

SUMMARY
Classic Hsp70 chaperones assist in diverse processes of protein folding and translocation, and Hsp110s had seemed by sequence to be distant relatives within an Hsp70 superfamily. The 2.4Å-resolution structure of Sse1 with ATP shows that Hsp110s are indeed Hsp70 relatives, and it provides insight into allosteric coupling between sites for ATP and polypeptide-substrate binding in Hsp70s. Subdomain structures are similar in intact Sse1(ATP) and in the separate Hsp70 domains, but conformational dispositions are radically different. Interfaces between Sse1 domains are extensive, intimate and conservative in sequence with Hsp70s. We propose that Sse1(ATP) may be an evolutionary vestige of the Hsp70(ATP) state, and an analysis of 64 mutant variants in Sse1 and three Hsp70 homologs supports this hypothesis. An atomic-level understanding of Hsp70 communication between ATP and substrate-binding domains follows. Requirements on Sse1 for yeast viability are in keeping with the distinct function of Hsp110s as nucleotide exchange factors.
INTRODUCTION
The 70-kD heat shock proteins (Hsp70s) are ubiquitous and abundant molecular chaperones. Namesake Hsp70s are induced in response to cellular stresses, such as heat shock or oxidative stress, whereupon they impede protein aggregation and assist in the refolding of misfolded proteins. Hsp70s also participate in diverse processes in normal cellular conditions, including the folding of newly synthesized proteins, protein translocation across membranes, disassembly of protein assemblages, and protein degradation. These chaperones have roles in disease as well; notably, Hsp70s are complicit in cancer and protective in neurodegenerative diseases. All Hsp70s act in ATP-dependent cycles of binding and release of substrates, typically exposed hydrophobic polypeptide segments. The Hsp70 family is conserved across forms of life and cellular compartments (>40% pairwise sequence identity), and each member comprises an N-terminal nucleotide-binding domain (NBD) with ATPase activity and a C-terminal substrate-binding domain (SBD) (Hartl and Hayer-Hartl, 2002; Mayer and Bukau, 2005).
The binding functions of NBD and SBD are separable, but Hsp70 chaperone activity strictly requires that these linked portions interact, albeit transiently. NBDs bind nucleotides with relatively high affinity, but they have low intrinsic hydrolytic activity. Isolated SBDs or ADP-complexed Hsp70s bind tightly to appropriate polypeptides. ATP binding by an Hsp70 dramatically decreases the SBD affinity for polypeptide substrates, accelerating both on and off rates. Reciprocally, substrate binding stimulates ATP hydrolysis, whereupon substrates again exchange slowly. Nucleotide exchange reinitiates the cycle. Two types of co-chaperones modulate the Hsp70 chaperone cycle. Hsp40s stimulate ATP hydrolysis by Hsp70s and help to target them to substrates, and nucleotide exchange factors (NEFs) facilitate the release of ADP and rebinding of ATP, which in turn promotes substrate release (Hartl and Hayer-Hartl, 2002; Mayer and Bukau, 2005).
Two exclusively eukaryotic families of chaperone proteins join with the classic Hsp70s into a divergent superfamily (Easton et al., 2000). Both the cytosolic Hsp110/Sse1 family and the ER-localized Grp170/Lhs1 family show unmistakable NBD similarity to Hsp70s (~35% and ~30/20% sequence identity, respectively) and C-terminal halves, although in the twilight zone by sequence (12–15% Hsp70 identity), are putative SBDs by predicted secondary structures. Both Hsp110- and Grp170-type proteins are distinguished by larger size due to acidic SBD insertions in most and C-terminal extensions in many (Fig. 1A). It is thought that the two non-classic Hsp70s function similarly in their respective compartments. Hsp110 proteins are expressed broadly in mammalian tissues, and at especially high levels in brain. Sse1 is an Hsp110 counterpart in budding yeast Saccharomyces cerevisiae.
Figure 1. Overall structure of Sse1.

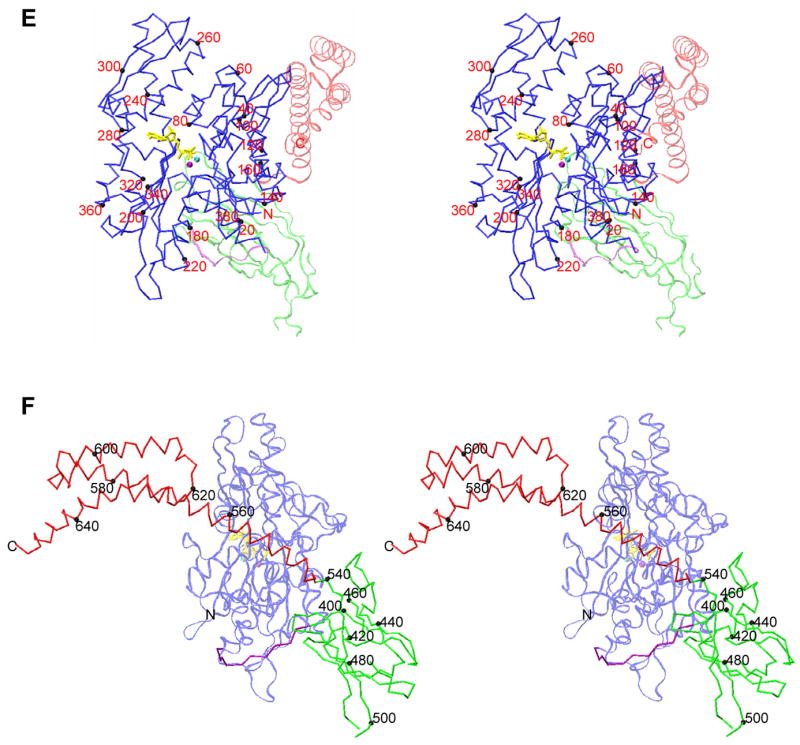
(A) Schematics of Hsp70 and Hsp110 sequences and prototypic Hsp70 domain structures. Coloring is NBD (blue), interdomain linker (purple), SBDβ (green), SBDα (red). (B) Ribbon diagram of the dimer. Protomer A is left and B is right. Missing loops are dotted. Protein coloring is as in A. ATP molecules have sticks for bonds (yellow) and balls for associated metal ions: Mg2+ (purple) and K+ (cyan). (C) Ribbon diagram of protomer B in the canonical, front-face NBD view. (D) Orthogonal view of the protomer drawn as in C, but from the right side. (E) Stereo drawing of a Cα trace of protomer B. NBD is in stick representation (full intensity) and SBD is in a continuous fine ribbon (subdued). View and coloring are as in C. Every 20th NBD Cα is marked with a black sphere and labeled. (F) Stereo drawing as in E, but viewed and colored as in D except that now SBD is at full intensity and NBD is subdued.
Cellular functions of Hsp110s are poorly characterized, but recent biochemical analyses suggest that Hsp110s may be the principal NEFs for cytosolic Hsp70s. In particular, it was shown that Sse1 is a potent NEF for yeast Hsp70s Ssa1 and Ssa2 (Dragovic et al., 2006; Raviol et al., 2006b) as are human Hsp110 for porcine Hsc70 (Dragovic et al., 2006) and Sse1 for human Hsp70 (Shaner et al., 2006). These studies also found synergistic effects of Hsp110 and Hsp40 co-chaperones on Hsp70 refolding activity. The discovery of NEF activity followed earlier evidence that Hsp110s interact physically and functionally with counterpart Hsp70s (Yamagishi et al., 2004; Shaner et al., 2005; Yam et al., 2005) and the finding that Lhs1 is an NEF for the ER-Hsp70 Kar2 in yeast (Steel et al., 2004). Hsp110s may also have other functions. They can block aggregation of denatured proteins in vitro, but they do not actively cause refolding (Oh et al., 1999); hence description as ‘holdases’. Intrinsic ATPase activity is very low for Hsp110s (Raviol et al., 2006a).
Crystal structures of individual NBD and SBD domains of Hsp70s provide a framework for biochemical understanding of Hsp70 chaperone activity (Fig. 1A). The prototype NBD structure is that from bovine Hsc70 (bHsc70) (Flaherty et al., 1990). It comprises four subdomains (IA, IB, IIA and IIB) built up from two structurally similar lobes (I and II). Adenosine nucleotides bind at the interface between lobes, making contacts with all four subdomains. The prototype SBD structure is that from DnaK of Escherichia coli (Zhu et al., 1996). It has a compact β-sandwich subdomain (SBDβ) followed by an extended α-helical subdomain (SBDα). A substrate peptide is bound to DnaK in an extended conformation through a channel defined by loops from the β subdomain and covered by the α-helical subdomain.
Findings from the initial Hsp70 domain structures were extended in following x-ray and NMR structural studies (Mayer and Bukau, 2005); and measurements by tryptophan fluorescence, small-angle x-ray scattering, NMR spectroscopy, infrared spectroscopy, and proteolytic susceptibility show that NBD and SBD interact upon ATP binding with consequent conformational changes in each (Buchberger et al., 1995; Wilbanks et al., 1995; Zhang and Zuiderweg, 2004; Moro et al., 2006; Swain et al., 2007). NBD-SBD associations are labile and difficult to maintain for structural analysis, but two recent studies do capture interdomain interactions (Jiang et al., 2005; Revington et al., 2005). Neither is in the ATP state and each is a mutant variant, however, and the two have very different SBD-to-NBD domain dispositions. These and other concerns have led to questions about relevance to physiology (Vogel et al., 2006b; Swain et al., 2007).
