

Summary
Long-distance organelle transport toward axon terminals, critical for neuron development and function, is driven along microtubules by kinesins [1, 2]. The biophysics of force production by various kinesins is known in detail. However, the mechanisms of in vivo transport processes are poorly understood, because little is known about how motor-cargo linkages are controlled. A c-Jun N-terminal kinase (JNK) interacting protein (JIP1) has been identified previously as a linker between kinesin-1 and certain vesicle membrane proteins, such as Alzheimer's APP protein and a reelin receptor ApoER2 [3, 4]. JIPs are also known to be scaffolding proteins for JNK pathway kinases [5, 6]. Here we report evidence that a Drosophila ubiquitin specific hydrolase and a JNK signaling pathway that it modulates, can regulate a JIP1-kinesin linkage. The JNK pathway includes a MAPKKK (Wallenda/DLK), a MAPKK (Hemipterous/MKK7), and the Drosophila JNK homolog Basket. Genetic tests indicate that those kinases are required for normal axonal transport. Biochemical tests show that activation of Wallenda (DLK) and Hemipterous (MKK7) disrupts binding between kinesin-1 and APLIP1, which is the Drosophila JIP1 homolog. This suggests a control mechanism in which an activated JNK pathway influences axonal transport by functioning as a kinesin-cargo dissociation factor.
Results and Discussion
Maintaining proper distributions of protein complexes, RNAs, vesicles, and other organelles in axons is critical for the development, function, and survival of neurons. The primary distribution mechanism relies on long-distance transport driven by microtubule motor proteins [2]. Components newly synthesized in the cell body, but needed in the axon, bind kinesin motors that carry them toward microtubule plus-ends and the axon terminal (anterograde transport). Components in the axon that require transport to the cell body, such as neurotrophic signals and endosomes, bind dynein motors that carry them toward minus ends (retrograde transport). The importance of these processes is highlighted by the observation that mutation of motors and other transport machinery components can cause neurodegenerative diseases in humans and analogous phenotypes in model organisms [1, 7, 8].
Two key questions are 1) how do cargoes link to particular motors? and 2) how are such linkages regulated to ensure appropriate pick-up and drop-off dynamics? For kinesin-vesicle linkages, scaffolding proteins have emerged as key connectors. For example, the cargo-binding kinesin light chain (Klc) subunit of kinesin-1 binds not only the kinesin-1 heavy chain (Khc), but also JNK interacting proteins (JIPs) [4, 5, 9]. Vertebrate JIPs can bind multiple components of the JNK signaling pathway, e.g. JNK itself, upstream activating kinases (MAPKKs), and regulatory kinases (MAPKKKs) [5, 6]. JIPs can also bind vesicle-associated membrane proteins, such as ApoER2, which is a reelin receptor, and APP, a key factor in Alzheimer's disease [4, 10, 11]. Therefore, JIP scaffolding proteins likely link JNK pathway kinases and kinesin-1 to vesicles carrying these membrane proteins. This raises an interesting question: Are the JNK pathway kinases simply passive hitchhikers on the kinesin-1/JIP/vesicle complex, or can they actively regulate its transport [12]?
We conducted a genetic screen for factors that control kinesin-JIP linkage during axonal transport. The screen was based on the previous observation that neuron specific overexpression of Aplip1, which encodes the Drosophila JIP1, causes synaptic protein accumulation in axons, larval paralysis, and larval-pupal lethality [13], which are the classic axonal transport disruption phenotypes caused by Khc and Klc mutations [8, 14]. Why might overexpression of the JIP1 cargo linker for kinesin-1 disrupt axonal transport? The disruptive effect requires APLIP1 (JIP1)-Klc binding [13]. It may be that excess APLIP1 (JIP1) competes with other Klc-binding proteins; for example, different linkers that may attach kinesin-1 to other cargoes. In search of factors that can disrupt or antagonize APLIP1 (JIP1)-Klc binding, we screened for genes that can suppress the axonal transport phenotypes when co-overexpressed with Aplip1. We screened an “EP” collection of fly strains capable of the targeted overexpression of endogenous Drosophila genes [15, 16] and identified P{EP}fafEP381, a line that overexpresses fat facets (faf) [17], as a strong suppressor of the APLIP1 (JIP1)-induced lethality and other neuronal overexpression phenotypes (Fig. 1A to C, and Table S1 in Supplemental Data, available online).
