

Abstract
Structural and functional analyses for many mammalian systems depend on having abundant supplies of recombinant multi-protein complexes that can be produced best, or only, in mammalian cells. We present an efficient fluorescence marking procedure for establishing stable cell lines that overexpress two proteins in co-ordination, and we validate the method in the production of monoclonal antibody Fab fragments. The procedure has worked without fail on all seven of seven trials on Fabs, which are being used in the crystallization of G-protein coupled receptors. This manner of efficient selection may readily be adapted for the co-production of other complexes of two or more proteins.
Keywords: antibodies, protein overexpression, mammalian cells, protein complexes, fluorescence
INTRODUCTION
Recent proteomic studies have strengthened the concept that functional units within a cell typically consist of macromolecular assemblages as opposed to individual proteins [1]. Accordingly, there is a justified and increasing interest in structure determination and biophysical characterization of macromolecular complexes [2]. These interests require the production of stable complexes, which for the most part depends on recombinant expression in appropriate hosts. Since reconstitution from separately expressed proteins is often frustrated by instability of the isolated components, there are obvious advantages for simultaneous over-expression of multiple proteins in the same call. Bacteria, given their cost-effectiveness and robustness, have been by far the most successful hosts for protein production to support biophysical studies, and strategies for the co-expression of genes in Escherichia coli are available [2,3]. Bacteria, and yeast or insect cells for that matter, might be poorly suited for the production of mammalian proteins [4,5]. These proteins may be toxic to the host, may require a better matched folding machinery, and may need post-translational modifications unavailable in the heterologous host.
Expression of recombinant proteins in mammalian cells can be achieved through transient transfection, viral infection or stable integration of expression constructs into the host genome. Transient co-transfection of separate plasmids carrying the individual genes represents possibly the most frequently employed technique for functional studies. Unfortunately, this has yet to translate into a general approach for producing abundant amounts of material as required for applications such as structure determination. Stable cell lines have obvious advantages for amplified recombinant protein production. However, the integration of the expression constructs into the genome of the host cell inevitably leads to a wide spectrum in protein synthesis levels within the same batch of transfected cells, depending primarily on the number of integrants and their sites of integration. The method therefore relies on being able to select for cells capable of producing the most protein.
Through coupling of the expression of each polypeptide chain with that of different antibiotic-resistant markers, targeting a specific genomic locus for integration, or incorporating all the components in a single plasmid one can increase the frequency of stable transfectants able to synthesize all the desired components. These provide only marginal relief, however, in the task of identifying the best expressing cells, which has typically involved time-consuming rounds of screening to yield lines with the required characteristics [6].
Here we present and validate procedures for the rapid selection of mammalian cells that co-express multiple proteins. We do this by coupling the expression of each protein chain of a complex to a separate fluorescent marker, and we test the system in applications to the high-level expression of antibody Fab fragments. Proper functionality of the expressed complexes was demonstrated by assessing correct assembly of the antibody fragments and their ability to recognize the antigen, a 5HT2c serotonin receptor.
MATERIALS AND METHODS
Cloning of Fv chains
Cloning of Fv regions was performed with appropriate kits and degenerate primers (Novagen) following standard procedures and guidelines. The amplified fragments were cloned into pGEM-T vector (Promega). A second PCR reaction was used to introduce appropriate restriction sites for cloning into expression vectors.
Construction of expression vectors
pFM1.2 [7] was used to generate two vectors for the separate expression of the two Fab chains. For light chain expression, GFP was substituted for RFP, taken from pIRES2-DsRed-Express (Clontech). The CH-His6 and CL regions of D1.3 anti-lysozyme antibody were removed from pASK84 [8], and cloned into the MCS regions of the respective vectors. The heavy and light chains of the Fv regions were then cloned into the matching vectors to generate the final expression constructs. The vectors for expression of heavy and light chains were named pFMFabH and pFMFabL respectively.
