

Abstract
Oxalate decarboxylase (OxDC) catalyzes the conversion of oxalate into CO2 and formate using a catalytic mechanism that remains poorly understood. The Bacillus subtilis enzyme is composed of two cupin domains, each of which contains Mn(II) coordinated by four conserved residues. We have measured heavy atom isotope effects for a series of Bacillus subtilis OxDC mutants in which Arg-92, Arg-270, Glu-162, and Glu-333 are conservatively substituted in an effort to define the functional roles of these residues. This strategy has the advantage that observed isotope effects report directly on OxDC molecules in which the active site manganese center(s) is (are) catalytically active. Our results support the proposal that the N-terminal Mn-binding site can mediate catalysis, and confirm the importance of Arg-92 to catalytic activity. On the other hand, substitution of Arg-270 and Glu-333 affected both Mn(II) incorporation and the ability of Mn to bind to the OxDC mutants, thereby precluding any definitive assessment of whether the metal center in the C-terminal domain can also mediate catalysis. New evidence for the importance of Glu-162 in controlling metal reactivity has been provided by the unexpected observation that the E162Q OxDC mutant exhibits a significantly increased oxalate oxidase and a concomitant reduction in decarboxylase activities relative to wild type OxDC. Hence the reaction specificity of a catalytically active Mn center in OxDC can be perturbed by relatively small changes in local protein environment, in agreement with a proposal based on prior computational studies.
Keywords: Oxalate, Decarboxylation, Manganese, Metalloenzymes, Evolution of Enzyme Catalysis, Enzyme Mechanism, Heavy Atom Isotope Effects
Oxalate decarboxylase (E.C. 4.1.1.2) (OxDC) catalyzes the decarboxylation of oxalic acid to yield formic acid and carbon dioxide (Equation 1) [1], which is a chemically interesting transformation [2, 3]. Efforts to delineate the catalytic mechanism employed by the enzyme [4] have been facilitated by the isolation of a native, bacterial form of OxDC from Bacillus subtilis [5]. Subsequent identification of the gene encoding the enzyme then permitted the development of methods to obtain catalytically active, wild type enzyme in large amounts by expression in Escherichia coli [6].
Equation 1.

Two high resolution X-ray crystal structures of Bacillus subtilis OxDC have been reported [7, 8], and these show that the enzyme is a bicupin [9], in agreement with predictions from primary sequence alignment studies [10]. The OxDC monomer is composed of two cupin domains, each of which contains a metal-binding site (Figure 1A). The resting form of OxDC contains Mn(II), and kinetic experiments have shown that catalytic activity is Mn-dependent and requires the presence of dioxygen (the enzyme is inactive under anaerobic conditions) even though the overall transformation is a disproportionation reaction [6]. Heavy-atom isotope effect (IE) measurements [4] have lead to a working model for the mechanism of OxDC-catalyzed C-C bond cleavage (Scheme 1).
Fig. 1. (A) Cartoon representation of the Bacillus subtilis OxDC monomer.
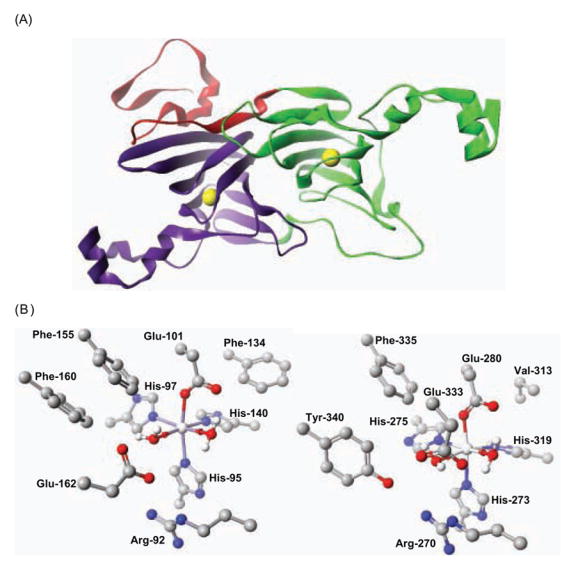
The N- and C-terminal cupin domains are colored green and purple, respectively, and the N-terminal segment that contributes to the secondary structure of the C-terminal domain is colored red. The yellow spheres show the locations of the two Mn centers. (B) Residues defining the Mn-binding (and putative catalytic) sites in the N-terminal (1UW8) (left) and C-terminal (1P58) (right) domains of OxDC. Residue numbering is for the enzyme encoded by the OxdC gene in Bacillus subtilis. For clarity, hydrogen atoms bound to carbon atoms are omitted. Atom coloring: C, black; H, white; N, blue; O, red; Mn, silver. These structures were visualized using the CAChe Worksystem Pro V6.5 software package (Fujitsu America Inc., Beaverton, OR).