Figure 1.

Suppression of Aplip1 (Jip1) overexpression-induced vesicle accumulation in axons by a ubiquitin hydrolase and a MAPKKK.
The distribution of a synaptic vesicle glutamate transporter, DVGLUT, is shown in nerves of third instar Drosophila larvae by immunostaining and confocal fluorescence microscopy. Each nerve contains 60-80 motor and sensory axons. Up arrows reflect GAL4-UAS driven expression of transgenes. (A) Wild-type nerves (wt) with DVGLUT in small punctae consistent with its concentration in membranes of axonal transport vesicles. (B to D) Expression of a transgenic FLAG-tagged Aplip1 (Aplip1(F)) was induced in neurons with an elav-GAL4 driver. (B) Over-expression of Aplip1 caused dramatic accumulation of DVGLUT, consistent with organelle accumulation in focal axonal swellings [8]. (C) Suppression of Aplip1-induced accumulation by transgenic co-overexpression of faf, a ubiquitin hydrolase. (D) A 50% reduction of the gene dosage for a MAPKKK (wnd) prevented faf suppression of Aplip1. (E to G) Expression of a transgenic GFP-tagged Aplip1, Aplip1(G), was induced in motoneurons with the OK6-Gal4 driver. (E) DVGLUT accumulations caused by overexpression of Aplip1. (F) Suppression of the accumulations by co-expression of wild-type wnd. (G) No suppression of the accumulations by co-expression of a kinase-dead version of wnd (wndKD). The wndKD transgene was expressed at a higher level than the wild-type wnd transgene (not shown). Full genotypes are noted in Table S1 (Scale bar = 20μm).
Faf protein antagonizes ubiquitination and proteasome-mediated degradation of its target proteins [18, 19]. Interestingly, Faf was recently reported to stimulate a Drosophila neuronal JNK signaling pathway that is regulated by the MAPKKK Wallenda (Wnd) [20], a homolog of dual leucine zipper-bearing kinase (DLK) which is known to bind JIP1 [6, 21]. Overexpression of faf leads to increased levels of Wnd (MAPKKK) protein, which causes excessive synaptic sprouting through a pathway that requires the Drosophila JNK homolog Basket (Bsk) [20]. We found that mutating just one copy of wnd blocked the suppression of Aplip1 overexpression by P{EP}fafEP381 (Fig. 1D and Table S1). This suggests that faf overexpression suppresses APLIP1 (JIP1)-Klc interaction by elevating the level of Wnd (MAPKKK). Consistent with this, direct overexpression of wnd in neurons using a wild-type transgene (UAS-wnd) was as effective as P{EP}fafEP381 in suppressing UAS-Aplip1-induced axonal accumulation of synaptic proteins (Fig. 1F, Table S1). Equivalent expression of a “kinase-dead” mutant transgene (UAS-wndKD) did not suppress the defects (Fig. 1G, Table S1). Thus, Wnd (MAPKKK) and its downstream phosphorylation targets may actively regulate APLIP1 (JIP1)-Klc binding in neurons.
If Wnd (MAPKKK) signaling plays a role in normal axonal transport, disrupting its function should cause axonal transport phenotypes. Consistent with this, wnd loss-of-function mutations (wnd1/wnd2) in an otherwise wild-type background caused accumulation of synaptic proteins in axons (Fig. 2B, Table S1). The accumulation phenotype was rescued by motoneuron expression of the wild-type wnd transgene, but not by equivalent expression of the kinase-dead mutant transgene (Fig. 2C and D, Table S1). The likely target of Wnd (MAPKKK) kinase activity is the Drosophila homolog of MKK7, Hemipterous (Hep), a MAPKK that activates Bsk (JNK) [22, 23]. Mutation of hep also caused axonal accumulations, as did neuronal expression of a dominant-negative mutant bsk transgene (Fig. 2E and F, Table S1). The results of these genetic inhibition tests combined with those of the Aplip1 overexpression suppression tests suggest that a Wnd (MAPKKK)-activated JNK pathway influences fast axonal transport by regulating APLIP1 (JIP1)-Klc binding.