Cell culture and generation of stable lines
HEK293 cells were maintained at 37°C, in a humidified environment enriched with 5% CO2. HEK293 cells were grown in DMEM (Chemicon) supplemented with 10% FBS (Hyclone), Penicillin, Streptomycin, L-glutamine (Pen/Strep/L-glu; SIGMA). 293 GntI− cells [9] carrying stable integration of an inducible expression cassette for 5HT2c, were grown in DMEM/F12 (Gibco) supplemented with 10% FBS, Pen/Strep/L-glu, 4μg/mL blasticidin (Invitrogen) and 500μg/mL G418. A plasmid carrying resistance to puromycin was mixed with the two expression vectors in a 1:5:5 ratio prior to transfection with lipofectamine (Invitrogen). Stable integrants were selected by addition of 5μg/mL puromycin to the growth medium. Production cell lines from single double fluorescent colonies were selected either by FACS sorting in Autoclone mode and subsequent visual inspection of the resulting clones, to identify single, highly fluorescent colonies, or by manual picking of the most intense double fluorescent colonies after antibiotic selection.
FACS sorting
Data were collected using a Beckman Coulter Altra flow cytometer equipped with Autoclone. Untransfected (control) and stably-integrated cells were passed through the cell sorter. The viable cell population was determined using the forward and side scatter characteristics of the cells. The fluorescent cells were excited at the 488nM line of a krypton-argon laser. RFP emission was detected using a 590/20 nm band pass filter. GFP emission was detected with a 525/30 nm band pass filter. 100,000 viable cells were collected for each pool, according to their fluorescence profile. Typically, the top fluorescent cells of the double positive population (corresponding to 0.1% of the viable cell population) were chosen for cloning to single cell purity. This was achieved with the flow cytometer in Autoclone mode, in 96 well plates.
Fab purification
NaHepes pH7.5, NaCl and Imidazole were added to the harvested medium to 20, 400 and 10mM final concentrations respectively. The medium was added to equilibrated Ni-NTA (Qiagen) on a column. The column was washed with 10 column volumes of 20mM NaHepes pH7.5, 400mM NaCl, 25mM Imidazole. Purified Fab was eluted with 200mM Imidazole in the same buffer.
Confocal microscopy on fluorescent cells
Cells were plated on 8-well CC2 treated glass slides (Nalge Nunc Int.) at a density of 1×105. The next day, the cells were fixed with 4% Paraformaldehyde in 0.1M Phosphate buffer for 10 minutes at room temperature. Cells were then washed 3 times with PBS and incubated for 30 minutes with the nuclear stain TOTO-3 iodide (Invitrogen) diluted at 1:2000 in PBS. Stained cells were viewed and photographed on a Zeiss confocal microscope at 40X magnification in 3 separate emission channels, at 520nM (GFP), 570nM (RFP) and 633nM (TOTO-3).
Immunocytochemistry
293 GntI− cells carrying stable integration of an inducible expression cassette for 5HT2c were induced with tetracycline (2μg/mL) and sodium butyrate (5mM). Two days later, induced and uninduced (control) cells were plated on 8-well CC2 treated glass slides (Nalge Nunc Int.) at a density of 1×105. After 24 hours, cells were fixed with 4% Paraformaldehyde in 0.1M Phosphate buffer for 10 minutes at room temperature. The fixed cells were blocked with 5% goat serum in PBS and incubated for 1 hour with hybridoma supernatants (for full length Ab), and medium from Fab-expressing cells (for Fab). The secondary antibody, used at a dilution of 1:1000, was Cy3-conjugated anti-mouse IgG (Jackson Immunoresearch). TOTO-3 iodide (Invitrogen) was used diluted at 1:2000 as a nuclear stain. Labelled cells were visualized and photographed by confocal microscopy as described in the previous paragraph.
