been measured thus far in Drosophila (~400 bp) and mouse (<533 bp) models (15, 20, 21). We report here the synthesis of this substrate and show evidence supporting its proposed structure.
EXPERIMENTAL PROCEDURES
The Purification of the Enzymes
Reverse gyrase (22) and Cre recombinase (23) were purified from overproducing strains as described previously.
The Synthesis of the DNA Substrates and Marker Molecules
The plasmids used to synthesize the DHJS were created by ligating four small, oligonucleotide based dsDNA fragments into pBluescript SK+ and pBluescript SK−. These inserts contained the two loxP sites along with the balance of the homologous region of the DHJS (supplemental Fig. 1). Sequence A or B was then cloned into the center of the homologous region to create four plasmids: pDHJS AN+, pDHJS AN−, pDHJS BN+, and pDHJS BN−. Sequence A was created by amplification of a 300-bp region of the Top3μα gene centered around the BamHI restriction site, and sequence B was created by amplification of a 251-bp region of pBR322 centered around the BamHI restriction site in the tetracycline resistance gene. Sequences A and B are not homologous to each other.
These plasmids were then transformed into Escherichia coli strain XL2 MRF′ and phagemids were produced as described (24). Single stranded DNA was then purified as described (25) with the following modification. After the phagemids were precipitated and pelleted from the culture supernatant, the pellets were resuspended in 50 mM MOPS (pH 7.0), 10 mM EDTA, and SDS was added to 0.5%, and the solution was heated to 50 °C for 10 min to denature the protein coat of the phagemid. KCl was added to 500 mM, and the solution was cooled on ice for 15 min to then precipitate SDS and protein. The precipitate was removed by centrifugation at 20,000 × g for 20 min, and the cleared solution containing the DNA was loaded onto a Qiagen DEAE column, and the column was washed and developed according to the published protocol.
Plus and minus strands were annealed and linked together using Archaeoglobus fulgidus reverse gyrase as described (24). AN+ ssDNA was linked with BN−, and BN+ ssDNA was linked with AN− to create the large heterodimers. After the reactions had cooled to 37 °C, XhoI (New England Biolabs) was added directly to the reactions at 1.5 units/μg of DNA, and the digestions were incubated at 37 °C for 1 h. The digested DNAs were then diluted out to 10 μg/ml DNA into Cre reaction buffer (50 mM Tris, pH 7.5, 33 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol) and incubated with an empirically determined amount of Cre recombinase at 37 °C for 30 min. The Cre was then heat-inactivated, and the reactions were cleaned up and concentrated over Qiagen DEAE columns. The small heterodimers were then gel-purified from the other Cre reaction products, electroeluted, butanol-extracted to remove the ethidium bromide, and concentrated using a Qiagen DEAE column. After quantitation, the AN+/BN− and BN+/AN− small heterodimers were annealed and linked with reverse gyrase as described earlier.
The marker molecules were prepared by digesting plasmid DNA, pDHJS AN+ and pDHJS BN+, with XhoI, reacted with Cre recombinase, and the small, circular DNAs were gel-purified and processed as the DHJS had been.
The DHJS was cross-linked by incubating the substrate (10 ng/μl) with psoralen (1 ng/μl) in 10 mM Tris, pH 7.9, 0.1 mM EDTA, 10% ethanol and exposing the reactions to long wavelength ultraviolet light from above for 30 min. The DHJS was then butanol-extracted to remove unreacted psoralen and ethanol-precipitated. Mock reactions that excluded psoralen were carried out in parallel and served as the “−psoralen” controls in the experiments described here.
DHJS Resolution Reactions
For resolution reactions T7 endonuclease I (New England Biolabs) was incubated with the DHJS in the recommended reaction buffer at 0.1 unit/100 ng DHJS in a 10-μl total reaction volume at 37 °C for 1 h. Reactions were then stopped with 1% SDS, 20 mM EDTA, 100 μg/ml Proteinase K (final concentrations) and incubated at 50 °C for 30 min. The reactions were then phenol/chloroform-extracted, and the DNA was ethanol-precipitated and dissolved in 10 mM Tris, pH 7.9, 0.1 mM EDTA. The DNA was either loaded directly on a gel or was digested by the restriction enzymes indicated before loading on a gel.