Figure 2.
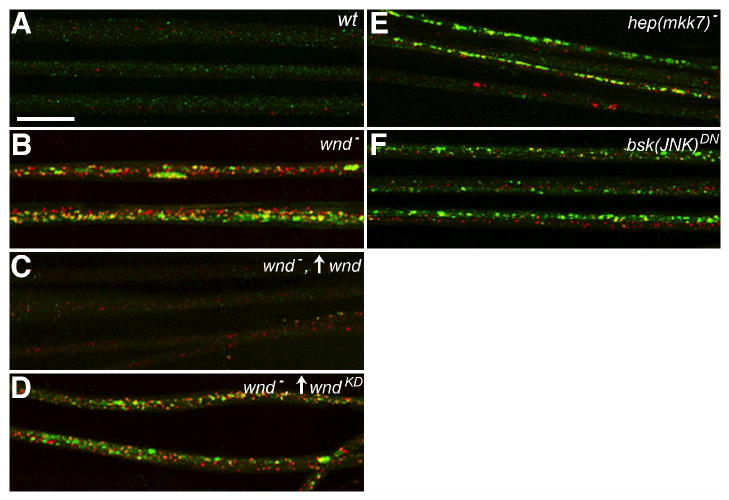
Wnd (MAPKKK) and downstream JNK pathway signaling components are required for normal axonal transport.
The distribution of DVGLUT (green) and an independent presynaptic terminal protein, Bruchpilot (red) is shown in larval nerves imaged by immunostaining and fluorescence confocal microscopy. Up arrows reflect GAL4-UAS driven expression of transgenes. (A) In wild-type nerves (wt), the two proteins appear in different punctae, consistent with their transport in separate axonal cargo complexes. (B) Mutation of wnd caused accumulation of both synaptic proteins in nerves, consistent with defective axonal transport. (C) The accumulation phenotype was suppressed by OK6-GAL4-driven motoneuron expression of transgenic wild-type wnd. (D) Accumulations were not suppressed by equivalent expression of a transgenic kinase-dead mutant (wndKD). (E) Inhibition of Hep, an MKK7-like MAPKK, by hemizygous mutation (hep(mkk7)) caused axonal accumulations. (F) Inhibition of Bsk, the Drosophila JNK homolog, by GAL4-UAS induced neuronal expression of a dominant-negative transgene (bsk(JNK)DN) also caused axonal accumulations. Full genotypes are noted in Table S1 (Scale bar = 20μm).
Is a Wnd (MAPKKK)-Hep (MAPKK)-Bsk (JNK) signaling module bound by APLIP1 (JIP1)? While all three components of the homologous vertebrate module (DLK-MKK7-JNK) bind JIP1 [5, 6], APLIP1 (JIP1) lacks a conserved JNK binding domain, and it does not bind directly to Bsk (JNK) [3]. However, APLIP1 (JIP1) does bind Hep (MAPKK), Klc, and the Drosophila APP homolog APPL [3]. To determine if Wnd (MAPKKK) associates with Hep (MAPKK) and/or APLIP1 (JIP1), we performed co-expression and immunoprecipitation tests in Drosophila S2 cultured cells. Wnd (MAPKKK) did not co-precipitate with APLIP1 (JIP1) (Fig. 3A). However, Hep (MAPKK) did co-precipitate with APLIP1 (JIP1), and Wnd (MAPKKK) coprecipitated with Hep (MAPKK) (Fig. 3B and C). Thus, Wnd (MAPKKK) may bind and influence the APLIP1 (JIP1)-kinesin complex via Hep (MAPKK).
Figure 3.