Fab-antigen complex reconstitution and analysis
Purified Fab was concentrated to ~2mg/mL. 5HT2c expressed in E. coli as a fusion to periplasmic maltose-binding protein (MBP) was purified on an immobilized streptavidin column by means of an SBP epitope tag [10] fused to the C-terminus of the receptor, by prior solubilization of the isolated membranes at 5mg/mL membrane protein concentration with 1% (w/v) n-Dodecyl-α-D-Maltopyranoside (αDDM; Anatrace) and 0.2% Cholesteryl Hemisuccinate, (CHS; Anatrace). MBP was removed by cleavage of the fusion with TEV protease [11] at an engineered site in the linker region. Purified 5HT2c was concentrated to ~1mg/mL. For reconstitution, detergent was added to the concentrated Fab to 0.1% final concentration, mixed with 5HT2c in approximately a 2:1 molar ratio and incubated on ice for 2 hours. Samples were run on a 7.5mm × 60cm TSKgel G3000SW column (TOSOH Bioscience, LLC) at a flow rate of 0.3mL/min in 20mM NaHepes pH7.0, 200mM NaCl, 1mM EDTA, 0.025% αDDM.
RESULTS
We first adapted a previously described fluorescence-selection vector for the high-level production of proteins in mammalian cells [7] for use with a second color. This vector carries a strong, constitutive promoter sequence, derived from cytomegalovirus (CMV [12]. Target gene expression is coupled via an IRES element [13] to that of green fluorescent protein (GFP [14]), and was modified to replace the coding sequence for GFP with that for red fluorescent protein (RFP [15]). The target genes for our current application are those for antibody Fab fragments corresponding to monoclonal antibodies (MAbs) against a rat G-protein coupled receptor (GPCR) for serotonin (5HT2c [16]), which were generated as previously described [17]. In this instance, we further modified the GFP- and RFP-containing vectors to include coding sequences for the conserved heavy-chain CH1 and light-chain CL domains from the extensively characterized, anti-lysozyme antibody D1.3 [18] ahead of the respective IRES elements. CH1 and CL domains don’t confer antigen specificity to the antibody, and are interchangeable, even between species [6,19]. These provided the scaffold to express cloned VH and VL domains as Fab fragments. Six histidine residues were genetically fused to the C-terminus of CH1 for purification purposes [8]. Finally, coding sequences for VH and VL domains, inclusive of signal peptide sequences, were cloned from our hybridoma cells [17] using established protocols and inserted in-frame into vectors with matching C regions. A schematic of the two resulting expression constructs, one for each chain of the Fab fragment, is shown in Fig 1A.
Fig. 1.

Co-expression of two polypeptide chains with fluorescent markers. (A) Schematic of the expression constructs. Expression is driven by the cytomegalovirus (CMV) promoter. The gene for the heavy chain of the Fab is in blue colors (VH and CH1 D1.3), followed by a hexa-histidine tag (H6) fused to its C-terminus. The light chain, in a separate vector is shown in yellow (VL) and orange (CL D1.3). Expression of the heavy and light chains is coupled to that of green (GFP) and red (RFP) protein respectively by an internal ribosome entry site (IRES). (B) Fluorescence profile of sorted cells. Fluorescence data from 300,000 viable HEK293 cells expressing 2A11 after antibiotic selection are shown. Fluorescence is plotted in logarithmic scale for RFP (vertical axis) and GFP (horizontal axis). The orange lines are representative of the 4×4 grid used to select negative, +, ++, and +++ cells, for RFP fluorescence (horizontal lines from bottom to top, in increasing order) and for GFP fluorescence (vertical lines left to right, in increasing order). (C) Correlation of Fab 2A11 expression levels with fluorescence. Cells were sorted according to their fluorescence profile, and the pools photographed by confocal microscopy in the emission channels corresponding (from top to bottom) to RFP, GFP, and the nuclear stain TOTO-3. The resulting Fab is shown under the respective columns, run out, after purification, on a Coomassie blue-stained SDS-PAGE gel. (D) Fluorescence profile of a production cell line. A clone cell line expressing 2A11 Fab was analyzed by FACS as described for panel (B).