Frustrated by the transience of events, prior studies on Hsp70s have left unanswered the key question of how allosteric coupling happens. In the case of Sse1, however, separated halves do interact stably, both in vivo and in vitro (Shaner et al., 2004), and ATP stabilizes this Hsp110 as it does Hsp70 proteins (Raviol et al., 2006a; Shaner et al., 2006; Supplemental Text S1). These findings suggest probable NBD-SBD interactions in the ATP state, which prompted us to undertake a crystallographic study of Sse1. Given the probable evolutionary relationship of Hsp110s to Hsp70s (Fig. 1A), we reasoned that structural features in Sse1(ATP) might preserve vestiges of the ATP state in Hsp70s, even though functions have diverged apart.
Here, we first report the structure of Sse1 at 2.4Å resolution as a complex with ATP. Structures within pieces of this holo Hsp110 are highly similar to those in isolated NBD and SBD portions of Hsp70s, but dramatic conformational rearrangements distinguish each half from its Hsp70 counterpart. This establishes that Hsp70s and Hsp110s are indeed evolutionary relatives throughout. Thus, an Hsp70-like enzyme is now at last seen in the ATP state and intact. The interface between NBD and SBD is extensive and intimate, and it shows striking sequence conservation with classic Hsp70s. We then test the hypothesis that Sse1(ATP) reflects the Hsp70(ATP) state as an evolutionary vestige. Heat-stress responses of a battery of analogous mutants of Hsp70s Ssa1 and DnaK and of Sse1 itself were evaluated in corresponding yeast and bacterial hosts. The very interfaces found in the Sse1(ATP) structure prove to be relevant for in vivo function of Hsp70s. The resulting model for the Hsp70(ATP) state provides a comprehensive atomic basis for understanding allosteric communication between the ATP and peptide binding sites.
RESULTS
Crystal structure of Sse1
We crystallized Sse1(2-659), shortened C-terminally (Δ34) but less so than fully functional Sse1(Δ44) (Shaner et al., 2004), as a complex with ATP/Mg2+/K+. The structure was solved (Text S2) by MAD phasing of selenomethionyl Sse1 (Fig. S1) and refined at 2.4Å resolution to an R-value of 19.5% (Rfree=23.5%). Sse1(ATP) is a labile dimer in solution (Text S3), and it is quasi-diad symmetric in the crystal (Fig. 1B, Table S1). The dimer interface is substantial (1976Å2), but shape complementarity is low (Table S2).
Sse1 is modular, built up from Hsp70-like subdomain pieces. Thus, Hsp110s do indeed constitute a superfamily with Hsp70s despite highly divergent SBD domains (12.7% and 19.1% identity for Sse1 vs. DnaK SBDβ and SBDα, respectively). Sse1 β-strands and α-helices align closely with those in DnaK domains (Fig. S2), but Sse1 subdomains are disposed very differently than in Hsp70 prototypes (see below). NBD and SBD domains associate intimately in the Sse1(ATP) protomer, burying a large (4001Å2) interface of uniformly high shape complementarity (Table S2). SBDβ protrudes behind NBD and SBDα extends forward from it (Fig. 1, C and E; canonical NBD view), whereby the molecule is elongate in profile (Fig. 1, D and F). The Hsp110 insertion is disordered between SBD strands β7 and β8.
Comparison of Sse1 to SBD of DnaK
The SBD portion of Sse1(ATP) is rearranged radically from the conformation in SBD of DnaK bound to heptapeptide NRLLLTG (Zhu et al., 1996). SBDα in Sse1 is peeled away from association with SBDβ as in DnaK, and helices αA and αB are straightened into one long helix (residues 544-588; cf. DnaK 509-553) called αB1-αB2 (Fig. S2). Sse1 surfaces corresponding to the interface between αA and SBDβ in DnaK are now buried into interfaces with NBD (Figs. 2A and 2B), αA between subdomains IA and IB and SBDβ between IA and IIA. Helix αB1 in Sse1 is rotated by ~160° from αA in DnaK (Fig. 2C) and the helix straightening entails a rotation of ~80° (Fig. 2D).
Figure 2. Comparison of SBD conformations in Sse1 and DnaK.
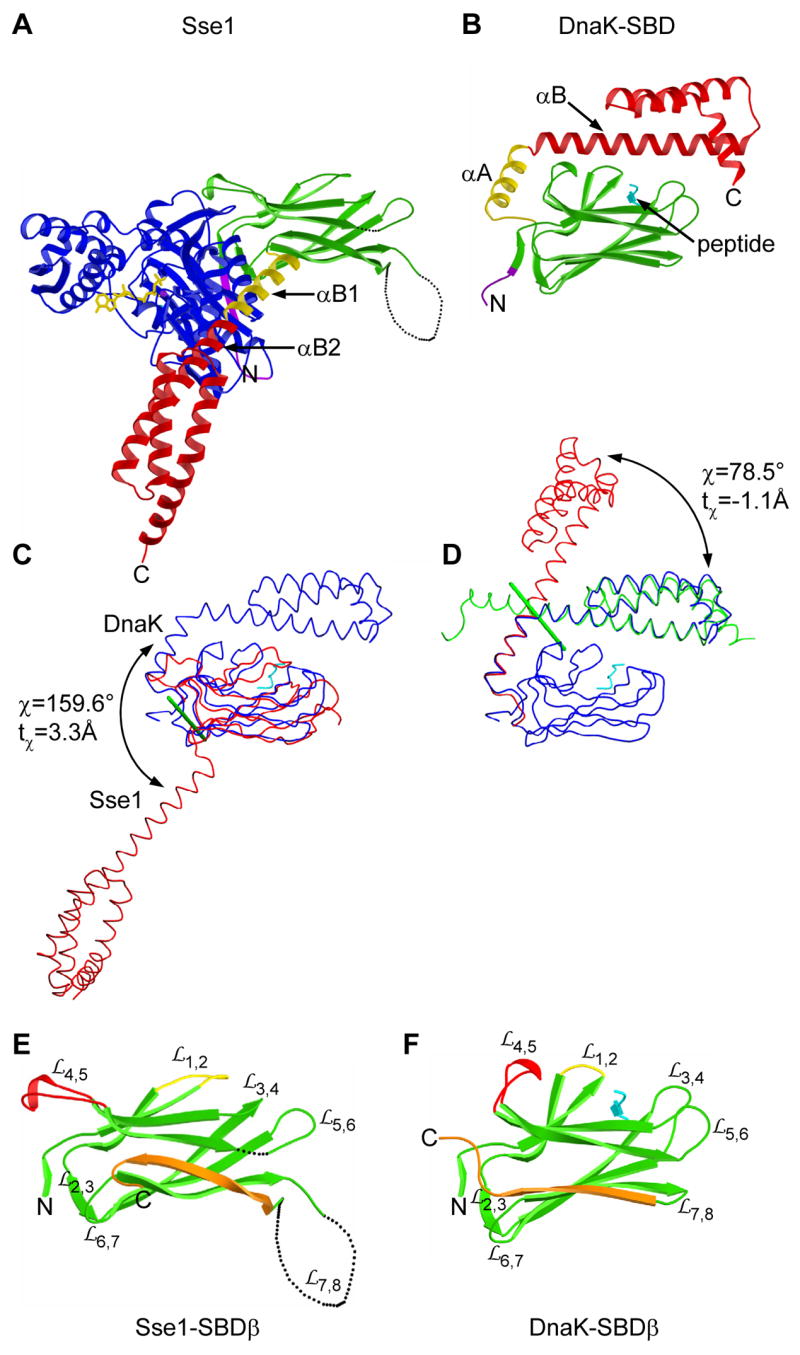
(A) Ribbon diagram of Sse1 drawn and colored as in 1B except that helix αB1 is now yellow. Orientation is after superposition of its SBDβ onto that of DnaK as in 2B. (B) Ribbon diagram of the DnaK SBD complexed with a heptapeptide (cyan). Linker and SBD coloring is as in A. (C) Superposition of SBD from Sse1 (red) with that of DnaK (blue) complexed with a heptapeptide (cyan) as based on SBDβ Cαs. Cα traces are oriented as in A and B. Rotation of α-subdomains is about the green rod. (D) Superposition of the DnaK SBD (blue) with SBDα from Sse1 as based on helix αA/αB1 (red) or helices αB/αC/αD/αE (green). Rotation is about the green rod. (E) Ribbon diagram of Sse1 SBDβ oriented as in A. Conformationally distinct features are highlighted: (yellow), (red), and β8 through β-α connector (orange). (F) Ribbon diagram of DnaK SBDβ oriented and colored as in E, adding the heptapeptide (cyan).
The α-helical lid domains (αB-αE) from Sse1 and DnaK have identical topology and are structurally very similar (1.45Å Cα r.m.s.d.). The two SBDβs are also topologically the same, and strands β1′-β7 have nearly identical extents (Fig. S2) and similar conformations (1.82Å Cα r.m.s.d.). Strand β8 is lengthened in Sse1 and its conformation is twisted to take part in both β-sheets. Thereby, SBDβ in Sse1 is barrel-like compared with the β-sandwich of DnaK (Fig. 2, E and F).