For the complete digestion of the DHJS, 1 unit of T7 endonuclease I/100 ng of DHJS was used, and the reactions were incubated at 25 °C for 2 h. The reactions were stopped with 1% SDS, 20 mM EDTA before electrophoresis.
RESULTS AND DISCUSSION
Design and Synthesis of the DHJS
The DHJS is created from sequences encoded on four phagemid vectors. Two of these vectors are illustrated in Fig. 2A, step I. The vectors are derivatives of pBluescript SK+ and pBluescript SK−, in which two tandem loxP sites are separated by sequence that includes either sequence A or B. Sequences A and B were chosen to be ~50% of base composition in GC, with no unusual features predicted. Each contain a BamHI site near the center of the sequence that will be utilized later in this study. The resulting phagemids are pDHJS AN+, pDHJS AN−, pDHJS BN+, and pDHJS BN−. Although sequences in A and B are not homologous, sequences outside of A and B are homologous in these vectors.
FIGURE 2. Synthesis of a topologically constrained double Holliday Junction substrate.

A, schematic of synthesis. The substrate is derived from four phagemid vectors, containing tandem loxP sites and differing with either sequence A or B, and with the f1 origin in both the plus and minus orientations for each. For simplicity, reactions for only two of the plasmids are shown (I). Using the helper phage M13KO7, ssDNA circles are expressed, purified (II), mixed, and linked using reverse gyrase to yield a large heterodimer intermediate (III). The large heterodimer is linearized by digestion with XhoI (IV), and the small heterodimer was excised and circularized utilizing Cre recombinase and the loxP sites flanking the A/B bubble (V ). The small heterodimer is gel-purified away from the other reaction products of the Cre reaction and is mixed with the complementary small heterodimer created from the other two plasmids in a parallel reaction (VI). The small heterodimers are linked together with reverse gyrase to yield the double Holliday junction substrate (VII). This illustration is not drawn to scale. B, agarose gel electrophoresis of intermediates of DHJS synthesis. The first lane contains a size marker, with the size of the bands denoted to the left of the gel in base pairs. The roman numerals below the lanes correspond to the stages of synthesis in A. C, agarose gel electrophoresis of the marker molecules. Lane 1 contains the DHJS; lane 2, Marker A; lane 3, Marker B. Diagrams of the structure of these dsDNA molecules are shown to the left and right of the gel.
Single stranded DNA circles were expressed from each of these phagemids by infecting E. coli harboring the phagemid with the helper phage, M13K07. The ssDNA was purified from the supernatants of these cultures using a modified protocol from Brégeon and Doetsch (25) (Fig. 2A, step II). In parallel reactions, BN+ ssDNA was annealed and linked with AN− (Fig. 2A, step III), and AN+ ssDNA was annealed and linked with BN− using reverse gyrase from the hyperthermophile A. fulgidus. The products of these reactions are relaxed, heterodimeric DNA circles containing two denatured DNA bubbles, termed the large heterodimers (Fig. 2A, step III). The first bubble is the result of placing sequence A opposite the non-homologous sequence B and will be important for the formation of the DHJS. The second bubble arises from the nature of the f1 origin region; the sequence on the opposing strands is identical, and not complementary, in this region. To liberate the DHJS sequences from the plasmid sequences and the f1 bubble, the large heterodimers are first linearized with XhoI (Fig. 2A, step IV) and then incubated with Cre recombinase to excise small DNA circles, termed the small heterodimers, containing 165bp of duplexed DNA and the A/B bubble (Fig. 2A, step V). The small heterodimers are gel-purified away from the other products of the Cre reaction (Fig. 2A, step VI). The complementary sequences of the purified small heterodimers, BN+/AN− and AN+/BN−, are then annealed and linked with reverse gyrase to create the DHJS (Fig. 2A, step VII). When analyzed, the intermediates of the DHJS synthesis show the expected electrophoretic mobility (Fig. 2B).