After co-transfection of the two plasmids into HEK293 cells, together with that for a selectable marker for antibiotic resistance, stable integrants were selected. As expected, the resulting antibiotic-resistant colonies displayed a wide spectrum of fluorescence levels, due to the presence of both RFP and GFP. Firstly, then, we wanted to determine if the cells with both red and green fluorescence also actually secreted antibody fragments. Secondly, we were interested in verifying the extent to which fluorescence intensity correlated with Fab expression. In other words, we wanted to test the predictive value of our fluorescent markers for the production of desired protein products.
Using a fluorescence-activated cell sorter (FACS), we classified cells according to their two-color fluorescence profiles into a 4×4 grid of − ++ and +++ for each color. The vast majority of cells after antibiotic selection were non-fluorescent, or only green, and populations fell off progressively with intensity for each color (Fig 1B). Pools of cells with pertinent characteristics were selected and analyzed. Cells from these selected pools were visualized by confocal microscopy for fluorescence from RFP, GFP and a nuclear stain, and Fabs secreted from these different pools were purified by metal affinity chromatography and observed by SDS-PAGE. Results from such experiments are shown in Fig 1B for 2A11, an Fab fragment from an anti-5HT2c MAb [17]. We find that while negative cells (RFP−/GFP−) showed no detectable antibody fragments, Fab expression was always associated with fluorescence. Moreover, robust Fab expression could only be detected in double-positive cells, in amounts that were correlated to the levels of GFP and RFP fluorescence.
Each production cell line was cloned from a single RFP+++/GFP+++ progenitor cell and chosen from a few such cultures to assure that maximal fluorescence was maintained (Fig 2A). For these experiments, the RFP+++/GFP+++ grid was typically set to include only 0.1% of the cells. FACS analysis of a production cell line for