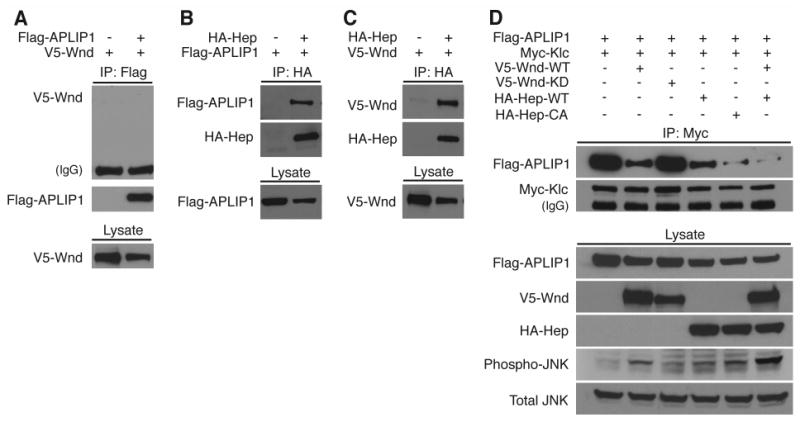
Wnd (MAPKKK) and Hep (MAPKK) associate and their activities inhibit APLIP1 (JIP1)-Klc binding.
Drosophila S2 cells were used for transfection and expression of epitope-tagged constructs as noted across the top of each panel. The presence of proteins was detected in cell lysates (Lysate) or in immunoprecipitation pellets (IP) by western blotting. Proteins detected with specific antibodies are noted to the left of each blot panel. (A) Co-precipitation of Wnd (V5-Wnd) was not detected with APLIP1 (FLAG-APLIP1). (B) APLIP1 co-precipitated with Hep (HA-Hep). (C) Wnd co-precipitated with Hep. (D) APLIP1 co-precipitation with kinesin-1 light chain (Myc-Klc) was partially inhibited by expression of Wnd (V5-Wnd), but not by a kinase-dead mutant Wnd (V5-Wnd-KD). Co-precipitation of APLIP1 with Klc was partially inhibited by expression of wild-type Hep (HA-Hep), and was more completely inhibited by a constitutively active mutant Hep (HA-Hep-CA). Near-complete inhibition was also observed with co-expression of wild-type Wnd and wild-type Hep. Wnd-Hep kinase activity increased the phosphorylation level of endogenous Bsk (compare Total JNK with Phospho-JNK).
Can Wnd (MAPKKK) and Hep (MAPKK) control the binding of APLIP1 (JIP1) to Klc? When expressed in S2 cells, APLIP1 (JIP1) and Klc exhibit strong binding, as assessed by co-immunoprecipitation (Fig. 3D, lane 1) [13]. Co-expression of wild-type Wnd (MAPKKK) partially inhibited that APLIP1 (JIP1)-Klc binding, but co-expression of a kinase-dead mutant Wnd (MAPKKK) did not (Figure 3D, lanes 2 and 3). Wild-type Hep (MAPKK) also caused a partial inhibition of APLIP1 (JIP1)-Klc binding, and a constitutively active mutant Hep (MAPKK) caused nearly complete inhibition (Fig. 3D, lanes 4 and 5). Finally, co-expression of wild-type Wnd (MAPKKK) and Hep (MAPKK) together caused an almost complete inhibition of APLIP1 (JIP1)-Klc binding (Fig. 3D, lane 6). In addition to inhibiting APLIP1 (JIP1)-Klc binding, Wnd-Hep activation in S2 cells increased the level of Bsk (JNK) activation (Fig. 3D). Hence, there is a correlation between decreased levels of APLIP1 (JIP1)-Klc binding and elevated levels of Bsk (JNK) activation. This suggests that, despite the lack of a known JNK binding site on APLIP1 (JIP1), Bsk (JNK) may be the kinase that disrupts the APLIP1 (JIP1)-Klc complex. These results suggest that Wnd (MAPKKK) activation of Hep (MAPKK), and perhaps also Hep (MAPKK) activation of Bsk (JNK), can regulate the linkage between kinesin-1 and a cargo complex via the JIP1-like scaffolding protein, APLIP1 (Fig. 4).
Figure 4.