Scheme 1. Hypothetical catalytic mechanism and minimal kinetic model for oxalate decarboxylase, showing the putative roles of active site arginine and glutamate residues.
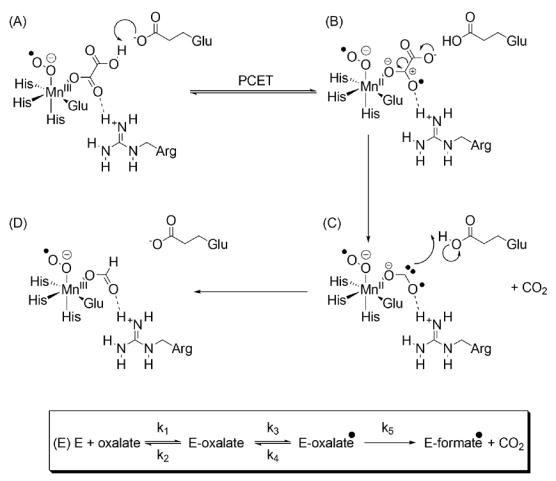
(A) Mono-protonated oxalate and dioxygen, which is required for activity, bind to the Mn(II) center. Electron transfer then takes place to give Mn(III) and metal-bound superoxide [18]. The Mn oxidation state in this and subsequent structures, however, is hypothetical and remains to be directly determined. (B) The active site glutamate than abstracts a proton permitting electron transfer from the substrate to give the Mn-bound oxalate radical anion, one C-O bond of which may be polarized by hydrogen bonding to the active site arginine (dashed line). (C) Decarboxylation by heterolytic C-C bond cleavage then gives a metal-bound formyl radical anion, which is drawn to be consistent with the C-O bond orders computed from experimental heavy-atom IEs. (D) The active site glutamic acid then re-protonates the radical anion with concomitant electron transfer to yield formate and regenerate Mn(III). (E) The kinetic model used in the quantitative analysis of heavy-atom IEs. Here, the Michaelis complex is converted reversibly (Keq3 = k3/k4) to the oxalate radical anion prior to decarboxylation (k5).
Hence, in the first step, mono-protonated oxalate and dioxygen bind to the metal. Oxidation of the active site Mn(II) (present in the resting state of the enzyme) to Mn(III) is then thought to take place with concomitant formation of Mn-bound superoxide (Scheme 1A). An active site glutamate residue can then mediate proton-coupled electron transfer (PCET) in the initial complex to give a Mn(II)-bound oxalate radical anion, which then undergoes heterolytic bond cleavage to release CO2 (Scheme 1B). After decarboxylation, a Mn(II)-bound radical anion of formate is produced (Scheme 1C), which can then be protonated by the active site glutamate, again with electron transfer from the metal, to yield Mn(III)-bound formate (Scheme 1D). The irreversibility of this protonation step has recently been demonstrated using Fourier Transform IR spectroscopy [11]. A quantitative analysis of the 13C and 18O IEs observed for the OxDC-catalyzed reaction suggests that the carbonyl group of the substrate was polarized by the side chain of an adjacent residue [4], which has been tentatively identified as an arginine on the basis of the X-ray crystal structure [7] (Scheme 1B). Because heavy-atom IEs only report on changes in the substrate structure during reaction, we note that the nature of any interaction between bound Mn(II) and dioxygen should be regarded as speculative and is the subject of ongoing study. Indeed, direct experimental evidence for the participation of higher oxidation states of manganese in catalysis has yet to be reported [12].
The observation that OxDC contains Mn in both the N- and C-terminal cupin domains, however, raises the question of whether catalysis takes place in only one or both of the two Mn-binding sites. We [4], and others [7], initially assigned the putative catalytic arginine and glutamate residues to be Arg-270 and Glu-333, which are both located in the Mn-binding site of the C-terminal cupin domain. Work published after the appearance of our mechanistic hypothesis, however, revealed that a loop segment in the N-terminal cupin domain of the enzyme (comprising residues 161–165) can adopt different conformations in OxDC [8], and in one of these the side chains of Arg-92 and Glu-162 are also correctly positioned for performing the functional roles initially assigned to Arg-270 and Glu-333 (Figure 1B) [8]. These results therefore raise the possibility that both the N- and C-terminal Mn-binding sites may be responsible for catalyzing the breakdown of oxalate.