Fab 2A11, after having been maintained for several months in continuous culture is shown in Fig 1D. Cells were grown in adherent monolayers, and for Fab 2A11 the yield was quantified to be above 30mg of purified protein per liter from the endpoint of a time course experiment (Fig 3) and from subsequent scale-ups (data not shown). Comparable yields have also been obtained from each of six other Fab productions attempted with this system, three reactive to 5HT2c, and three to a different GPCR. Relative yields were judged from the relative intensities of coomassie blue-stained bands on gels. Three of these, all from antibodies against 5HT2c, are shown in Fig 2. Ultimate production levels (Fig 2B) seem to correlate with maximal fluorescence across these proteins (Fig 2A) much as Fab production correlates with fluorescence for a given protein (Fig 1C).
Fig. 2.
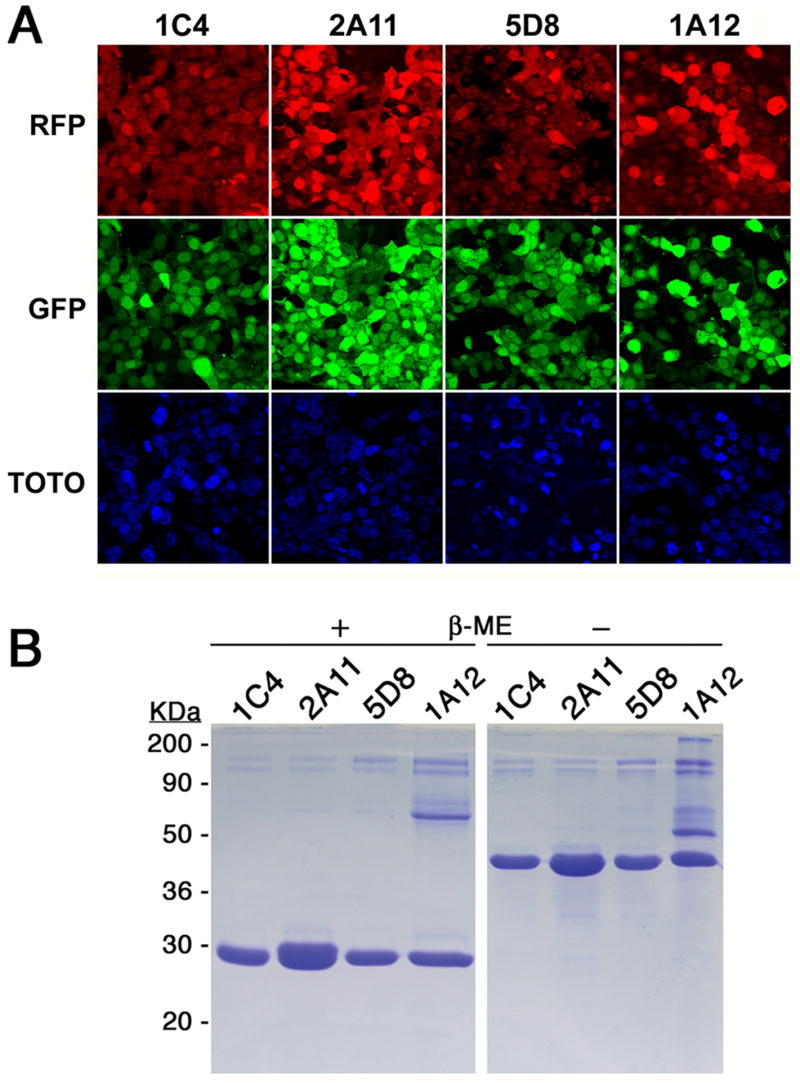
Fluorescence levels, yields and folding of expressed Fabs from production cell lines. (A) Cells secreting a specific Fab fragment, 1C4, 2A11, 5D8 or 1A12, as indicated on the top of each corresponding column, were visualized by confocal microscopy for fluorescence from RFP (top row), GFP (middle row) and TOTO-3 (bottom row). (B) The different Fabs were run, after purification, on an SDS-PAGE denaturing gel after treatment with loading dye with (+) and without (−) β-mercaptoethanol. The identity of each Fab is shown above the corresponding lane.
Fig. 3.
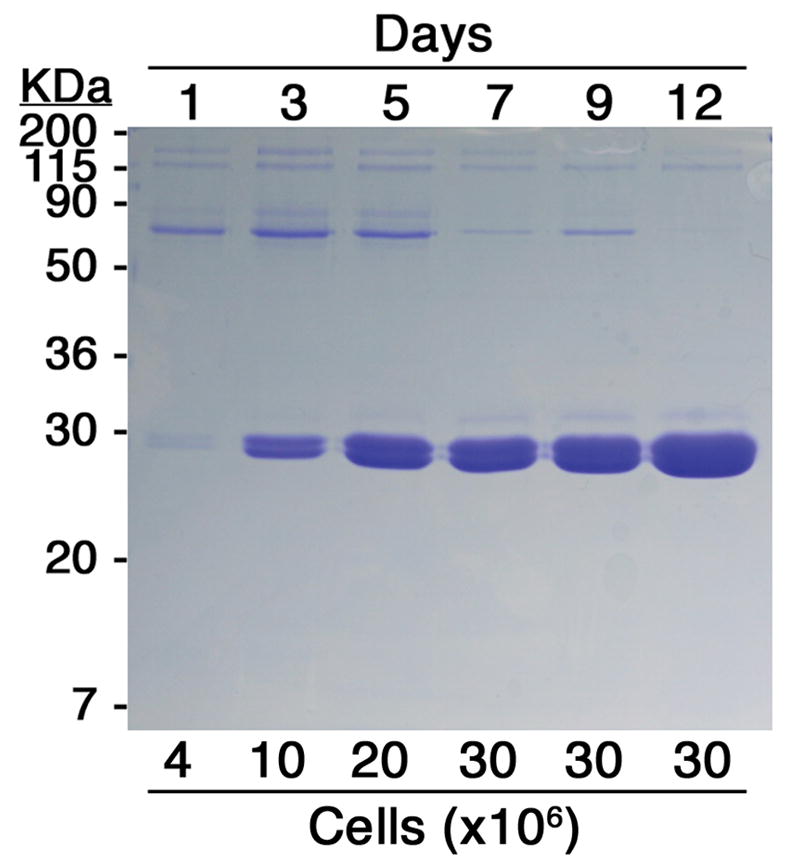
Time course of Fab expression. 18×106 cells expressing Fab 2A11 were equally divided into 6, 10cm-diameter dishes, in 10mL of medium. At each time point, the medium from a plate was removed, cells counted, and the Fab purified. 1/10th of each purified sample was run on an SDS-PAGE gel, stained with Coomassie blue. The length of each incubation period is indicated above the matching lane, in days, and the corresponding number of cells, in millions, below. The yield of Fab resulting from a 12day incubation period was quantified by absorbance reading at 280nM.