Loops and of SBDβ, located at the NBD interface in Sse1, are similar in Sse1 and DnaK; but most of the other loops differ substantially. In particular, the site of peptide binding in DnaK is empty in Sse1 with its loops and pressed against one another; and buttressing loop is flipped out toward the NBD in Sse1, while retaining its local tip conformation. Nevertheless, features used in peptide binding to DnaK have analogs in Sse1 (Text S4A).
Comparison of Sse1 to NBDs of Hsp70s
The NBD of Sse1(ATP) is superficially similar to other NBDs, and subdomain topologies are virtually identical. The relative orientations of subdomains differ substantially, however (Text S4B). Compared with the NBD of human Hsp70 (hHsp70) in the ADP state (Sriram et al., 1997), lobe I is rotated markedly relative to lobe II in Sse1, into the plane from the canonical view (Fig. 3A) and counterclockwise when viewed from above this into the IB/IIB tips (Fig. 3B). This distinction arises primarily from differences at the IA-IIA interface, whereas IIB mainly rides along with IIA and IB rides along with IA. By contrast hHsp70(ADP) and bHsc70(ADP) are virtually identical, and all known nucleotide complexes of Hsp70s have this conformation (Flaherty et al., 1990; Johnson and McKay, 1999). Only nucleotide-free states differ significantly, and these simply have subdomain IIB rotated open by 13–16° (Harrison et al., 1997; Sondermann et al., 2001; Jiang et al., 2005). Structure within Sse1 subdomains is quite similar to that within hHsp70 (Cα r.m.s.d.s of 1.08, 1.14, 1.35, and 0.76Å for IA, IB, IIA and IIB, respectively).
Figure 3. Comparison of NBD conformations in Sse1 and Hsp70s.
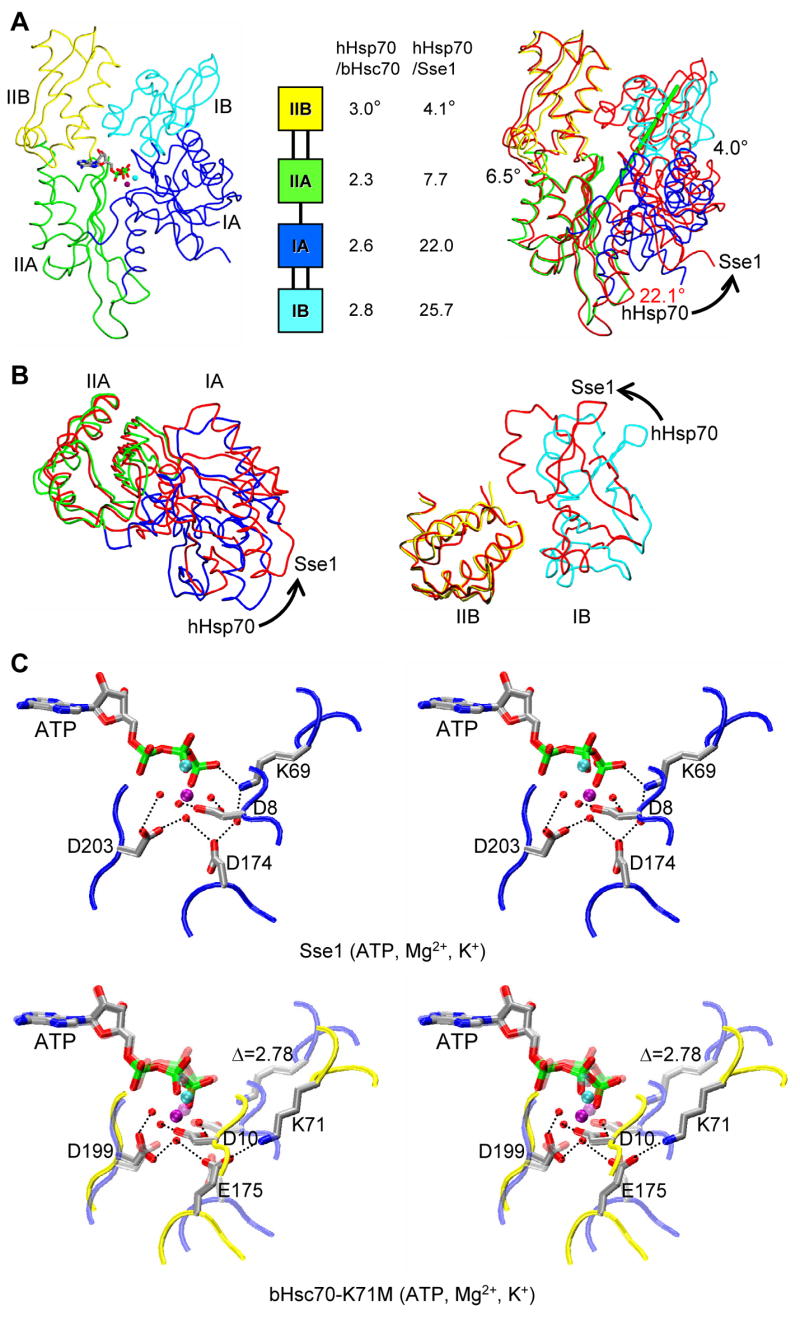
(A) Cα traces of Sse1 (left) and Sse1 superimposed onto hHsp70 (right, based on Cαs in subdomain IIA), and relative domain orientations (middle, based on superpositions of adenine-ribose-Pα atoms). NBDs are oriented in the canonical, front-face view. Sse1 subdomains (left) and hHsp70 subdomains (right) are colored: IA (blue), IB (cyan), IIA (green) and IIB (yellow). Sse1 (right) is red. ATP is drawn and colored as in 3C. Rotation relating IA and IIA is about the green rod (right). (B) Superpositions of Sse1 and hHsp70 NBD subdomains viewed from above A, rotated 90° around the horizontal. Coloring is as in A (right). Superpositions of both IA/IIA (left) and IB/IIB (right) are based on Cαs in lobe II subdomains. (C) Stereodrawing of ATP catalytic sites in Sse1 (above) and bHsc70 (below). The view is changed slightly from 3A. Adenine-ribose-Pα atoms are superimposed. The image for bHsc70(ATP) is a composite from bHsc70(ATP, K71M) (PDB 1KAX) and side-chain atoms of Lys71 translated into Cα superposition from bHsc70(ATP, D199N) (PDB 1NGF); a subdued version of Sse1 is superimposed. Side chains are shown for four residues shown for bHsc70 to be most critical for ATPase function and for counterparts in Sse1. Coloring for ATP, ions and side chains has the pattern: carbon (grey), nitrogen (blue), oxygen (red), phosphorous (green), Mg2+ (purple) and K+ (cyan); backbone traces are bHsc70 (yellow) and Sse1 (red). Hydrogen bonds are drawn as black dotted lines.
Comparison of ATP interactions in Sse1 and Hsp70s
ATP is bound to Sse1 much as nucleotides are bound to bHsc70 and hHsp70 (Text S4C). Contacts with the adenine-ribose-α-phosphate moiety, all from lobe II, are essentially identical. Interactions at the β- and γ-phosphate positions (Pβ, Pγ) are similar but necessarily differ since ATP was hydrolyzed to the products ADP and phosphate (Pi) in bHsc70 and hHsp70 (Flaherty et al., 1990; Sriram et al., 1997). We focus our comparison here on the ATP-Mg2+-K+ complex with the NBD of K71M bHsc70 (O’Brien et al., 1996), which is proposed to represent the “precatalytic” state poised for in-line nucleophilic attack on Pγ (Flaherty et al., 1994; Johnson and McKay, 1999). ATP in Sse1 has this same conformation, it coordinates Mg2+ in the same bidentate manner with positioning by Asp 8 and Asp203 as by 10 and 199 in bHsc70, and it binds K+ as at bHsc70 site #1 (Fig. 3C). Lys69 and Asp174, counterparts of key catalytic residues in Hsp70s, are displaced from Hsc70-like positions due to the altered orientation for lobe I in Sse1(ATP), and accordingly ATP remains intact.
Sse1(ATP) is stabilized by conformation-specifying interactions at interfaces between NBD lobes (Text S4D). The front-face cleft (canonical view, Fig. 3A) is widened in Sse1(ATP) relative to hHsp70(ADP) and that on the back side is narrowed. Two hydrogen-bonded triplets of residues unique to Hsp110s participate: Gln368 mediates one between IA and IIA on the front side (Fig. S3A), and Arg235 mediates another between IB and IIB on the back side (Fig. S3B). These triplets specifically affect the placement of catalytic residues Asp174 and Lys71, respectively. Other hydrogen-bonded interactions also stabilize subdomain dispositions at the catalytic center relative to ATP (Figs. S3C and D). Altogether, Sse1(ATP) has 16 inter-lobe hydrogen bonds as compared with 11 in hHsp70(ADP), of which 4 are in common. We find by modeling that NBD of hHsp70 should be able to adopt the Sse1(ATP) conformation, and vice versa provided that SBD is displaced from engagement.
Interfacial Contacts and Mutational Analysis
NBD surfaces that become buried in interfaces within Sse1 are exceptionally well conserved with Hsp70s, and notably so for NBD-linker, NBD-SBDβ and NBD-SBDα contacts (Fig. 4, A–C vs. D–F). In contrast, NBD sequence conservation between Sse1 and classic Hsp70s is relatively modest overall (28–34% identity vs. 45–88% among Hsp70s). Segments of the linker and SBD from the complementary surface of interaction are also conservative in sequence, and an examination of the particular contacts in Sse1 (Fig. 4, G–I) reveals interactions that could have counterparts in Hsp70 proteins.