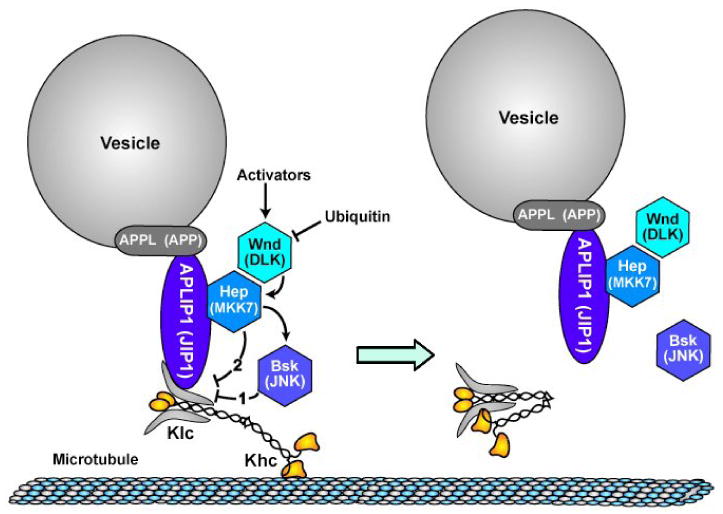
A model for Wnd (MAPKKK) pathway control of APLIP1 (JIP1)-linked kinesin-1 cargo transport. Components are labeled with Drosophila names and parenthetically with names of vertebrate homologs. Lines with arrowheads indicate activation influences and lines with crossbars indicate inhibition influences. Wnd (MAPKKK), whose levels can be modulated by ubiquitination, is activated by unknown upstream signals. Wnd activates Hep (MAPKK) by phosphorylation and activated Hep (MAPKK) then causes dissociation of APLIP1 (JIP1) from Klc, probably by phospho-activation of Bsk (JNK), which then directly or indirectly modifies the linkage complex (pathway 1). It is also possible that phosphorylation of Hep (MAPKK) causes a conformational change in the linkage complex that inhibits APLIP1 (JIP1)-Klc binding independent of Bsk (pathway 2). Disruption of the APLIP1 (JIP1)-Klc linkage may allow kinesin to adopt an inactive, folded conformation that does not bind to microtubules [29, 30].
Hep (MAPKK) may regulate the APLIP1 (JIP1) complex either by activating JNK (Fig. 4, pathway 1) or by a mechanism independent of JNK (Fig. 4, pathway 2). Our observations that motoneuron specific inhibition of Bsk (JNK) caused transport defects similar to those caused by mutations in wnd and hep (Fig. 2B, E and F) and that decreased APLIP1 (JIP1)-Klc binding in S2 cell lysates coincided with increased phosphorylated Bsk (JNK) (Fig. 3D) support pathway 1, i.e. Hep (MAPKK) activation of Bsk (JNK), which then directly or indirectly inhibits APLIP1 (JIP1)-Klc binding. Pathway 2 employs an alternative mechanism in which activated Hep (MAPKK) does not need Bsk (JNK) to inhibit APLIP1 (JIP1)-Klc binding. There is little current evidence that Hep or its vertebrate MAPKK homolog MKK7 have phosphorylation targets other than Bsk (JNK) [22, 24]. However, that does not exclude the possibility that activated Hep propagates a direct conformational change to APLIP1 (JIP1) that causes Klc dissociation. Regardless of how Hep (MAPKK) disrupts binding, when kinesin-1 is not attached to cargo via JIP1, it can fold into a compact form that does not interact with microtubules [25]. Hence the activated Wnd (MAPKKK) pathway could both inhibit APLIP1 (JIP1)-Klc binding and it could cause dissociation of kinesin-1 from microtubules (Fig. 4). Consistent with this, recent studies report that stimulation of JNK pathways in cultured cells or axoplasm can disrupt the association of kinesin-1 with microtubules [26, 27].
From a broader perspective on axonal transport regulation, it is interesting to consider that there are multiple types of kinesin-1 cargoes [2], there are various JIPs that could be specific for different cargoes [4, 5, 9], and different MAPKKKs can associate with different JIPs [5, 6]. By sitting at the top of a classic signaling cascade, MAPKKKs like Wnd are in a good position to differentially control the transport of specific subsets of anterograde kinesin-1 cargoes in response to specific cellular signals. It is known in mammals that other MAPKKKs such as MLK, ASK1, and MEKK1 can bind JIP scaffolding proteins [28]. It will be interesting to determine if they too influence kinesin-cargo interactions.