To confirm that the substrate possesses the predicted structure, we characterized the substrate using restriction and structure-specific endonucleases. To aid in the correct identification of the products of these reactions, separate A and B circles were also prepared. This was accomplished by incubating the plasmids (Fig. 2A, step I) with XhoI and Cre recombinase, and then gel-purifying the circles away from the other reaction products (Fig. 2C). These small circles are referred to as markers A (465 bp) and B (416 bp).
Characterization of the DHJS by Restriction Digestion
We first probed the structure of the DHJS using restriction endonuclease sites engineered into the molecule. Both sequences A and B were chosen to contain an internal BamHI site (Fig. 3A). As predicted, digestion of the substrate by BamHI results in a single band with reduced electrophoretic mobility relative to the undigested substrate (Fig. 3B, lane 2). As shown in Fig. 3A, digestion of the DHJS by both BamHI and NheI would result in two DNA molecules each containing a single HJ. If left unconstrained, these HJs would be free to migrate off the ends of the DNA, resulting in four linear DNAs of various sizes. Two of these linear DNAs are very similar in length, and would therefore co-migrate during electrophoresis. The double digest of the DHJS resulted in three bands, which co-migrate with the bands generated from the double digest of markers A and B and are of the predicted sizes (Fig. 3B, compare lane 3 with lanes 4–6). This analysis has been carried out using several other restriction enzymes (Fig. 4 and data not shown), and thus far all of the results have been consistent with the predicted structure of the substrate.
FIGURE 3. Double digest of the DHJS is consistent with the predicted structure.

A, digestion of the DHJS with BamHI and NheI should separate the two HJs, allowing them to spontaneously migrate. The migration of the HJs off the ends of the DNA would result in four linear DNAs, 266, 265, 200, and 150 bp in size. B, the double digest of the DHJS yields the predicted fragments. Digestion of the DHJS by BamHI (lane 2) produces a single band with reduced electrophoretic mobility compared with the undigested DHJS (lane 1). DHJS digested with BamHI and NheI (lane 3) produces four bands, which co-migrate with the double digest of marker A (lane 4) and marker B (lane 5). Either the two circular DNAs linked by dHJ or its linearized derivatives are topologically constrained in between HJs thus allowing topoisomer formation, which may account for the appearance of species with more than a single electrophoretic mobility in lanes 1 and 2. A DNA size marker was run in lane 6, with the size of the bands denoted to the right of the gel in base pairs.
FIGURE 4. Digestion of psoralen cross-linked DHJS is consistent with the predicted structure.

A, the predicted cross-linking sites of psoralen within the homologous region. Psoralen preferentially binds and cross-links at TA sequences, with the repeated TA sequence being particularly good sites. TA dinucleotides are indicated with red arrows, with double arrows showing the location of the TATA sequences in the loxP site. B, the predicted outcomes of BamHI and XmnI digestion of the cross-linked DHJS. Psoralen cross-links are represented by red dots in the center of the homologous region. The XmnI, BamHI double digest should produce 236-, 234-, 231-, and 180-bp bands if the HJs are left unconstrained. However, if the DHJS is cross-linked in the loxP sequence, then this double digest will show a diminished ~235-bp heterogeneous band with the concomitant appearance of a slower migrating species. C, the predicted outcomes of BamHI and EcoRI digestion of the cross-linked DHJS. The EcoRI, BamHI double digest should produce 266-, 212-, 204-, and 199-bp bands if the HJs are left unconstrained. However, if the DHJS is cross-linked in the loxP sequence, then this double digest will show a nearly eliminated 266-bp band and diminished ~200-bp heterogeneous band, with the appearance of a slower migrating species. D, double digestion of the cross-linked DHJS shows the predicted pattern. Lane 1 contains a DNA size marker, with the sizes of the relevant bands denoted to the left of the gel in base pairs. Digestion of the DHJS by BamHI is not effected by the cross-linking (compare lane 3 with lane 2). The XmnI, BamHI double digest of the cross-linked DHJS shows a shifted banding pattern consistent with cross-linking within the loxP sequence (compare lane 4 with lane 5). The EcoRI, BamHI double digest of the cross-linked DHJS also shows a shifted banding pattern consistent with cross-linking within the loxP sequence (compare lane 6 with lane 7).