In contrast to our 100% success rate so far from two-color selection in HEK293 cells, alternative methods have proved far less reliable for us. We attempted Fab production by digestion of the intact IgG with papain, but only one of four different antibody-secreting hybridoma cell lines tried could be adapted to grow in serum-free medium and scaled-up for Fab production. We also attempted to express recombinant Fabs in E. coli following established procedures (reviewed in [20]), but achieved yields in micrograms per liter for the two antibodies tested. Reported expression levels of Fabs in E. coli [20,21] seem highly variable, while in the yeast Pichia pastoris, proper Fab assembly seems to be the major rate limiting factor [22]. In both these hosts, high-density cell growth in appropriate fermenters appears to have been essential for very high yields [20,22]. Indeed, if required, yields from our cell lines could most likely be improved dramatically by optimizing cell growth conditions, as in current practice for industrial production of antibodies in mammalian cells [6].
The light and heavy chains of purified Fab appeared to be disulfide-bond linked, in agreement with what is typical for a correctly folded antibody (Fig 2B). Furthermore, functionality of the expressed Fab was assessed by immunohistochemistry on antigen-expressing cells, showing a staining profile essentially identical to that observed with the intact IgG antibody derived from hybridomas (Fig 4A). Finally, in preparation for crystallization trials, the Fab-5HT2c complex was successfully reconstituted with purified components as shown by size-exclusion chromatography (Fig 4B). All these data are consistent with proper assembly and function of the expressed Fab.
Fig. 4.
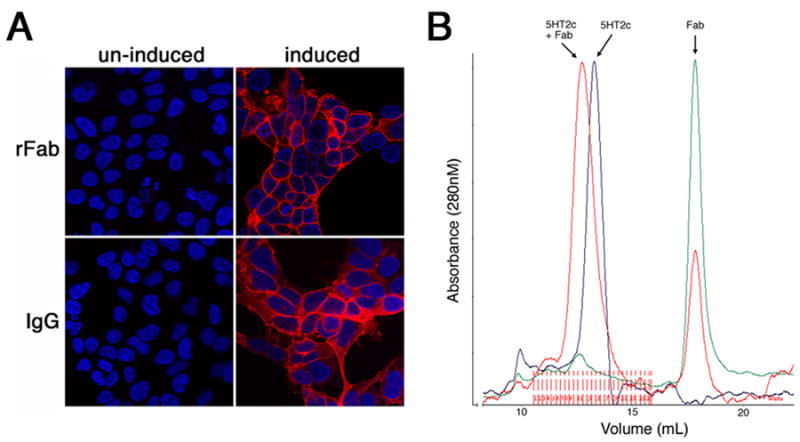
Functionality of the expressed Fab (A) Immunocytochemistry using recombinant Fab 2A11 (rFab) and intact antibody (IgG) on cells, before and after induction of 5HT2c expression. (B) Overlay of elution profiles from size-exclusion chromatography, monitored by absorbance at 280nM, of purified 5HT2c (blue trace), purified Fab (green trace) and reconstituted 5HT2c-Fab complex (red trace).