Two structural observations support a recent proposal that the N-terminal Mn-binding site is catalytically active while the C-terminal Mn-binding domain in OxDC is important for maintaining enzyme structure [8]. First, the N-terminal domain contains a channel, which can exist in an “open” or “closed” form as a result of the conformational rearrangement in residues 161–165, along which oxalate can diffuse from solution [8]. Second, formate ion coordinates the N-terminal manganese ion in one of the OxDC crystal structures [7]. The identification of the first bicupin variant of a homologous enzyme, oxalate oxidase (OxOx), which oxidizes oxalate to CO2 with concomitant reduction of dioxygen to hydrogen peroxide (Equation 2) [13], also provides somewhat indirect evidence for this hypothesis [14]. On the other hand, OxDC activity was decreased by mutations to Arg-270 and Glu-333 in the C-terminal Mn-binding site, suggesting that this domain is important for catalytic activity [7]. Unfortunately, any unambiguous interpretation of the kinetic properties of the OxDC mutants used in these prior studies is complicated by the facts that (i) all of the recombinant enzymes contained polyhistidine purification tags, and (ii) no quantitative information on their Mn content was reported [7, 8].
Equation 2.

Resolving the number and location of the active site(s) in OxDC is an important problem because if only a single site mediates the OxDC-catalyzed reaction, legitimate questions are raised concerning the function (if any) of the second Mn-binding domain, and the extent to which local protein structure in each domain can cause the differential reactivity of the two metal centers. We have therefore reexamined the kinetic properties and Mn content of a series of Bacillus subtilis OxDC mutants that do not contain polyhistidine tags in which Arg-92, Arg-270, Glu-162 and Glu-333 are conservatively substituted (Table 1). We have also performed the first determinations of heavy atom isotope effects (IEs) for this series of OxDC mutants [15–17] because observed isotope effects report directly on OxDC molecules in which the active site manganese center(s) is (are) catalytically active. Our results support the proposal that the N-terminal Mn-binding site can mediate catalysis, and confirm the importance of Arg-92 to catalytic activity. On the other hand, effects on Mn(II) incorporation caused by substitution of Arg-270 and Glu-333 do not exclude the possibility that catalysis also takes place at the C-terminal Mn-binding site. In addition, we show that the E162Q OxDC mutant unexpectedly exhibits ten-fold increased oxalate oxidase activity, suggesting that the reaction specificity of the metal center in the enzyme can be perturbed by changes in the local protein environment, in agreement with a proposal based on prior computational studies [18].
Table 1.
Structural and steady state kinetic properties of recombinant, wild type OxDC and OxDC mutants.
| % Activitya | KM (mM)b | Mn (mol/monomer) | Quaternary Structurec | |
|---|---|---|---|---|
| WT OxDC | 100 | 5 ± 1 | 2 | hexamerd |
| R92K | 1.6 | 10 ± 0.2 | 1.7 | -d |
| E162D | 51 | 3.1 ± 0.7 | 1.1 | hexamerd |
| E162Q | 0.5 | 10 ± 0.2 | 0.5 | hexamerd |
| R270K | 7 | 2.6 ± 0.6 | < 0.01e | trimerd |
| E333D | 13 | 7 ± 1 | < 0.01e | trimerd |
| E333Q | 1.3 | 1.7 ± 0.2 | < 0.01e | ndf |
| R92K/R270K | 1.3 | nd | < 0.01e | nd |
| E162Q/E333Q | 2.3 | 0.6 ± 0.2 | < 0.01e | nd |
The percentage activity is reported for each OxDC mutant relative to that of wild type OxDC purified by the same protocol. Hence, the activities of N-terminal domain mutants (R92K, E162D, E162Q) are compared to 79 U/mg. The activities of the remaining OxDC mutants are compared to 30 U/mg.
KM values were determined from assays in which the oxalate concentration was varied over a range of 0.2–80 mM.
The quaternary structure is computed from molecular weight estimates obtained from size-exclusion chromatography and the molecular weight of an OxDC monomer
Aggregate forms of the enzyme with molecular weights greater than that expected for a hexameric complex were also observed by size-exclusion chromatography.
Mn content was below the limits of detection – this value, however, was obtained from a sample treated with EDTA to remove non-specifically bound metal. Based on the values of specific activity, the metal content of the OxDC mutant used in the kinetic experiments is higher than that reported here.
nd. Value was not determined.
MATERIALS AND METHODS
Materials
All chemicals and reagents were purchased from Sigma-Aldrich, unless otherwise stated, and were of the highest available purity. 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) was obtained from Vector Laboratories (Burlingame, CA). All DNA primers were obtained from Integrated DNA Technologies, Inc. (Coralville, IA), and DNA sequencing was performed by the core facility in the Interdisciplinary Center for Biotechnology Research (ICBR) at the University of Florida. Protein concentrations were determined by the Lowry method using standard curves constructed using bovine serum albumin [19].