The previous analysis was performed without constraining the migration of the HJs predicted to be in the DHJS. We repeated this analysis while constraining the migration of the HJs with interstrand cross-links that were introduced into the DHJS before digestion with restriction enzymes. The DHJS was cross-linked with psoralen, a chemical that creates interstrand cross-links primarily at TA sequences, once the psoralen-DNA complex is irradiated with long wavelength ultraviolet light (26, 27). This prevents the migration of a HJ beyond the cross-link, and has commonly been used to maintain endogenous HJs in previous studies (7–9). The homologous region of the DHJS contains 12 TA dinucleotides, with 4 of them occurring in pairs within the loxP sequence at the center of the homologous region (Fig. 4A). Repeated TA sequences have been shown to be particularly efficient cross-linking sites (27). Based on this, we predicted that the bulk of the cross-links created within the homologous sequence would be located in the loxP sequence, and chose restriction sites bordering the loxP sequence for the analysis for the cross-linked DHJS.
Psoralen cross-linking of the DHJS, and the subsequent inhibition of HJ migration, will change the electrophoretic pattern of the double digest if the substrate possesses the predicted structure. Without psoralen treatment, digestion of the DHJS with BamHI and XmnI, followed by migration of HJs across the homologous regions, will produce four linear fragments. Three of the four linear molecules are of similar length and co-migrate as a single band ~235 bp in size, with the fourth fragment at 180 bp (Fig. 4B). However, if the substrate is cross-linked within the loxP sequence before the double digest, then two of the three molecules that make up this band would contain a trapped HJ and would be electrophoretically retarded on the gel, diminishing the intensity of the 235-bp band. This is precisely the pattern that is observed when the cross-linked DHJS is digested with BamHI and XmnI (Fig. 4D, compare lane 5 with lane 4). The intensity of the 235-bp band present on the cross-linked DHJS lane is diminished relative to the control lane, with the appearance of a new band with retarded migration relative to the linear fragments.
The cross-linked DHJS was also analyzed in a similar fashion by digestion with BamHI and EcoRI. Without cross-linking, the double digest yields four linear fragments after branch migration. Three of the fragments ~200 bp in size are expected to co-migrate during electrophoresis, with the fourth 266-bp fragment migrating as a resolved band (Fig. 4C). With psoralen treatment prior to double digest, the HJ trapped by cross-linking within the loxP sequence should involve the unique, resolved band as well as one of the three molecules contained within the heterogeneous band. When the psoralen cross-linked DHJS is digested by BamHI and EcoRI, the homogeneous 266-bp band almost completely disappears, and the intensity of the 200-bp heterogeneous band diminishes relative to the control digest with the appearance of a new band with retarded migration relative to the linear fragments (Fig. 4D, compare lane 7 with lane 6). Taken together, the data from the two double digests is consistent with the predicted structure of the DHJS, as well as the predicted location of the psoralen cross-links.