Figure 4. Characteristics of interdomain interfaces.


(A, B, C) Mapping of NBD sequence conservation onto the NBD surface from Sse1. Three orthogonal views are presented: (A) front view as in 1B, (B) side view as in 1C, and (C) back view, rotated 180° from A about the vertical axis. Sequence similarity is quantified from across the Hsp70 superfamily and mapped by ConSurf (http://consurf.tau.ac.il/) into colors of increasing purple intensity for conservation and increasing aqua intensity for variability (key under C), relative to average conservation (white). Cα traces are overlayed from SBD components color coded as linker (purple), SBDβ (green) and SBDα (red). (D, E, F) Mapping of NBD surfaces as buried into linker and SBD interfaces. Views are as in A, B and C, respectively. Surface imprints are shown colored: SBDα (red), SBDβ (green), interdomain linker (purple), and apposing protomer (cyan). Cα traces are overlayed as in A, B and C, adding the apposing protomer in cyan for D. (G, H, I) Stereodrawings of interdomain interfaces featuring residues targeted for mutagenesis. Main-chain ribbons are colored as in 1 and side-chain atoms are colored as in 3C. (G) NBD-linker interface. (H) NBD-SBDβ interface. (I) NBD-SBDα interface.
The evolutionary relationship between Hsp70s and Hsp110s (Fig. 1A) makes it plausible to consider structural features in one as vestiges from the other, despite functional divergence. Especially given the high conservation at contacts between domains, Hsp70s may well assume Sse1(ATP)-like states. We therefore entertain the hypothesis that, during the chaperone cycle, classic Hsp70s make ATP-induced conformational changes to engage interfacial contacts like those found in Sse1(ATP). To test this hypothesis and certain other conformational features, we constructed a battery of structure-based mutations in Sse1 and at homologous sites in two Hsp70s, yeast cytosolic Ssa1 and E. coli DnaK.
Mutant variants were tested for heat-stress responses by expression in appropriate null backgrounds for the respective yeast and bacterial hosts. Since Ssa1-4 exhibit redundancy and it is lethal to delete all four, we used a temperature-sensitive yeast strain, ssa1ts ssa2 ssa3 ssa4 (Becker et al., 1996), for our Ssa1 tests. Cell growths at heat-shock temperatures were compared with those at a permissive temperature (30°C) (Figs. 5A and S4–S6), and phenotypic responses were scored for colony morphology and culture viability (Fig. 5C).
Figure 5. Mutational analysis.
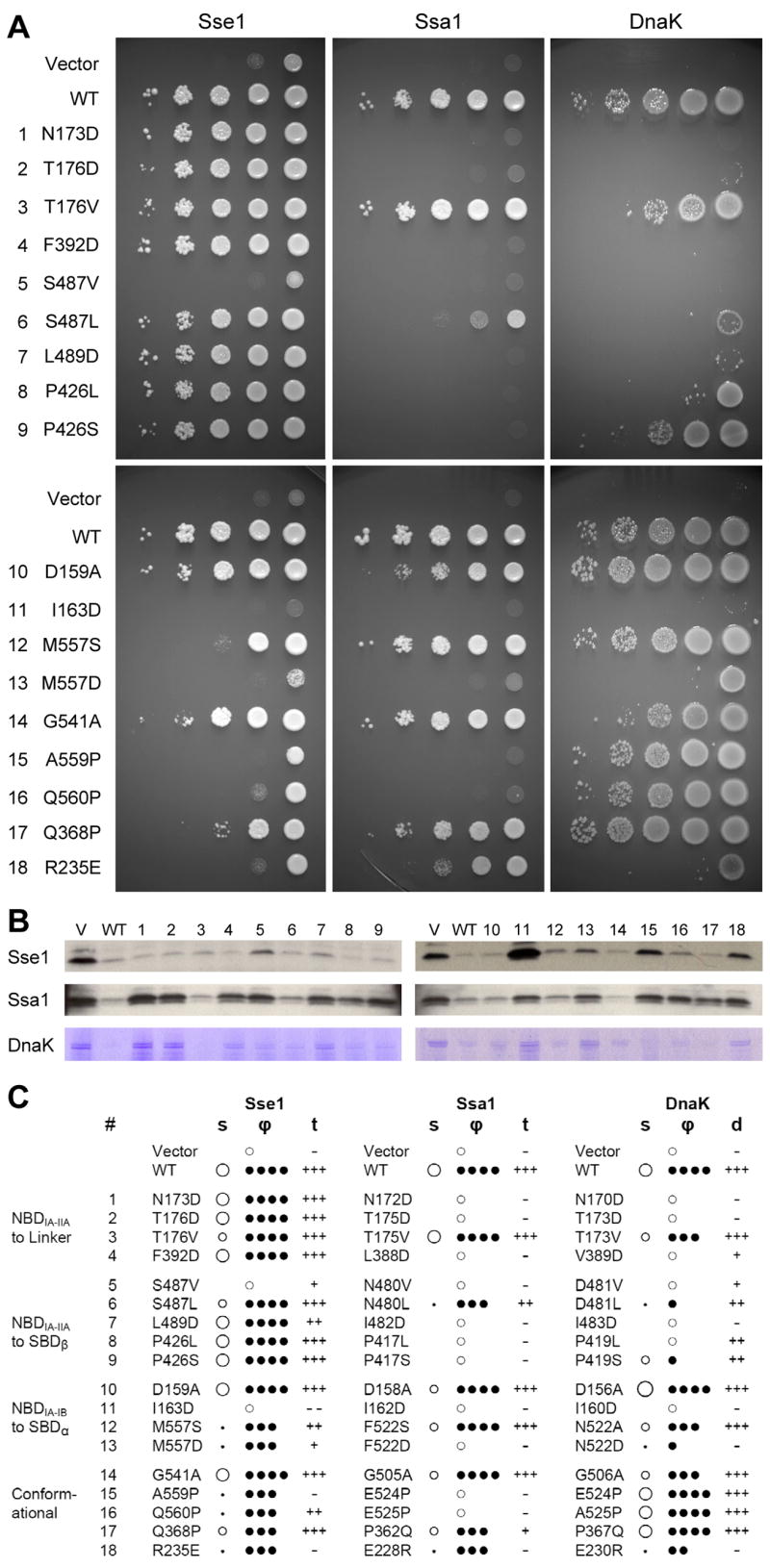
(A) Growth tests of mutant variants of Sse1, Ssa1 and DnaK. Expression plasmids mutated as indicated for Sse1, or homologously for Ssa1 or DnaK, were tested in appropriate null backgrounds. For each test, 10-fold serial dilutions (10−1 to 10−5) from overnight cultures were spotted (right to left) onto agar plates containing appropriate growth media. Plates were incubated at heat-stress temperatures of 40°C, 37°C, and 40°C, respectively. (B) Tests of specific heat-shock chaperone activities. Sse1 and Ssa1 panels test translocation activity, showing accumulation of unprocessed ppαF by Western blot analysis. DnaK panels test folding activity, showing residual aggregation of RNA polymerase β andβ ′ chains after heat shock and return to permissive temperature. Mutant variants (identified by numbers cited in A) are compared to vector (V) and wild type (WT) controls. (C). Phenotypic analyses of heat-stress responses. Mutant variants were analyzed microscopically for colony morphology (s) and culture viability (ϕ) in growth response at heat-stress temperatures. Colony morphology (s) is scored based primarily on size as larger than wild type (○), wild-type size (○), small (●), point-like (·) or not visible (blank). Culture viability (ϕ) is scored according to the fractions of colonies formed at 40°C or 37°C in comparison to those at 30°C: ϕ=100% (● ● ● ●), 10% ≤ϕ<100% (● ● ●),1%≤ϕ<10% (● ●),0%<ϕ<1% (●), 0% (●). Translocation activity (t) and disaggregation activity (d) are scored as wild type (+++), impaired (++), seriously impaired (+), vector control level (−), impaired beyond control (− −). Each phenotypic designation is based on at least three replicates of experiments such as those shown in A and B, together with associated controls.
Each mutation was also tested in a specific assay for heat-shock chaperone activity. We used accumulation of prepro-α factor (ppαF) to evaluate the roles of both Ssa1 (Becker et al., 1996) and Sse1 (Shaner et al., 2005) in protein translocation into the ER, and we used aggregation of RNA polymerase subunits β and β′ to assess the role of DnaK in protein folding (Hesterkamp and Bukau, 1998). Results from these assays (Figs. 5B and S7) were scored for phenotypic responses (Fig. 5C). Specific chaperone deficiencies correlate well with growth defects, as expected from the essentiality of the tested processes for cellular vitality and replication, although there were a few notable distinctions (Fig. 5C).
The mutational tests provide overwhelming support for Sse1(ATP)-like interfaces being essential in Hsp70 chaperone activity. Mutations at 8 of 9 tested contact sites, including 10 of 13 specific mutations, had severe phenotypes in both Ssa1 and DnaK. Only 1 of 26 mutants had no observable defect. Some heat-shock sensitive mutants also had negative effects even at the permissive temperature (Fig. S4). For Sse1, only the NBD-SBDα interfaces proved comparably sensitive in these tests; evidently, Sse1 function does not require high fidelity at its NBD-linker and NBD-SBDβ interfaces. Tests of specific conformational features have varying results, showing some to be critical and others not. Nothing tested at the Sse1 dimer interface proved sensitive.