Expression and purification of “untagged” recombinant, wild type OxDC and site-specific OxDC mutants
The expression and purification of recombinant, wild-type Bacillus subtilis OxDC from transformed BL21(DE3) Escherichia coli cells was carried out according to literature procedures [4]. Mutagenic primers were designed using the GeneRunner package (GeneRunner for Windows 3.05, www.generunner.com), and a pET-9a plasmid containing the oxdC gene insert encoding wild type OxDC was used as a template for PCR with the mutagenic primers and reagents in the QuikChange® Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). The mutated pET-9a plasmids were isolated from XL-1 Blue supercompetent cells (Stratagene, La Jolla, CA) using standard alkaline lysis, chloroform extraction and PEG precipitation procedures. The presence of the desired mutations in the insert encoding the OxDC mutant enzyme was verified by DNA sequencing, and plasmids containing the correct DNA sequences were then transformed into BL21(DE3) competent cells (Novagen, Madison, WI). All N-terminal OxDC mutants were expressed and purified following identical procedures to that reported for the recombinant, wild type enzyme [4]. For OxDC mutants in which C-terminal, Mn-binding domain residues were mutated, however, purification on phenyl-Sepharose did not improve specific activity, and hence these proteins were purified using the two anion exchange chromatography steps. We note that the omission of the phenyl-Sepharose purification step yields wild type OxDC with a reduced specific activity of 30 ± 3 U/mg rather than the 79 ± 2 U/mg obtained for the enzyme when purified by the standard three-step procedure [4].
Enzyme assays
The rate of oxalate decarboxylation was assayed using formate dehydrogenase (FDH) to determine formate production in an end-point assay [20]. Activity at saturating substrate concentrations was measured in reaction mixtures containing OxDC (5 μg), or the appropriate OxDC mutant, in 50 mM potassium acetate buffer, pH 4.2, containing 50–80 mM potassium oxalate, 0.2% Triton X-100, and 0.5 mM ortho-phenylenediamine (o-PDA) (100 μL total volume). Experiments to determine the Michaelis constant, KM, for wild type OxDC and each OxDC mutant were carried out under identical conditions except that the final potassium oxalate concentration in the reaction mixture was varied from 0.2–80 mM. Turnover was initiated by the addition of substrate, and the reaction mixtures incubated at ambient temperature (23 °C) for 1–2 min before being quenched by the addition of 1 M NaOH (20 μL). An aliquot (50 μL) was then taken, and the amount of formate quantified by addition to an assay mixture containing FDH (0.4 U/mg) and 0.9 mM NAD+ in 50 mM potassium phosphate buffer, pH 7.8 (1 mL total volume). After incubating overnight at 37 °C, the absorbance at 340 nm was determined and formate quantitated through comparison to a standard curve generated by spiking assay solutions with known amounts of formate. All measurements of either specific activity or steady-state kinetic parameters were performed in triplicate, and initial rate data were analyzed to obtain V and KM values by curve fitting with standard computer-based algorithms [21]. For experiments involving the E162Q OxDC mutant, 0.5 mM o-PDA was omitted from the reaction mixture because control experiments showed that this reagent, in contrast to wild type OxDC and all other site-specific mutants, did not increase the decarboxylase activity of this OxDC mutant.
Experiments to determine the pH profiles for oxalate decarboxylation by wild type OxDC and the E162Q OxDC mutant were carried out in 50 mM potassium citrate buffer over a pH range of 4.2–6.7. In these experiments (total volume 100 μL), however, the higher buffer capacity of citrate required the addition of 20 μL of 1 N NaOH to quench the enzyme-catalyzed reaction.
As well as measuring the rate at which formate was produced by the enzyme-catalyzed decarboxylation of oxalate, we routinely examined the oxidative activity of wild type OxDC and the OxDC mutants using a spectroscopic assay involving the dye ABTS [22]. Thus, the appropriate enzyme (final concentration 15–30 μg/mL) was incubated at 23°C with 5 mM ABTS and 50 mM potassium oxalate in 50 mM potassium acetate, pH 4.2 (total volume 1 mL). The rate of dye oxidation was determined in the absence or presence of horseradish peroxidase (HRP) (1000 U/mg; final concentration 25 μg/mL). Reactions were initiated by the addition of oxalate, and the initial rate of ABTS oxidation was measured by monitoring the change in solution absorbance at 650 nm as a function of time. Blank solutions were identical except that OxDC was omitted from the assay.