DISCUSSION
Both the tight correlation that we observe between production levels of functional antibody fragments and two-color fluorescence of transfected HEK293 cells (Fig 1C) and the high reliability of the procedure (seven successes in seven trials) provide a compelling validation of this selection method. Our application to monoclonal antibody production itself has broad potential. Monoclonal antibodies are important tools for structural studies on membrane proteins [23]. Indeed, Fabs described here are now being used in our crystallization of the 5HT2c receptor. Monoclonal antibodies are also gaining an increasingly relevant role in clinical applications [6,19]. This two-color system should have other applications as well, however, as we have already shown that our previous GFP system works for other secreted proteins and for membrane proteins such as GPCRs [7]. Moreover, given the availability of several different fluorescent proteins [24], one could anticipate being able to extend this methodology to studies of multi-component protein complexes.
In contrast to time-consuming multi-step conventional amplification systems for generating stably transfected mammalian cell lines, this fluorescence-based approach is simple and rapid. Fluorescence surveillance is also convenient for monitoring and maintaining the continued health of an established cell line. We hope that these procedures may aid in finally bridging the gap between multi-gene expression in mammalian cells for functional studies, which is commonplace and routine, and the use of mammalian cells for biophysical studies, which has had very limited popularity even as the structural community is increasingly interested in macromolecular assemblages.
Acknowledgments
We are grateful to Richard Axel for discussions. We thank Susan Brenner-Morton for advice and help with cloning of the antibody fragments and Yonghua Sun for excellent technical assistance. This work was supported in part by NIH grants GM68671 and GM75026.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gavin AC, Superti-Furga G. Protein complexes and proteome organization from yeast to man. Curr Opin Chem Biol. 2003;7:21–27. doi: 10.1016/s1367-5931(02)00007-8. [DOI] [PubMed] [Google Scholar]
- 2.Romier C, Ben Jelloul M, Albeck S, Buchwald G, Busso D, Celie PH, Christodoulou E, De Marco V, van Gerwen S, Knipscheer P, Lebbink JH, Notenboom V, Poterszman A, Rochel N, Cohen SX, Unger T, Sussman JL, Moras D, Sixma TK, Perrakis A. Co-expression of protein complexes in prokaryotic and eukaryotic hosts: experimental procedures, database tracking and case studies. Acta Crystallogr D Biol Crystallogr. 2006;62:1232–1242. doi: 10.1107/S0907444906031003. [DOI] [PubMed] [Google Scholar]
- 3.Tan S. A modular polycistronic expression system for overexpressing protein complexes in Escherichia coli. Protein Expr Purif. 2001;21:224–234. doi: 10.1006/prep.2000.1363. [DOI] [PubMed] [Google Scholar]
- 4.Geisse S, Gram H, Kleuser B, Kocher HP. Eukaryotic expression systems: a comparison. Protein Expr Purif. 1996;8:271–282. doi: 10.1006/prep.1996.0101. [DOI] [PubMed] [Google Scholar]