Characteristics of the individual interfaces are described below in the context of our mutational tests and functional analyses by others.
NBD-linker contacts
NBD and SBD domains are connected by a linker segment (residues 386-396 in Sse1) that is conservative at its C-terminal end (sequence PFKFED in Sse1; DLLLLD typical for Hsp70s) but variable at its N-terminus (Fig. S2). The linker is flexible in the ADP state (trypsin cleaves both Sse1 and DnaK apart at exactly homologous linker residues (Buchberger et al., 1995; Text S1). Here in the ATP state, the linker (1) extends C-terminally into SBD β1′ (as in the ‘out’ conformer of DnaK SBD (Zhu et al., 1996)), adding parallel to the exposed edge of NBD β14 (Fig. S1B) to form an interdomain β-sheet (↓β12-↑β13-↓β14-↓1/β1′-↑β2′). The variable N-terminal end of the linker, which contains the Arg388 proteolysis site (Text S1), makes little contact and is disordered in protomer A.
Accommodation of the linker strand and its connection back to NBD helix αO requires that NBD subdomains IA-IIA be rotated and SBD helix αA/αB1 be displaced as in Sse1. Phe392 and Phe394 on the linker then make hydrophobic contacts with conserved pockets from IA/IIA residues (Ile184-Phe378-Leu221 and Thr176-Val180-Leu221, respectively), and Asp396 hydrogen bonds to Thr176 and to Asn173 supported by Arg154 (Figs. 4G and S2).
Chaperone functions of Ssa1 and DnaK are abolished for three of our four mutations at this interface (excepting only T176V homologs). Similarly, DnaK linker mutants VLLL389-392AAAA and LL390-391DD are unresponsive to ATP (Laufen et al., 1999) and DnaK mutations D389A/R (Asp396 in Sse1) are highly disruptive as well (Vogel et al., 2006b). Moreover NMR chemical shift (Revington et al., 2005; Swain et al., 2007) and amide hydrogen exchange (Rist et al., 2006) results from two DnaKs are also consistent with involvement of an Sse1-like interface in inter-domain communication. Our NBD-linker mutations have little impact on Sse1 itself, however (Fig. 5), suggesting that this interface is not strictly essential for Sse1 function or perhaps too robust to be disrupted by a single mutation.
NBD-SBDβ contacts
SBDβ binds to the backside of NBD at the IA/IIA interface. As for the NBD-linker interface, contacts between NBD and SBDβ require displacement of SBD αA/αB1 and closure of the NBD IA-IIA interface relative to prototypic Hsp70 domains. From the NBD side, direct contacts are made by strand β11 and helix αF from IA and by strand β14 from IIA. From the SBDβ side, three loops ( and ) and both termini (N-terminal β1′ and the rearranged β-α connector) contribute. Loop takes part only via the linker, making backbone hydrogen bonds and van der Waals contacts from Phe425 to Asp396. Loop buttresses this interface without making direct contacts, but is heavily involved.
Loop has the same conformation in Sse1 as in DnaK, whereby Ser 487 Oγ is in position to hydrogen bond with Ile171 N and the side chain of Leu489 is fitted into a hydrophobic pocket formed by aliphatic portions of NBD helix αF residues Glu151, Arg154 and Tyr155 (Fig. 4H). Nearby, Phe540 and Leu542 from the β-α connector segment also associate with αF. Hydrophobic contacts between Ile397 and Val223 further brace SBD β1′ against IIA strand β14.
All five mutations at this interface led to severe heat-stress phenotypes in each of Ssa1 and DnaK (Fig. 5). Notably, conserved hydrophobic Ile483 is surface-exposed in DnaK SBD (Zhu et al., 1996), and yet neither I483D DnaK nor I482D Ssa1 showed any chaperone activity. These striking results, together with those at the associated NBD-linker interface, strongly implicate such interfaces in Hsp70 function. Moreover, the Sse1 model is consistent with the adverse impact on interdomain coupling found from mutations YND145,147,148AAA (Gässler et al., 1998) and R151A/K (Vogel et al., 2006a) in DnaK (Trp148,Thr150,Glu151,Arg154 in Sse1). Sse1 function was severely disrupted by S487V, but not otherwise. As at NBD-linker contacts, Sse1 is relatively insensitive here.
NBD-SBDα contacts
The straightened-out fusion of SBD helices αB1 and αB2, as displaced in Sse1, packs between NBD subdomains IA and IB. Fully conserved Asp159 hydrogen bonds to the backbone amide of Leu542, but two sets of hydrophobic interactions predominate in this interface: (1) Ile163 on IA meshes with a cluster of Leu547, Leu550 and Ile551 from αB1, and (2) Met557 at the start of αB2 fits into a pocket formed by Arg47, Phe106 and Phe113 from IB and Met121 from IA (Fig. 4I). The first set is absolutely conserved as hydrophobic; all superfamily Hsp70s have Ile for Ile163 and all classic Hsp70s have Ile/Met/Val for Leu547/Leu550/Ile551. These latter αA/αB1 side chains are buried into the β-subdomain interface in the structure of DnaK SBD (Zhu et al., 1996) where they interact with Leu397. The second set of hydrophobic interactions is also preserved in character across the superfamily, although less exactly so, and with DnaK as an exception.
We tested this interface with mutations at the Ile163 and Met557 positions, which are both free and open in isolated Hsp70 subunits, and avoided the conserved hydrophobic partners of Ile163 because their engagements with SBDβ would muddle interpretation. The NBD-SBDα interface proves highly sensitive to mutation for Sse1 as well as for Ssa1 and DnaK (Fig. 5), although D159A and homologs had little effect. Mutations homologous with I163D completely abolish chaperone activity. Tests at the Met557 site also affirm the significance of this interface.
DnaK has Asn522 in place of Met557 and replacements in the Met557 pocket as well, but N522D is severely impaired nevertheless. Trp102 is partially exposed (26%) nearby to this site in DnaK, and, upon Sse1-like superposition of SBD αA, conserved Leu507 and Met515 are each within 7Å of Trp102 indole. This is consistent with the shifted tryptophan fluorescence that accompanies ATP binding to DnaK (Buchberger et al., 1995; Moro et al., 2003).
Sse1 function depends on integrity of this NBD-SBD interface. The pattern of defects from mutations here in Sse1 is strikingly similar to those for Ssa1 and DnaK (Fig. 5), suggesting that details of the molecular interaction may also be similar in all three. Notably, Sse1 I163D accumulates ppαF even more abundantly than the vector control (Figs. 5B and C).
Conformational tests
We also tested conformational features that distinguish Sse1(ATP) from known Hsp70 structures. The fusion of SBD αA and αB into one straightened helix is such a distinction. By modeling, the αA-αB kink in DnaK can accommodate proline substitutions for Glu524 and Ala525 but, of course, a straight helix cannot. Proline residues at DnaK-like kink positions lead to severely disrupted function in Sse1 and Ssa1, consistent with required access to straight helices in these chaperones. By contrast, there is no impairment of function for DnaK variants E524P or A525P (Fig. 5). Since mitochondrial and bacterial Hsp70s are close relatives, we thought they might behave similarly and, indeed, yeast mitochondrial Hsp70 Ssc1 does tolerate proline replacements (Fig. S5).
Another distinction lies in the β-α connector where Gly506 in DnaK has a conformation restricted to glycine (ϕ=107°, ψ=−152°) whereas analogous Gly541 in Sse1 has a generally accessible conformation; however, a glycine-specific state does not appear to be essential for function. G506A and the analogous mutations had only slight effects in heat-shock tests (Figs. 5 and S5) although overexpression of this DnaK mutant was lethal even at the permissive temperature (Fig. S4).
The hydrogen-bonded triplets at NBD IB-IIB and IA-IIA interfaces in Sse1 (Figs. S3A and S3B), which distinguish Sse1(ATP) from NBD(ADP) conformations of Hsp70s, were also tested by mutation. The inversions of R235E in Sse1 and E→R in Ssa1(228) and DnaK(230) each severely disrupted function. Inversions Q368P in Sse1 and P362Q in Ssa1 also had an appreciable impact although P367Q in DnaK did not (Fig. 5). This sensitivity is consistent with a role for an Sse1(ATP)-like NBD conformation in Hsp70 activity. It is also consistent with the Sse1(ATP) state being required for NEF activity (Raviol et al., 2006b; Shaner et al., 2006).
Finally, we also tested function of acidic loop in Sse1 with deletions. We found no detectable effect from eliminating the disordered segment (Fig. S8). This contrasts with reported loop-deletion impairments in hHsp110 (Oh et al., 1999), but that deletion wrongly extended through all of Sse1 β8 by our alignment (Fig. S2). Previous experiments had already shown that C-terminal deletion of Sse1 by Δ44, to residue 649 within terminal helix αE, is fully tolerated in yeast (Shaner et al., 2004). Thus, neither the Hsp110 insertion nor the Hsp110 extension (Fig. 1A) is essential for the Sse1 heat-stress response.
Dimer contacts
The Sse1 dimer features tip-to-tip contacts between NBDs and contact of each α-helical lid domain with its partner NBD. At the tip-tip interface, Asp294 of one NBD hydrogen bonds to Asn260′ and Arg266′ on its partner, NBD′. The α-helical lid domain interacts through an electronegative face with the conserved electropositive front face of NBD′. Specific interactions include the Glu586-Glu587 pair with Asn367′ and Arg34′ and the Glu609-Glu610 pair with Arg346′. All of these were tested with prospectively disruptive mutations, but none presented an obvious phenotype (Fig. S8). Sse1 homodimers may not be functionally important; indeed, effectively all Sse1 in yeast extracts is in heterodimeric associations with Ssa or Ssb (Shaner et al., 2005; Yam et al., 2005).