Resolution of the DHJS by T7 Endonuclease I
We next tested the structure of the substrate using the HJ resolvase, T7 endonuclease I, to specifically cleave at the HJs. This enzyme has been well characterized and robustly cleaves HJs, DNA mismatches, and to a lesser degree nicked DNA (28). Fig. 5A shows the predicted products of the digestion of the DHJS with T7 endo I. As in the model of Szostak et al. (3), the dHJ can be cleaved by the resolvase in two different ways, parallel (two DNA strands are cut twice; Fig. 5A, V+V and H+H) or perpendicular (all four DNA strands are cut once; Fig. 5A, V+H and H+V). If the substrate is digested in a perpendicular fashion, analogous to gene conversion with exchange of flanking sequences, then the resulting molecule would be a large circle containing two BamHI sites, one from sequence A and another from sequence B (Fig. 5A, molecule I). Although this large circle contains the same number of base pairs as the DHJS, it would be expected to migrate slower during electrophoresis because of its more extended structure. If the substrate is resolved by the parallel pathway, analogous to gene conversion without exchange of flanking sequences, then the products of the reaction would be two small circles, A and B (Fig. 5A, molecules II and III).
FIGURE 5. Digestion of the DHJS by T7 endonuclease I.

A, digestion of the DHJS substrate by T7 endonuclease I can proceed by two different pathways. In this diagram H represents a horizontal cut, whereas V represents a vertical cut of a HJ. If the HJs of a given molecule are cut in a perpendicular fashion, as shown in the right pathway, then the products of the reaction are predicted to be two smaller DNA circles, 465 and 416 bp. If both HJs of a given molecule are digested in a parallel fashion, as shown in the left pathway, then the expected product of the reaction would be a large DNA circle 881 bp in size. This large DNA circle would be expected to migrate slower than the DHJS, whereas the small circles would be expected to migrate faster. Digestion of the 881-bp circle by BamHI would result in two linear bands, 470 and 411 bp in size, which would be expected to co-migrate with the linearized smaller circles 466 and 416 bp in length. B, T7 endonuclease I digests the DHJS in the predicted fashion. A DNA size marker was run in lanes 5 and 10, with the size of the bands denoted to the right of the gel in base pairs. Digestion of the DHJS by T7 endo (lane 2) produces both faster and slower migrating species when compared with the DHJS (lane 1). Two of these bands co-migrate with marker molecules for the A circles (lane 3) and B circles (lane 4). Digestion of the products of the T7 endo reaction with BamHI convert all DNA species into two linear bands (lane 7) which co-migrate with linearized marker A (lane 8) and marker B (lane 9). C, T7 endonuclease I can convert a HJ to a double strand break with an excess of enzyme. T7 endo I digests a HJ efficiently to yield two dsDNA strands, each containing a nick at the site of the HJ. T7 endo I has a less efficient, secondary activity of cleaving a nicked DNA strand at the site of the nick, thus converting a HJ to a double stranded break. D, an excess of T7 endonuclease I should convert the DHJS to four linear molecules. If the HJs are near the refractory boundaries, then the molecules should be 300, 251, and 165 bp in size. E, the HJs are located near the ends of the homologous region. Complete digestion of the DHJS by T7 endo I produces five prominent linear bands (lane 1). The lower three bands are calculated to be 170, 253, and 302 bp in size, which correlates well with the predicted location of the HJs. See the text for a discussion of the upper two bands. Lane 2 contains a DNA size marker, with the size of the bands denoted to the right of the gel in base pairs. This is a 15% polyacrylamide gel stained with SYBR gold.
Upon incubation of the DHJS with T7 endo I, products are generated that are consistent with these predictions. T7 endo I creates a pair of faster migrating bands, relative to the unreacted substrate, which migrate with similar mobilities as the undigested markers A and B, as well as a slower migrating band, which is consistent with the large dimer circle (Fig. 5B, lanes 1–4). There are other bands present in Fig. 5B, lane 2 that are consistent with the linearization of the resolved circles, which is likely a result of secondary cleavage of the DNA at a nick by T7 endo I (Fig. 5C). The identities of the electrophoretically retarded species generated by T7 endo I can be confirmed by an additional digestion by the restriction enzyme BamHI. When the T7 endo I products are digested by BamHI, they collapse down to two bands which co-migrate with the digested markers A and B (Fig. 5B, lanes 6–9), which is consistent with the model presented in Fig. 5A.