The relative rates of the “oxidase” and the “decarboxylase” activities of the E162Q OxDC mutant were determined by measuring the oxalate/formate stoichiometry. Thus, the E162Q OxDC mutant (0.24 mg/mL final concentration) was incubated with 20 mM potassium oxalate at 23 °C in 50 mM acetate buffer, pH 4.2, containing 0.2% Triton X-100 (1 mL total volume). The reaction was initiated by the addition of substrate and allowed to proceed for a total time of 300 min. Aliquots (50 μL) were removed at regular intervals and quenched by the addition of 1 N NaOH (10 μL). The concentrations of formate and residual oxalate were measured in each aliquot using the FDH-based assay and an oxalate detection kit (Trinity Biotechnology, Bray, Ireland), respectively. The amount of oxalate consumed in the oxidase reaction was then obtained by subtracting the concentrations of formate and residual oxalate from 20 mM.
Size Exclusion Chromatography
A BIOSEP SEC-S2000 column (300 × 7.8 mm with 75 × 7.8 mm guard column) (Phenomenex, Torrance, CA) was equilibrated with 100 mM potassium phosphate at pH 7 containing 100 mM KCl and samples were run at 1.0 mL/min flow rate. The column was calibrated with molecular weight standards: lysozyme (14.4 kDa), carbonic anhydrase (29.0 kDa), peroxidase (44.0 kDa), bovine serum albumin (66.0 kDa), alcohol dehydrogenase (150 kDa), amylase (200 kDa), apoferritin (443.0 kDa) and thyroglobuline (669.0 kDa). The void volume (Vo) of the column was measured by injecting blue dextran, and log10(MW) was plotted against Ve/Vo, where Ve corresponds to the volume required to elute a given reference protein of molecular weight (MW), to obtain a calibration curve. Samples of wild type OxDC and various OxDC mutants (10–50 μg) were loaded onto the column and their molecular weights estimated by interpolation using the calibration curve.
Determination of metal content
Samples of purified wild type OxDC or the OxDC mutants were treated to remove adventitiously bound surface metals by incubating the enzyme (6–10 mg/mL) with 2 μL of 100 mM EDTA, pH 11, prepared in Milli-Q water (200 μL total volume) for 15 min. The resulting solution was loaded onto a G-25 column in which the resin had been pre-treated with 1 M EDTA, pH 11 [23], and then equilibrated with Milli-Q (metal-free) water (resistivity 18.3 Ω). The enzyme was eluted with Milli-Q water and fractions showing activity in a qualitative, o-PDA-based assay [6] were pooled. After dilution of the protein solution with metal-free 1% aq. HNO3 (10 mL final volume), the samples were analyzed using inductively coupled plasma-mass spectrometry (ICP-MS) [24] at the Soil and Plant Analysis Laboratory (Madison, WI). Metal concentrations were determined by comparison to “blank” solutions containing only buffer that had been treated identically to the enzyme samples.
Isotope effect nomenclature
The nomenclature used in this work to describe the heavy-atom 13C and 18O IEs is that due to Northrop [25], in which 13(V/K) represents the Vmax/KM value for the 12C-containing substrate relative to the 13C-containing substrate, and 18(V/K) is the similar ratio for 16O and 18O.
13C and 18O isotope effect measurements
Natural abundance, heavy-atom 13C and 18O IEs were measured for a subset of the OxDC mutants under steady-state conditions, following an identical procedure to that reported previously for wild type OxDC [4]. These experiments employed reaction times sufficient for consumption of 17–51% or 100% of the oxalate substrate. For OxDC mutants that were not stable over these extended times (as evaluated by a non-linear rate of formate production) fresh portions of enzyme were added at intervals to maintain activity. The quantitative analysis of the observed IEs was performed on the basis of our standard kinetic model [4] (Scheme 1E).
RESULTS AND DISCUSSION
Expression and purification of the “untagged” site-specific OxDC mutants
A series of OxDC active site mutants were prepared by heterologous expression in Escherichia coli using an optimized procedure developed in our laboratory that yields wild type enzyme containing approximately 2.0 Mn/monomer [4]. Given that we considered both Mn-binding sites to be capable of catalyzing the decarboxylation reaction, we examined paired OxDC mutants in each of the cupin domains. Thus, Glu-162 (N-terminal) and Glu-333 (C-terminal) were replaced by either aspartate (E162D and E333D) or glutamine (E162Q and E333Q). We also sought to characterize the importance of the guanidine moiety in the catalytic mechanism by modifying Arg-92 and Arg-270 to lysine (R92K and R270K). In addition, double mutants were prepared in which cognate changes were made to cognate residues in both of the Mn-binding sites (R92K/R270K and E162Q/E333Q). Although some of these OxDC mutants have been studied previously [7, 8], the earlier work employed mutant enzymes engineered to contain a polyhistidine tag at either the N- or C-terminus. The presence of this tag appears to affect enzyme activity, perhaps because of its effects on the extent of Mn(II) incorporation or OxDC stability under the assay conditions. For example, the N-terminally tagged, wild type OxDC is reported to exhibit a specific activity of 21 U/mg [8] rather than the value of 79 U/mg that is reproducibly observed by us for the non-tagged enzyme [4].