DISCUSSION
Implications for Hsp70 Chaperone Activity
Although the evolutionary relationship is somewhat cryptic in sequences, an Hsp70 superfamily that embraces both Hsp110s and classic Hsp70s is clear from structure. Vestiges of this relatedness are found in striking sequence conservation at interfaces within Sse1(ATP), despite limited sequence similarity overall and radically different conformations. We hypothesized that although Sse1(ATP) does not itself undergo an Hsp70 conformational cycle, it may reflect an Hsp70(ATP) state. Our mutational analyses support this hypothesis that classic Hsp70s make conformational changes during the chaperone cycle to engage inter-domain contacts like those found in Sse1(ATP). It emerges that, upon ATP binding, the two NBD lobes rotate against one another by ~25° to expose binding sites for the interdomain linker and SBDβ. We envision a conformational equilibrium within SBD such that the α- and β-subdomains dissociate to expose conserved binding surfaces that had matched one another within SBD but can also match complementary surfaces on NBD. The proximal end of SBDβ inserts between NBD lobes as they close down to sandwich a conserved linker segment within an inter-domain β-sheet, and relocated SBD helix αA/αB1 binds alongside NBD between lobe I subdomains. Disengagement of the α-helical lid from SBDβ favors disassociation of polypeptide substrates (Pellecchia et al., 2000) and, in turn, we expect substrate-free SBDβ to have enhanced affinity for NBD.
ATP is not as stably maintained in classic Hsp70s as in Sse1. Nevertheless, steady-state complexes of ATP with bHsc70 could be analyzed by solution small-angle x-ray scattering (SAXS) (Wilbanks et al., 1995). The SAXS-deduced elongated shape of bHsc70(ATP) is consistent with the atomic structure of Sse1(ATP). Notably, the pair-distribution function and radius-of-gyration simulated from Sse1(ATP) compare well with those measured from bHsc70(ATP) (Text S5, Fig. S9). Moreover, from NMR residual dipolar couplings, the NBD of bHsc70 in solution shows subdomain rotations (Zhang and Zuiderweg, 2004) about similar axes as for Sse1 (Fig. 3, A and B). On the other hand, NBD-SBD interactions in a truncated and mutated bHsc70 (bHsc70ΔC, residues 1-554) are very different (Jiang et al., 2005). It seems unlikely, however, that this conformation of bHsc70ΔC could support allosteric communication: its interface between domains is relatively spare and poorly complementary (Table S2), it is nucleotide-free, its LLLL-linker segment is solvent-exposed and poorly ordered, the ATP-sensitive Trp102(DnaK) site is completely open, its NBD interacts exclusively with SBDα whereas NBD-SBDβ alone shows allosteric coupling (Pellecchia et al., 2000), its interface includes only one (Hsc70 N174) of our eight sites of severely deleterious Hsp70 mutation, and it is inconsistent with recent NMR data from DnaK(ATP) (Swain et al., 2007).
ATP binding to Hsp70s promotes exchange of polypeptide substrates, and substrate binding stimulates ATP hydrolysis. What is the mechanistic basis for this allosteric coupling? The Sse1 structure, taken together with other studies, provides insight into linkage between the two sites. These sites are far apart (43.7Å from ATP Pγ in Sse1 to Leu0 Cα in the NRLLLTG peptide as superimposed from DnaK, Fig. 2), but the path between them is direct (Fig. 6A). An intricate network of hydrogen bonds links the ATP in Sse1 to one end of the β-subdomain, which then connects through β-strands to the peptide site at the other end (Fig. 6B). First, Pγ is coordinated through magnesium hydration to catalytically crucial Asp174, and then flanking residues hydrogen bond through Thr176 and Arg154-buttressed Asn173 to Asp396 in linker-strand β1′ and through the backbone of Ile171 to Ser487. All of these are highly sensitive interactions from our mutational analysis of Ssa1 and DnaK. In turn, β1′ hydrogen bonds to strand β2′; specifically, the backbone at Asp396 makes dual hydrogen bonds to β2′ at position 425.
Figure 6. Mechanistic implications.

(A) Structural linkage between binding sites for ATP and polypeptide substrates. Outlines of structural subdomains from Sse1, oriented as in 1C, are superimposed with selected atomic details. Outlines are colored: NBD (blue), linker (purple), SBD β-subdomain (green), SBD α-subdomain (red), polypeptide substrate (cyan); sidechains and ATP are colored as in 3C. (B) Hydrogen-bonding details of the SBDβ-linker-NBD interface. The view is as in 6A but rotated clockwise by ~40°. Cα atoms are black; other coloring is as in 6A. (C) Homology model of DnaK(ATP) based on the Sse1 template. The ribbon diagram is oriented and colored as for Sse1 in 1B. Trp102 is colored orange. (D) Schematic for the Hsp70 chaperone cycle in the eukaryotic cytosol. Component outlines are based on structures discussed in the text. Coloring is as in 1A and 6A, but with polypeptide substrates in black and Hsp40 and Hsp110 components in orange. Hsp110 is drawn in the Sse1(ATP) conformation. It is not clear where Hsp110 preferentially leaves the cycle as it may participate at multiple stages.
SBDβ loop connects through β2 and β3 to peptide-binding loops and , respectively. Loop has strictly conserved Pro426, which has a cis peptide bond in Sse1 as in DnaK, at this juncture. Strains of Ssc1, Ssa1 and DnaK selected for chaperone deficiencies all have mutations at this proline site (Becker et al., 1996; Burkholder et al., 1996; Kang et al., 1990), and those in both DnaK and Ssc1 have demonstrated peptide-binding defects (Burkholder et al., 1996; Liu et al., 2001). Moreover, DnaK1-507, truncated before the α-subdomain, retains coupling between ATP and peptide sites (Pellecchia et al., 2000). Thus, an Sse1-like model for allosteric communication has support from both structure and genetics.
Our mutational data show that whereas straightening of the SBD αA-αB junction is required for Sse1 and Ssa1, this is not so for either DnaK (Fig. 5) or Ssc1 (Fig. S5). Modeling of Ssa1(ATP) is obvious based on Sse1(ATP), but is this possible for DnaK(ATP) with its kinked α-subdomain? In fact, we can readily make such a model of intact DnaK(ATP), adjusting the αA-αB kink azimuthally by ~25° to optimize α-helical lid contacts while keeping the same inter-helix angle. Conserved front-face NBD elements are then engaged as in NEF complexes or the Sse1 dimer (Fig. 4D), but this time within a monomer (Fig 6C). Moreover, Trp102 is shielded in keeping with ATP-induced fluorescence shifts.
Implications for Functional Roles of Hsp40 and Hsp110
Our results also bear on the nature of Hsp40 and Hsp110 interactions with Hsp70s in the chaperone cycle. Hsp40s are well established as co-chaperones, and functional interaction of DnaJ with DnaK requires that NDB and SBD domains be linked together in the presence of ATP (Gässler et al., 1998). We find that Hsp70 residues identified by mutational analyses as essential for this interaction also map close to one another on the surface of Sse1(ATP). These include Arg170 from NBD, Lys393 and Glu395 exposed from the linker, and Pro426 from SBD (DnaK R167H is a suppressor of DnaJ D35N (Suh et al., 1998); DnaJ does not stimulate ATP hydrolysis in DnaK VLLL389-392AAAA (Laufen et al., 1999); and Hsp40-stimulated ATPase activity is severely defective in Ssc1 P442S (Liu et al., 2001)). This site may also be relevant to Hsp40 binding to Hsp110s; the yeast Hsp40 Sis1, although not Ydj1, stimulates ATP hydrolysis by Sse1 (Raviol et al., 2006a).
Hsp110s appear to be the primary NEF co-chaperones for cytosolic Hsp70s in eukaryotes (Dragovic et al., 2006; Raviol et al., 2006b), and it seems that they must be bound to ATP to associate with their Hsp70 targets (Shaner et al., 2006). Thus, this structure of Sse1(ATP) likely describes the active NEF species; indeed, given the high concentration of cellular ATP, this may be the main or only functional state of Sse1. Our mutations indicate that an intact NBD-SBDα interface is essential for function, but that Sse1 does not require high fidelity at its NBD interfaces with the linker and SBDβ.
NEF-Hsp70 complexes feature NEF contacts with the Hsp70 front face (Harrison et al., 1997; Shomura et al., 2005; Sondermann et al., 2001), which is highly conserved (Fig. 4A). The helical-lid domain from a partner protomer interacts with much of this same surface in the Sse1 homodimer (Fig. 4D), and we contemplate the possibility that Hsp110-Hsp70 heterodimers interact through similarly extended α-helical lids. Consistent with this hypothesis, the 1:1 Sse1-Ssa1/2 and Sse1-Ssb1/2 complexes seen in yeast extracts (Shaner et al., 2005; Yam et al., 2005) are mutually exclusive with Sse1 homodimers in vitro (Shaner et al., 2005), and helix-breaking mutations at the αB1-αB2 juncture severely impair function for both Sse1 and Ssa1 (Fig. 5). We do not expect Hsp110-Hsp70 heterodimers to be exactly like the Sse1 homodimers, however, since none of our dimer interface mutations showed a phenotype (Fig. S8).