- 5.Yin J, Li G, Ren X, Herrler G. Select what you need: a comparative evaluation of the advantages and limitations of frequently used expression systems for foreign genes. J Biotechnol. 2007;127:335–347. doi: 10.1016/j.jbiotec.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 6.Birch JR, Racher AJ. Antibody production. Adv Drug Deliv Rev. 2006;58:671–685. doi: 10.1016/j.addr.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 7.Mancia F, Patel SD, Rajala MW, Scherer PE, Nemes A, Schieren I, Hendrickson WA, Shapiro L. Optimization of protein production in mammalian cells with a coexpressed fluorescent marker. Structure (Camb) 2004;12:1355–1360. doi: 10.1016/j.str.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 8.Skerra A. A general vector, pASK84, for cloning, bacterial production, and single-step purification of antibody Fab fragments. Gene. 1994;141:79–84. doi: 10.1016/0378-1119(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 9.Reeves PJ, Callewaert N, Contreras R, Khorana HG. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc Natl Acad Sci USA. 2002;99:13419–13424. doi: 10.1073/pnas.212519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keefe AD, Wilson DS, Seelig B, Szostak JW. One-step purification of recombinant proteins using a nanomolar-affinity streptavidin-binding peptide, the SBP-Tag. Protein Expr Purif. 2001;23:440–446. doi: 10.1006/prep.2001.1515. [DOI] [PubMed] [Google Scholar]
- 11.Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 12.Thomsen DR, Stenberg RM, Goins WF, Stinski MF. Promoter-regulatory region of the major immediate early gene of human cytomegalovirus. Proc Natl Acad Sci USA. 1984;81:659–663. doi: 10.1073/pnas.81.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vagner S, Galy B, Pyronnet S. Irresistible IRES. Attracting the translation machinery to internal ribosome entry sites. EMBO Rep. 2001;2:893–898. doi: 10.1093/embo-reports/kve208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 15.Knop M, Barr F, Riedel CG, Heckel T, Reichel C. Improved version of the red fluorescent protein (drFP583/DsRed/RFP) Biotechniques. 2002;33:592–602. doi: 10.2144/02333rr02. [DOI] [PubMed] [Google Scholar]
- 16.Julius D, MacDermott AB, Axel R, Jessell TM. Molecular characterization of a functional cDNA encoding the serotonin 1c receptor. Science. 1988;241:558–564. doi: 10.1126/science.3399891. [DOI] [PubMed] [Google Scholar]
- 17.Mancia F, Brenner-Morton S, Siegel R, Assur ZSY, Schieren I, Mendelsohn M, Axel R, Hendrickson WA. Production and characterization of monoclonal antibodies sensitive to conformation in the 5HT2c serotonin receptor. Proc Natl Acad Sci USA. 2007;104:4303–4308. doi: 10.1073/pnas.0700301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amit AG, Mariuzza RA, Phillips SE, Poljak RJ. Three-dimensional structure of an antigen-antibody complex at 2.8 A resolution. Science. 1986;233:747–53. doi: 10.1126/science.2426778. [DOI] [PubMed] [Google Scholar]
- 19.Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol. 2005;23:1073–1078. doi: 10.1038/nbt0905-1073. [DOI] [PubMed] [Google Scholar]
- 20.Skerra A. Bacterial expression of immunoglobulin fragments. Curr Opin Immunol. 1993;5:256–262. doi: 10.1016/0952-7915(93)90014-j. [DOI] [PubMed] [Google Scholar]
- 21.Corisdeo S, Wang B. Functional expression and display of an antibody Fab fragment in Escherichia coli: study of vector designs and culture conditions. Protein Expr Purif. 2004;34:270–279. doi: 10.1016/j.pep.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 22.Gasser B, Maurer M, Gach J, Kunert R, Mattanovich D. Engineering of Pichia pastoris for improved production of antibody fragments. Biotechnol Bioeng. 2006;94:353–361. doi: 10.1002/bit.20851. [DOI] [PubMed] [Google Scholar]
- 23.Hunte C, Michel H. Crystallisation of membrane proteins mediated by antibody fragments. Curr Opin Struct Biol. 2002;12:503–508. doi: 10.1016/s0959-440x(02)00354-8. [DOI] [PubMed] [Google Scholar]
- 24.Lukyanov KA, Chudakov DM, Lukyanov S, Verkhusha VV. Innovation: Photoactivatable fluorescent proteins. Nat Rev Mol Cell Biol. 2005;6:885–891. doi: 10.1038/nrm1741. [DOI] [PubMed] [Google Scholar]