Importantly, in order to obtain samples of the untagged, wild type OxDC containing approximately 2.0 Mn/monomer that was used in all of our experiments, the recombinant enzyme had to be purified by a rigorous procedure involving the use of both phenyl-Sepharose and anion-exchange column chromatography [4]. More specifically, purification on phenyl-Sepharose fractionates the recombinant OxDC and all of the N-terminal site-specific OxDC mutants (R92K, E162D, E162Q), into three distinct forms, the least hydrophobic of which exhibits the highest specific activity. Only two forms were seen for the C-terminal OxDC mutants (R270K, E333D, E333Q) and double mutants (R92K/R270K, E162Q/E333Q) when purified on phenyl-Sepharose, however, each of which had a longer retention time than the most “hydrophilic” variant of wild type OxDC. This change in purification behavior suggests that the structure of the C-terminal domain is very sensitive to changes in the side chains defining the Mn-binding site; a finding that is consistent with previous hypotheses that the role of this domain is to maintain the bicupin fold and/or quaternary structure of the enzyme [8]. For comparison, we therefore prepared a sample of the recombinant, wild type enzyme omitting purification on phenyl-Sepharose. This modified procedure gave wild type OxDC with a reduced specific activity of 30 U/mg, which was used as a reference value when evaluating kinetic and heavy-atom IE measurements on OxDC mutants in which residues in the C-terminal Mn-binding site had been mutated, including the double mutants.
Quaternary structure and metal content of the site-specific OxDC mutants
The effects of these conservative side chain mutations on the quaternary structure of OxDC and Mn incorporation were examined using size-exclusion chromatography and ICP-MS measurements (Table 1). Under our standard gel-filtration assay conditions, wild type enzyme exhibiting high specific activity (79 U/mg) eluted from the column both as a hexamer, consistent with previous crystallographic observations [7, 8], and as a larger aggregate form with a molecular weight of approximately 430 kDa. This corresponds to a species containing approximately 9 OxDC monomers, perhaps corresponding to a quaternary structure that is composed of one of the trimeric and one of the hexameric motifs observed by X-ray crystallography [7]. We find that wild type OxDC forms aggregates that can precipitate when the ionic strength of the solution is decreased. These aggregates can be resolubilized on increasing the ionic strength, however, without any loss in catalytic activity. As for the wild type enzyme, each of the OxDC mutants formed aggregates of molecular weight greater than 260 kDa (approximately corresponding to that expected for the OxDC hexamer) that eluted from the gel filtration column. The mutant enzymes E162D and E162Q, in which Glu-162 in the N-terminal Mn-binding site had been substituted, eluted as a hexamer in addition to these higher molecular weight aggregates. Modifications in the C-terminal domain gave proteins that eluted as monomers and trimers (Table 1). These findings provide some support for the hypothesis that the C-terminal domain plays a key role in controlling the overall structure of OxDC, perhaps due to changes in the helical regions of this domain that seem to mediate monomer/monomer interactions [7, 8, 26] but this remains to be established using biophysical measurements.
Similar observations were made concerning the effects of the site-specific mutations on the extent of Mn incorporation and/or the stability of the Mn-containing form of the enzyme. For example, replacement of Arg-92 by lysine gave an OxDC mutant (R92K) that could be purified to homogeneity using the rigorous protocol and Mn binding was not affected to any significant extent, presumably because this change does not perturb the electrostatic properties of the Mn-binding site. This contrasts with the effects of cognate changes in the C-terminal domain (R270K), which yield OxDC mutants that either incorporate very low levels of Mn, or bind metal so weakly that it can be removed by EDTA treatment prior to ICP-MS analysis. As expected, modifying the overall charge of the N-terminal binding site did perturb Mn binding. Hence, removal of a (putative) negative charge when Glu-162 was substituted by glutamine reduced the Mn content of the recombinant enzyme even though this OxDC mutant behaved very similarly to the wild type enzyme during purification and in size-exclusion chromatography.