Hsp110s can also bind denatured polypeptides, acting as ‘holdases’ (Oh et al., 1999), and a role in directing substrates to the Hsp70 chaperone has been suggested (Dragovic et al., 2006), much as by Hsp40s. The Sse1 structure provides a plausible explanation for peptide binding, but we have no direct evidence of the kind now available from structural studies on Hsp40s (Li et al., 2006).
Hsp70 Chaperone Cycle
We can use our results to draw a refined picture of the cycle of Hsp70 chaperone activity (Fig. 6D). Hsp70s divide into three sequence subfamilies, each served by distinctive NEFs. Cytosolic Hsp70s in eukaryotes have Hsp110s as the most effective NEFs, although others also function (Dragovic et al., 2006; Raviol et al., 2006b). ER-localized Hsp70s use another non-classic Hsp70 as NEF, as characterized for Kar2 by Lhs1 (Steel et al., 2004). Mitochondrial and prokaryotic Hsp70s form the third subfamily, and these typically use GrpE-like NEFs. Hsp40s also divide into families, but these are not clearly separated into cellular compartments. We base our diagram for the Hsp110-Hsp40-Hsp70 system on NBD and SBD domain structures for ADP and apo states of Hsp70, an Sse1-based model for Hsp70(ATP), Sse1(ATP) for the Hsp110 NEF and Sis1(peptide) and J domain for the Hsp40.
EXPERIMENTAL PROCEDURES
Protein expression and purification
The yeast SSE1 gene was PCR-amplified from genomic S. cerevisiae DNA and various constructs were cloned into pSMT3 vectors for expression in E.coli as Smt3 fusion proteins with N-terminal His6-tags on Smt3. Following initial affinity purification, Smt3 was removed using Ulp1 protease. Proteins were further purified by size-exclusion and ion-exchange chromatographies. The NΔ1-CΔ34 truncated construct, Sse1(2-359), crystallized well and selenomethionyl (SeMet) Sse1(2-359) was produced for the structural analysis. Full SeMet incorporation was verified by mass spectrometry.
Crystallization, data collection and model building
SeMet Sse1(2-659) crystals were grown from 8–10% PEG6000 at pH6.5 in the presence of ATP, Mg2+ and K+. Crystals cryoprotected by addition of glycerol diffracted to 2.4Å Bragg spacings when frozen, and diffraction data were measured at NSLS beamline X4A (Table 1). Phases were evaluated from multiwavelength anomalous diffraction (MAD) measurements at three wavelengths (3Å data set), and the structure was developed and refined at 2.4Å resolution from a second data set.
Table 1.
Diffraction data and refinement
| Crystal1 | Crystal2
|
|||
|---|---|---|---|---|
| Seedge | Sepeak | Seremote | ||
| Crystal parameters | ||||
| Space group | C2 | |||
| a (Å) | 218.1 | |||
| b (Å) | 125.3 | |||
| c (Å) | 74.7 | |||
| β (°) | 98.7 | |||
| Za#/Solvent content (%) | 2/64 | |||
| Data collection | ||||
| Wavelength (Å) | 0.97923 | 0.97948 | 0.97917 | 0.96791 |
| Bragg spacings (Å) | 30–2.4 (2.49–2.4) | 20–3.0 | 20–3.0 | 20–3.0 |
| Total reflections | 1091762 | 799458 | 799638 | 800190 |
| Unique reflections | 75686 | 38936 | 38943 | 38988 |
| Rmerge (%)* | 5.9 (28.0) | 6.4 (22.3) | 6.4 (22.8) | 7.5 (28.4) |
| Average I/σ | 27.4 (4.0) | 22.2 (6.8) | 22.0 (6.7) | 19.6 (5.5) |
| Completeness (%) | 99.7 (99.7) | 99.7 (100) | 99.5 (100) | 99.8 (100) |
| Redundancy | 3.6 (3.3) | 3.7 (3.8) | 3.7 (3.8) | 3.8 (3.8) |
| Refinement | ||||
| Bragg spacings (Å) | 30–2.4 (2.48–2.4) | |||
| Rwork (%)† | 19.5 (24.5) | |||
| Rfree (%)† | 23.5 (28.7) | |||
| Averge B-factor (Å2) | 52.6 | |||
| Rmsd Bond/Angle ideality (Å/°) | 0.011/1.193 | |||
| Rmsd B-factor restraint (Å2) | ||||
| Main-chain bonds/angles | 0.431/0.880 | |||
| Side-chain bonds/angles | 1.503/2.608 | |||
| Ramachandran analysis‡ | ||||
| Favored/outlier (%) | 97.4/0.0 | |||
| Total atoms | 10062 | |||
| Protein residues/ATP:Mg2+:K+/H2O | 1228/2/355 | |||
Values in parentheses indicate the corresponding statistics in the highest resolution shell.
Za: number of molecules per asymmetric unit
Rsym = (Σ|Ih− <Ih>|)/ΣIh, where <Ih> is the average intensity over symmetry equivalent.
Rwork = Σ||Fo| − |Fc||/Σ|Fo|. Rfree is equivalent to Rwork, but calculated for a randomly chosen 5% of reflection, which were omitted from the refinement process.
Analysis from http://kinemage.biochem.duke.edu.
Site-directed mutagenesis and growth tests
Mutations were designed by visual inspection of the Sse1 structure and introduced by standard procedures into Sse1 and at homologous sites in Ssa1 and DnaK, and also in Ssc1 for some. Host-appropriate expression plasmids were then introduced into suitable null backgrounds in yeast and E. coli. Growth was evaluated both at a permissive temperature (30°C) and at a heat-stress temperature (37°C or 40°C) by plating in serial dilutions on agar plates constituted with appropriate media. Each test was run in triplicate. Expression levels of the mutant proteins were tested by Western blot analysis.
Tests of protein translocation in yeast
Appropriate yeast strains were transformed with mutant Ssa1 or Sse1 plasmids, grown to mid-log phase at a permissive temperature (25°C), and then shifted to a heat-stress temperature (33°C) for 30 min. After centrifugation, cell pellets were resuspended in buffer and broken open by vortexing with glass beads. Lysate samples were analyzed by SDS-PAGE and blotted with anti-ppαF antibody (R. Schekman, UC Berkeley) and with anti-phosphoglycerate kinase (PGK) monoclonal antibody (Molecular Probes, Eugene, OR) as a loading control. Tests were run in triplicate.
Tests of protein aggregation in bacteria
DnaK mutant plasmids were introduced into Δdnak E. coli. Cell cultures were grown to 0.6 O.D. at 30°C, shifted to 42°C for 1 hour, and then returned to 30°C for 1 hour. Cultures were cooled to 0°C, centrifuged, resuspended in lysis buffer, and sonicated in 1M guanidinium hydrochloride. The insoluble cell fraction was isolated by differential centrifugation and analyzed by SDS-PAGE with Coomassie staining.
Supplementary Material
Acknowledgments
We thank members of the Hendrickson laboratory for advice and discussions, especially Hui Xie, Erik Martinez-Hackert and Jonah Cheung; Lei Zhou and X4A staff for assisting with diffraction measurements; Leihai You for help with computing; Bill Burkholder and Betty Craig for bacterial and yeast strains, respectively; Randy Schekman for the prepro-α-factor antibody; and Betty Craig, Larry Shapiro and Max Gottesman for discussions. This work was supported in part by a Cancer Research Institute Postdoctoral Fellowship (Q.L.) and NIH grant GM34102 (W.A.H.). Beamline X4A at the National Synchrotron Light Source (NSLS), a DOE facility, is supported by the New York Structural Biology Center.
Footnotes
Supplemental Data
Supplemental Data include five elements of supplemental text, supplemental experimental procedures, eleven figures and two tables and can be found with this article online at xxx.