We decided to exploit the ability of T7 endo I to cleave opposite the nick left behind from its initial resolvase cut to help us map the locations of the HJs (Fig. 5D). The DHJS was incubated with an excess of T7 endo I, and the reaction products separated on a 15% polyacrylamide gel (Fig. 5E). There are five distinct bands that result from this reaction. Upon analysis of the gel, the three smallest bands are calculated to be 170, 253, and 302 bp in size. They correlate well with linearized fragments from the homologous and non-homologous sequence between the HJs, with predicted sizes of 165, 251, and 300 bp.
The sizes of the two larger bands, ~416 and ~465 bp, correspond to linearized A and B circles. One possible mechanism to generate them is that the initial cleavage at the HJ relieves the topological constraints, thus allowing the branch migration of the other HJ across the homologous region. Following the conversion of single strand nicks to double stranded breaks by T7 endo I, linearized A and B circles could then be produced.
Taken together, the restriction enzyme and T7 endo I analysis is entirely consistent with the DHJS containing two DNA circles conjoined by two HJs. The restriction enzyme analysis showed and the T7 endo I data suggest that the HJs are free to migrate within the homologous sequence once the topological restraint on the substrate is relieved. In addition, the complete digestion of the substrate by T7 endo I also indicates that the HJs are situated at the refractory boundaries created at the transition from the homologous to the non-homologous sequences in at least the majority of the molecules.
Concluding Remarks
The DHJS is most easily thought of as two DNA circles conjoined by two HJs. The HJs are separated by 165 bp of homologous sequence. This allows for the HJs to be mobile; however, the HJs cannot spontaneously migrate toward or away from each other because of the topology of a dHJ. If one views the substrate as two DNA circles conjoined by a dHJ, then the linking number between the two circles is a function of the distance between the HJs. Because the linking number cannot change without nicking or breaking the DNA, the distance between the two HJs cannot spontaneously change without inducing writhe within the substrate. Therefore, after synthesis, the two HJs are located near the refractory boundaries created by the borders of the homologous and non-homologous regions and remain there until acted upon by an enzyme that can relieve the topological constraint on the structure, such as an endonuclease or topoisomerase. Because of the distance between the HJs in this substrate (165 bp), the conjoined circles possess a linking number of ~30, providing a more rigorous test for enzymes that are believed to resolve this structure through collapse of the dHJ. This substrate, and derivatives of it, should prove useful for the analysis of protein complexes that are thought to process dHJs, whether they do so by a resolvase mechanism or through convergent migration of the HJs.
Supplementary Material
Acknowledgments
We are grateful to Drs. Chapin Rodriguez and Paul Sadowski for a kind gift of the expression vector of reverse gyrase and Cre recombinase, respectively. We also thank Carrie Reardon for excellent technical assistance and lab members and our colleagues for stimulating discussions.
Footnotes
This work was supported by Grant GM29006 from the National Institutes of Health.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
The abbreviations used are: dHJ, double Holliday junction; CO, crossover; NCO, non-crossover; DHJS, double Holliday junction substrate; HJ, Holliday junction; dsDNA, double stranded DNA; ssDNA, single stranded DNA; MOPS, 4-morpholinepropanesulfonic acid; endo, endonuclease.