To our knowledge, the work presented here provides the first demonstration that a kinesin and its transport functions can be influenced by a MAPKKK. More specifically, the MAPKKK Wnd and its downstream MAPKK Hep can regulate attachment of a JIP1 cargo linker to kinesin-1. Our results also provide the first indication that ubiquitination pathways, by way of MAPKKKs, could be important for proper regulation of axonal transport. Finally, our results suggest that JNK pathway kinases are not just hitchhikers on the axonal kinesin-1/JIP/cargo complex, rather they can actively regulate its transport dynamics.
Supplementary Material
Acknowledgments
This work was supported by predoctoral fellowships from the American Heart Association, Midwest Affiliate to D.H. and R.V.B, a postdoctoral fellowship from the Paralyzed Veterans of America to C.A.C., grants from the NIH (DA020812), Keck and McKnight foundations to A.D. and NIH grant GM46295 to W.M.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Duncan JE, Goldstein LS. The genetics of axonal transport and axonal transport disorders. PLoS Genet. 2006;2:e124. doi: 10.1371/journal.pgen.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirokawa N, Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci. 2005;6:201–214. doi: 10.1038/nrn1624. [DOI] [PubMed] [Google Scholar]
- 3.Taru H, Iijima K, Hase M, Kirino Y, Yagi Y, Suzuki T. Interaction of Alzheimer's beta -amyloid precursor family proteins with scaffold proteins of the JNK signaling cascade. J Biol Chem. 2002;277:20070–20078. doi: 10.1074/jbc.M108372200. [DOI] [PubMed] [Google Scholar]
- 4.Verhey KJ, Meyer D, Deehan R, Blenis J, Schnapp BJ, Rapoport TA, Margolis B. Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J Cell Biol. 2001;152:959–970. doi: 10.1083/jcb.152.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelkar N, Standen CL, Davis RJ. Role of the JIP4 scaffold protein in the regulation of mitogen-activated protein kinase signaling pathways. Mol Cell Biol. 2005;25:2733–2743. doi: 10.1128/MCB.25.7.2733-2743.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol Cell Biol. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chevalier-Larsen E, Holzbaur EL. Axonal transport and neurodegenerative disease. Biochim Biophys Acta. 2006;1762:1094–1108. doi: 10.1016/j.bbadis.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Hurd DD, Saxton WM. Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila. Genetics. 1996;144:1075–1085. doi: 10.1093/genetics/144.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowman AB, Kamal A, Ritchings BW, Philp AV, McGrail M, Gindhart JG, Goldstein LS. Kinesin-dependent axonal transport is mediated by the sunday driver (SYD) protein. Cell. 2000;103:583–594. doi: 10.1016/s0092-8674(00)00162-8. [DOI] [PubMed] [Google Scholar]
- 10.Inomata H, Nakamura Y, Hayakawa A, Takata H, Suzuki T, Miyazawa K, Kitamura N. A scaffold protein JIP-1b enhances amyloid precursor protein phosphorylation by JNK and its association with kinesin light chain 1. J Biol Chem. 2003;278:22946–22955. doi: 10.1074/jbc.M212160200. [DOI] [PubMed] [Google Scholar]
- 11.Matsuda S, Matsuda Y, D'Adamio L. Amyloid beta protein precursor (AbetaPP), but not AbetaPP-like protein 2, is bridged to the kinesin light chain by the scaffold protein JNK-interacting protein 1. J Biol Chem. 2003;278:38601–38606. doi: 10.1074/jbc.M304379200. [DOI] [PubMed] [Google Scholar]
- 12.Schnapp BJ. Trafficking of signaling modules by kinesin motors. J Cell Sci. 2003;116:2125–2135. doi: 10.1242/jcs.00488. [DOI] [PubMed] [Google Scholar]