Accession codes
Atomic coordinates and diffraction data are deposited into the Protein Data Bank with accession code NXXX.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Becker J, Walter W, Yan W, Craig EA. Functional interaction of cytosolic Hsp70 and a DnaJ-related protein, Ydj1p, in protein translocation in vivo. Mol Cell Biol. 1996;16:4378–4386. doi: 10.1128/mcb.16.8.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A, Theyssen H, Schroder H, McCarty JS, Virgallita G, Milkereit P, Reinstein J, Bukau B. Nucleotide-induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication. J Biol Chem. 1995;270:16903–16910. doi: 10.1074/jbc.270.28.16903. [DOI] [PubMed] [Google Scholar]
- Burkholder WF, Zhao X, Zhu X, Hendrickson WA, Gragerov A, Gottesman ME. Mutations in the C-terminal fragment of DnaK affecting peptide binding. Proc Natl Acad Sci U S A. 1996;93:10632–10637. doi: 10.1073/pnas.93.20.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragovic Z, Broadley SA, Shomura Y, Bracher A, Hartl FU. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J. 2006;25:2519–2528. doi: 10.1038/sj.emboj.7601138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton DP, Kaneko Y, Subjeck JR. The Hsp110 and Grp1 70 stress proteins: newly recognized relatives of the Hsp70s. Cell Stress Chaperones. 2000;5:276–290. doi: 10.1379/1466-1268(2000)005<0276:thagsp>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KM, DeLuca-Flaherty C, McKay DB. Three-dimensional structure of the ATPase fragment of a 70K heat-shock cognate protein. Nature. 1990;346:623–628. doi: 10.1038/346623a0. [DOI] [PubMed] [Google Scholar]
- Flaherty KM, Wilbanks SM, DeLuca-Flaherty C, McKay DB. Structural basis of the 70-kilodalton heat shock cognate protein ATP hydrolytic activity. II. Structure of the active site with ADP or ATP bound to wild type and mutant ATPase fragment. J Biol Chem. 1994;269:12899–12907. [PubMed] [Google Scholar]
- Gässler CS, Buchberger A, Laufen T, Mayer MP, Schroder H, Valencia A, Bukau B. Mutations in the DnaK chaperone affecting interaction with the DnaJ cochaperone. Proc Natl Acad Sci U S A. 1998;95:15229–15234. doi: 10.1073/pnas.95.26.15229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison CJ, Hayer-Hartl M, Di Liberto M, Hartl F, Kuriyan J. Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science. 1997;276:431–435. doi: 10.1126/science.276.5311.431. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Hesterkamp T, Bukau B. Role of the DnaK and HscA homologs of Hsp70 chaperones in protein folding in E.coli. EMBO J. 1998;17:4818–28. doi: 10.1093/emboj/17.16.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Prasad K, Lafer EM, Sousa R. Structural basis of interdomain communication in the Hsc70 chaperone. Mol Cell. 2005;20:513–524. doi: 10.1016/j.molcel.2005.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ER, McKay DB. Mapping the role of active site residues for transducing an ATP-induced conformational change in the bovine 70-kDa heat shock cognate protein. Biochemistry. 1999;38:10823–10830. doi: 10.1021/bi990816g. [DOI] [PubMed] [Google Scholar]
- Kang PJ, Ostermann J, Shilling J, Neupert W, Craig EA, Pfanner N. Requirement for Hsp70 in the mitochondrial matrix for translocation and folding of precursor proteins. Nature. 1990;348:137–143. doi: 10.1038/348137a0. [DOI] [PubMed] [Google Scholar]
- Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, Bukau B. Mechanism of regulation of Hsp70 chaperones by DnaJ cochaperones. Proc Natl Acad Sci U S A. 1999;96:5452–5457. doi: 10.1073/pnas.96.10.5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wu Y, Qian X, Sha B. Crystal structure of yeast Sis1 peptide-binding fragment and Hsp70 Ssa1 C-terminal complex. Biochem J. 2006;398:353–360. doi: 10.1042/BJ20060618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Krzewska J, Liberek K, Craig EA. Mitochondrial Hsp70 Ssc1: role in protein folding. J Biol Chem. 2001;276:6112–6118. doi: 10.1074/jbc.M009519200. [DOI] [PubMed] [Google Scholar]
- Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro F, Fernandez-Saiz V, Muga A. The allosteric transition in DnaK probed by infrared difference spectroscopy. Concerted ATP-induced rearrangement of the substrate binding domain. Protein Sci. 2006;15:223–233. doi: 10.1110/ps.051732706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro F, Fernandez V, Muga A. Interdomain interaction through helices A and B of DnaK peptide binding domain. FEBS Lett. 2003;533:119–123. doi: 10.1016/s0014-5793(02)03752-3. [DOI] [PubMed] [Google Scholar]
- O’Brien MC, Flaherty KM, McKay DB. Lysine 71 of the chaperone protein Hsc70 is essential for ATP hydrolysis. J Biol Chem. 1996;271:15874–15878. doi: 10.1074/jbc.271.27.15874. [DOI] [PubMed] [Google Scholar]
- Oh HJ, Easton D, Murawski M, Kaneko Y, Subjeck JR. The chaperoning activity of Hsp110. Identification of functional domains by use of targeted deletions. J Biol Chem. 1999;274:15712–15718. doi: 10.1074/jbc.274.22.15712. [DOI] [PubMed] [Google Scholar]
- Pellecchia M, Montgomery DL, Stevens SY, Vander Kooi CW, Feng HP, Gierasch LM, Zuiderweg ER. Structural insights into substrate binding by the molecular chaperone DnaK. Nat Struct Biol. 2000;7:298–303. doi: 10.1038/74062. [DOI] [PubMed] [Google Scholar]
- Raviol H, Bukau B, Mayer MP. Human and yeast Hsp110 chaperones exhibit functional differences. FEBS Lett. 2006a;580:168–174. doi: 10.1016/j.febslet.2005.11.069. [DOI] [PubMed] [Google Scholar]
- Raviol H, Sadlish H, Rodriguez F, Mayer MP, Bukau B. Chaperone network in the yeast cytosol: Hsp110 is revealed as an Hsp70 nucleotide exchange factor. EMBO J. 2006b;25:2510–2518. doi: 10.1038/sj.emboj.7601139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revington M, Zhang Y, Yip GN, Kurochkin AV, Zuiderweg ER. NMR investigations of allosteric processes in a two-domain Thermus thermophilus Hsp70 molecular chaperone. J Mol Biol. 2005;349:163–183. doi: 10.1016/j.jmb.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Rist W, Graf C, Bukau B, Mayer MP. Amide hydrogen exchange reveals conformational changes in Hsp70 chaperones important for allosteric regulation. J Biol Chem. 2006;281:16493–16501. doi: 10.1074/jbc.M600847200. [DOI] [PubMed] [Google Scholar]
- Shaner L, Sousa R, Morano KA. Characterization of Hsp70 binding and nucleotide exchange by the yeast Hsp110 chaperone Sse1. Biochemistry. 2006;45:15075–15084. doi: 10.1021/bi061279k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner L, Trott A, Goeckeler JL, Brodsky JL, Morano KA. The function of the yeast molecular chaperone Sse1 is mechanistically distinct from the closely related Hsp70 family. J Biol Chem. 2004;279:21992–22001. doi: 10.1074/jbc.M313739200. [DOI] [PubMed] [Google Scholar]
- Shaner L, Wegele H, Buchner J, Morano KA. The yeast Hsp110 Sse1 functionally interacts with the Hsp70 chaperones Ssa and Ssb. J Biol Chem. 2005;280:41262–41269. doi: 10.1074/jbc.M503614200. [DOI] [PubMed] [Google Scholar]
- Shomura Y, Dragovic Z, Chang HC, Tzvetkov N, Young JC, Brodsky JL, Guerriero V, Hartl FU, Bracher A. Regulation of Hsp70 function by HspBP1: structural analysis reveals an alternate mechanism for Hsp70 nucleotide exchange. Mol Cell. 2005;17:367–379. doi: 10.1016/j.molcel.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Sondermann H, Scheufler C, Schneider C, Hohfeld J, Hartl FU, Moarefi I. Structure of a Bag/Hsc70 complex: convergent functional evolution of Hsp70 nucleotide exchange factors. Science. 2001;291:1553–1557. doi: 10.1126/science.1057268. [DOI] [PubMed] [Google Scholar]
- Sriram M, Osipiuk J, Freeman B, Morimoto R, Joachimiak A. Human Hsp70 molecular chaperone binds two calcium ions within the ATPase domain. Structure. 1997;5:403–414. doi: 10.1016/s0969-2126(97)00197-4. [DOI] [PubMed] [Google Scholar]
- Steel GJ, Fullerton DM, Tyson JR, Stirling CJ. Coordinated activation of Hsp70 chaperones. Science. 2004;303:98–101. doi: 10.1126/science.1092287. [DOI] [PubMed] [Google Scholar]
- Suh WC, Burkholder WF, Lu CZ, Zhao X, Gottesman ME, Gross CA. Interaction of the Hsp70 molecular chaperone, DnaK, with its cochaperone DnaJ. Proc Natl Acad Sci U S A. 1998;95:15223–15228. doi: 10.1073/pnas.95.26.15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain JF, Dinler G, Sivendran R, Montgomery DL, Stotz M, Gierasch LM. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell. 2007;26:27–39. doi: 10.1016/j.molcel.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel M, Bukau B, Mayer MP. Allosteric regulation of Hsp70 chaperones by a proline switch. Mol Cell. 2006a;21:359–367. doi: 10.1016/j.molcel.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Vogel M, Mayer MP, Bukau B. Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. J Biol Chem. 2006b;281:38705–387011. doi: 10.1074/jbc.M609020200. [DOI] [PubMed] [Google Scholar]
- Wilbanks SM, Chen L, Tsuruta H, Hodgson KO, McKay DB. Solution small-angle X-ray scattering study of the molecular chaperone Hsc70 and its subfragments. Biochemistry. 1995;34:12095–12106. doi: 10.1021/bi00038a002. [DOI] [PubMed] [Google Scholar]
- Yam AY, Albanese V, Lin HT, Frydman J. Hsp110 cooperates with different cytosolic Hsp70 systems in a pathway for de novo folding. J Biol Chem. 2005;280:41252–41261. doi: 10.1074/jbc.M503615200. [DOI] [PubMed] [Google Scholar]
- Yamagishi N, Ishihara K, Hatayama T. Hsp105α suppresses Hsc70 chaperone activity by inhibiting Hsc70 ATPase activity. J Biol Chem. 2004;279:41727–41733. doi: 10.1074/jbc.M407947200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zuiderweg ER. The 70-kDa heat shock protein chaperone nucleotide-binding domain in solution unveiled as a molecular machine that can reorient its functional subdomains. Proc Natl Acad Sci U S A. 2004;101:10272–10277. doi: 10.1073/pnas.0401313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272:1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.