References
- 1.Paques F, Haber JE. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Resnick MA. J Theor Biol. 1976;59:97–106. doi: 10.1016/s0022-5193(76)80025-2. [DOI] [PubMed] [Google Scholar]
- 3.Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. Cell. 1983;33:25–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- 4.Alani E, Padmore R, Kleckner N. Cell. 1990;61:419–436. doi: 10.1016/0092-8674(90)90524-i. [DOI] [PubMed] [Google Scholar]
- 5.Bishop DK, Park D, Xu L, Kleckner N. Cell. 1992;69:439–456. doi: 10.1016/0092-8674(92)90446-j. [DOI] [PubMed] [Google Scholar]
- 6.Sun H, Treco D, Szostak JW. Cell. 1991;64:1155–1161. doi: 10.1016/0092-8674(91)90270-9. [DOI] [PubMed] [Google Scholar]
- 7.Collins I, Newlon CS. Cell. 1994;76:65–75. doi: 10.1016/0092-8674(94)90173-2. [DOI] [PubMed] [Google Scholar]
- 8.Schwacha A, Kleckner N. Cell. 1994;76:51–63. doi: 10.1016/0092-8674(94)90172-4. [DOI] [PubMed] [Google Scholar]
- 9.Schwacha A, Kleckner N. Cell. 1995;83:783–791. doi: 10.1016/0092-8674(95)90191-4. [DOI] [PubMed] [Google Scholar]
- 10.Hunter N, Kleckner N. Cell. 2001;106:59–70. doi: 10.1016/s0092-8674(01)00430-5. [DOI] [PubMed] [Google Scholar]
- 11.Heyer WD. Curr Biol. 2004;14:R56–58. doi: 10.1016/j.cub.2003.12.043. [DOI] [PubMed] [Google Scholar]
- 12.Heyer WD, Ehmsen KT, Solinger JA. Trends Biochem Sci. 2003;28:548–557. doi: 10.1016/j.tibs.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 13.Symington LS, Holloman WK. Science. 2004;303:184–185. doi: 10.1126/science.1093959. [DOI] [PubMed] [Google Scholar]
- 14.Borner GV, Kleckner N, Hunter N. Cell. 2004;117:29–45. doi: 10.1016/s0092-8674(04)00292-2. [DOI] [PubMed] [Google Scholar]
- 15.Guillon H, Baudat F, Grey C, Liskay RM, de Massy B. Mol Cell. 2005;20:563–573. doi: 10.1016/j.molcel.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 16.Nasmyth KA. Annu Rev Genet. 1982;16:439–500. doi: 10.1146/annurev.ge.16.120182.002255. [DOI] [PubMed] [Google Scholar]
- 17.Wang JC. Nat Rev Mol Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 18.Fu TJ, Tse-Dinh YC, Seeman NC. J Mol Biol. 1994;236:91–105. doi: 10.1006/jmbi.1994.1121. [DOI] [PubMed] [Google Scholar]
- 19.Wu L, Hickson ID. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 20.Blanton HL, Radford SJ, McMahan S, Kearney HM, Ibrahim JG, Sekelsky J. PLoS Genet. 2005;1:e40. doi: 10.1371/journal.pgen.0010040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hilliker AJ, Harauz G, Reaume AG, Gray M, Clark SH, Chovnick A. Genetics. 1994;137:1019–1026. doi: 10.1093/genetics/137.4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez AC, Stock D. EMBO J. 2002;21:418–426. doi: 10.1093/emboj/21.3.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaikh AC, Sadowski PD. J Biol Chem. 1997;272:5695–5702. doi: 10.1074/jbc.272.9.5695. [DOI] [PubMed] [Google Scholar]
- 24.Plank JL, Chu SH, Pohlhaus JR, Wilson-Sali T, Hsieh TS. J Biol Chem. 2005;280:3564–3573. doi: 10.1074/jbc.M411337200. [DOI] [PubMed] [Google Scholar]
- 25.Bregeon D, Doetsch PW. BioTechniques 37, 760–762. 2004;764:766. doi: 10.2144/04375ST01. [DOI] [PubMed] [Google Scholar]
- 26.Hearst JE. Annu Rev Biophys Bioeng. 1981;10:69–86. doi: 10.1146/annurev.bb.10.060181.000441. [DOI] [PubMed] [Google Scholar]
- 27.Zhen WP, Buchardt O, Nielsen H, Nielsen PE. Biochemistry. 1986;25:6598–6603. doi: 10.1021/bi00369a039. [DOI] [PubMed] [Google Scholar]
- 28.Dickie P, McFadden G, Morgan AR. J Biol Chem. 1987;262:14826–14836. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.