Steady-state kinetic characterization of the site-specific OxDC mutants
In preliminary experiments to evaluate which OxDC mutants might be employed in IE measurements, the steady-state kinetic behavior of each enzyme was determined by monitoring the rate of formate production [4]. Product formation was linear over extended times for the E162D, E333D and R270K OxDC mutants and, as for wild type enzyme, the reaction could be driven to completion. In contrast, the progress curves for the R92K, E162Q and E333Q OxDC mutants were non-linear over long reaction times, and oxalate could not be completely converted to formate, even with excess enzyme. Specific activities and oxalate KM values were measured for the complete set of OxDC mutants for times over which product formation was linear (Table 1). Turnover numbers (kcat) were, however, computed only for N-terminal domain variants that could be purified to homogeneity using a phenyl-Sepharose chromatographic step (Table 2).
Table 2.
Steady state kinetic properties of recombinant, wild type OxDC and N-terminal OxDC mutants.a
In these experiments, perhaps the most clear-cut result was the considerable reduction in kcat/KM for the R92K OxDC mutant relative to that of wild type enzyme even though both enzymes had similar levels of Mn incorporation (Table 2), and this result implicates Arg-92 as one of the key players in catalysis. To establish this conclusion more rigorously, we therefore expressed and characterized the OxDC mutant in which Arg-92 was replaced by alanine (data not shown). The catalytic activity of this OxDC mutant was so low, however, as to preclude its detailed characterization in our IE measurements.
The replacement of Glu-162 by aspartate or glutamine gave OxDC mutants exhibiting approximately 50% and 0.5% of the specific activity seen for the wild type enzyme, respectively, consistent with its hypothesized role in general acid/base catalysis. The decrease observed for the E162D OxDC mutant is probably a result of the reduced level of Mn incorporation (Table 1) rather than an effect of shortening the side chain on catalysis because the kcat and kcat/KM values, when normalized for Mn content, are close to those of the wild type enzyme (Table 2). In the case of the E162Q OxDC mutant, however, kcat and kcat/KM, when normalized for Mn content, are decreased about 50- and 100-fold, respectively, compared to wild type OxDC. This observation can be rationalized by assuming that (i) Glu-162 plays an important role in catalysis, or (ii) Mn preferentially binds to a site in which it is catalytically inactive, leading to an underestimation of the calculated kcat value. Evidence presented below supports the first of these hypotheses. We also note that the values of kcat and kcat/KM, when normalized for Mn content, observed for the R92K OxDC mutant, in which Mn incorporation is 1.7 Mn/monomer, are similar to those determined for E162Q. Changes to Glu-333, located in the C-terminal cupin domain of the enzyme, gave OxDC mutants for which approximately 10- and 100-fold decreases in specific activity were observed when this residue was modified to aspartate and glutamine, respectively (Table 1). Given the impact on Mn binding and quaternary structure of mutations in the C-terminal domain of OxDC, it seems likely that decreases in the specific activity of the R270K, E333D and E333Q OxDC mutants at least partially reflect changes in enzyme structure, severely complicating efforts to elucidate the functional role of Arg-270 and Glu-333. Examination of the R92K/R270K and E162Q/E333Q OxDC double mutants also showed (perhaps unsurprisingly) that their activities were very similar to those of the R270K and E333Q OxDC mutants, respectively.
Given the reduced catalytic activity of the E162Q OxDC mutant, which was substantially lower that expected based on the level of Mn incorporation, we examined the pH-dependence of Vmax using similar experiments to those performed on the wild type enzyme [4]. These studies showed that the dependence of log10(V) on pH for wild type OxDC and the E162Q OxDC mutant exhibited different slopes (Figure 2). Hence, at least qualitatively, removal of the glutamate side chain changes either the rate-limiting step for the decarboxylation reaction, or the pKa of an ionizable group controlling the rate-limiting step. The second of these possibilities is consistent with a model in which the glutamate side chain in wild type OxDC functions as an active site base (Scheme 1A).
Fig. 2.

pH-dependence of log10(V) for recombinant Bacillus subtilis OxDC (■) and the E162Q OxDC mutant enzyme (□).
Investigating the oxidative activities of the site-specific OxDC mutants
Despite the similarity in the Mn-binding sites of OxDC and OxOx (Figure 3), the Bacillus subtilis enzyme oxidizes oxalate at a steady state rate that is only 0.2% of that for decarboxylation [6]. We therefore characterized the OxDC mutants to evaluate whether modification of the arginine and glutamate residues had produced enzyme capable of oxidizing rather decarboxylating oxalate. The experiments employed a simple assay in which the ability of a given OxDC mutant to form hydrogen peroxide by oxidizing oxalate to two molecules of CO2 was determined by monitoring the rate of ABTS oxidation in the presence of HRP [22]. Because it had also been reported that ABTS oxidation can be mediated by wild type OxDC to a small extent [6], by an (as yet) undetermined mechanism, we also carried out these kinetic measurements in the absence of peroxidase. With the exception of the E162Q OxDC mutant, all of the OxDC mutants exhibited very low levels of ABTS oxidative activity in the presence or absence of peroxidase (Table 3); a finding consistent with previous reports [6, 8]. We note that (i) ABTS oxidation does not take place under our conditions when oxalate is omitted from the reaction mixture, and (ii) the rates of oxidation are always increased when peroxidase is present in solution (Table 3).