- 13.Horiuchi D, Barkus RV, Pilling AD, Gassman A, Saxton WM. APLIP1, a kinesin binding JIP-1/JNK scaffold protein, influences the axonal transport of both vesicles and mitochondria in Drosophila. Curr Biol. 2005;15:2137–2141. doi: 10.1016/j.cub.2005.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gindhart JG, Jr, Desai CJ, Beushausen S, Zinn K, Goldstein LS. Kinesin light chains are essential for axonal transport in Drosophila. J Cell Biol. 1998;141:443–454. doi: 10.1083/jcb.141.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G, Evans-Holm M, Hiesinger PR, Schulze KL, Rubin GM, et al. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics. 2004;167:761–781. doi: 10.1534/genetics.104.026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rorth P. A modular misexpression screen in Drosophila detecting tissue-specific phenotypes. Proc Natl Acad Sci U S A. 1996;93:12418–12422. doi: 10.1073/pnas.93.22.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiAntonio A, Haghighi AP, Portman SL, Lee JD, Amaranto AM, Goodman CS. Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature. 2001;412:449–452. doi: 10.1038/35086595. [DOI] [PubMed] [Google Scholar]
- 18.Chen X, Fischer JA. In vivo Structure/Function analysis of the Drosophila fat facets deubiquitinating enzyme gene. Genetics. 2000;156:1829–1836. doi: 10.1093/genetics/156.4.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 20.Collins CA, Wairkar YP, Johnson SL, DiAntonio A. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron. 2006;51:57–69. doi: 10.1016/j.neuron.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 21.Nihalani D, Merritt S, Holzman LB. Identification of structural and functional domains in mixed lineage kinase dual leucine zipper-bearing kinase required for complex formation and stress-activated protein kinase activation. J Biol Chem. 2000;275:7273–7279. doi: 10.1074/jbc.275.10.7273. [DOI] [PubMed] [Google Scholar]
- 22.Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- 23.Merritt SE, Mata M, Nihalani D, Zhu C, Hu X, Holzman LB. The mixed lineage kinase DLK utilizes MKK7 and not MKK4 as substrate. J Biol Chem. 1999;274:10195–10202. doi: 10.1074/jbc.274.15.10195. [DOI] [PubMed] [Google Scholar]
- 24.Stronach B. Dissecting JNK signaling, one KKKinase at a time. Dev Dyn. 2005;232:575–584. doi: 10.1002/dvdy.20283. [DOI] [PubMed] [Google Scholar]
- 25.Verhey KJ, Rapoport TA. Kinesin carries the signal. Trends Biochem Sci. 2001;26:545–550. doi: 10.1016/s0968-0004(01)01931-4. [DOI] [PubMed] [Google Scholar]
- 26.Morfini G, Pigino G, Szebenyi G, You Y, Pollema S, Brady ST. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat Neurosci. 2006;9:907–916. doi: 10.1038/nn1717. [DOI] [PubMed] [Google Scholar]
- 27.Stagi M, Gorlovoy P, Larionov S, Takahashi K, Neumann H. Unloading kinesin transported cargoes from the tubulin track via the inflammatory c-Jun N-terminal kinase pathway. Faseb J. 2006;20:2573–2575. doi: 10.1096/fj.06-6679fje. [DOI] [PubMed] [Google Scholar]
- 28.Whitmarsh AJ. The JIP family of MAPK scaffold proteins. Biochemical Society transactions. 2006;34:828–832. doi: 10.1042/BST0340828. [DOI] [PubMed] [Google Scholar]
- 29.Blasius TL, Cai D, Jih GT, Toret CP, Verhey KJ. Two binding partners cooperate to activate the molecular motor Kinesin-1. J Cell Biol. 2007;176:11–17. doi: 10.1083/jcb.200605099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stock MF, Guerrero J, Cobb B, Eggers CT, Huang TG, Li X, Hackney DD. Formation of the compact confomer of kinesin requires a COOH-terminal heavy chain domain and inhibits microtubule-stimulated ATPase activity. J Biol Chem. 1999;274:14617–14623. doi: 10.1074/jbc.274.21.14617. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.