Fig. 3. Structural representations of Hordeum vulgare oxalate oxidase (2ET1) [31], and Bacillus subtilis oxalate decarboxylase (1UW8) [8].
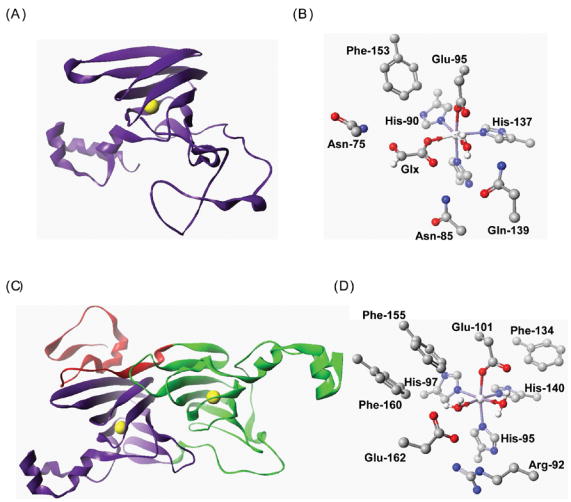
(A) Ribbon representation of the OxOx cupin domain showing the location of the Mn ion (yellow). (B) Structure of the Mn-binding site in OxOx showing coordination of Mn by His, His, His and Glu. The metal is also coordinated by glycolate (Glx) and water molecules. (C) Ribbon representation of the OxDC bicupin structure showing the location of the two Mn ions (yellow) in each cupin domain. In this structure, an N-terminal segment (red) forms part of the C-terminal cupin domain. (D) Structure of the N-terminal Mn-binding site in OxDC showing coordination of Mn by His, His, His, Glu and two water molecules. In figures (B) and (D) all non-hydrogen atoms are shown at their crystallographic coordinates, and hydrogens on protein residues are omitted for clarity. [Key: C – grey; H – white; N – blue; O – red; Mn - silver]
Table 3.
ABTS oxidation activity of recombinant, wild type OxDC and OxDC mutants.
| ABTS Oxidation Activity (U/mg) −Peroxidase | ABTS Oxidation Activity (U/mg) + Peroxidase | Specific Activity (U/mg) Decarboxylase Activity | |
|---|---|---|---|
| WT OxDC | 0.07 ± 0.01 | 0.12 ± 0.02 | 79 ± 2 |
| R92K | 0.04 ± 0.01 | 0.08 ± 0.02 | 1.3 ± 0.4 |
| E162D | 0.07 ± 0.01 | 0.12 ± 0.02 | 40 ± 3 |
| E162Q | 0.6 ± 0.1 | 1.16 ± 0.03 | 0.4 ± 0.1 |
| R270K | 0.02 ± 0.01 | 0.03 ± 0.01 | 2.4 ± 0.4 |
| E333D | 0.04 ± 0.01 | 0.08 ± 0.01 | 4 ± 1 |
| E333Q | 0.07 ± 0.01 | 0.12 ± 0.01 | 0.4 ± 0.1 |
| R92K/R270K | 0.04 ± 0.03 | 0.07 ± 0.01 | 0.4 ± 0.1 |
| E162Q/E333Q | 0.04 ± 0.03 | 0.05 ± 0.01 | 0.7 ± 0.1 |
In the case of the E162Q OxDC mutant, the specific activity of dye oxidation in the absence of peroxidase was 0.6 ± 0.1 U/mg, which is (i) comparable to that for the E162Q-catalyzed decarboxylation of oxalate (0.4 ± 0.1 U/mg) (Table 3), and (ii) an order of magnitude greater than the intrinsic ability of wild type OxDC to oxidize ABTS directly during catalytic turnover. This observation is in sharp contrast to the much lower rate of direct ABTS oxidation reported for the N-terminally tagged form of the E162Q OxDC mutant [8]. The basis for this discrepancy is likely due to the fact that peroxidase-coupled dye oxidation appears to be non-linear with time, implying that the correct initial rate was not measured in the previous studies.
The improved ability of the E162Q OxDC mutant to catalyze ABTS oxidation in the absence of peroxidase, raises a number of intriguing questions concerning the nature of the species that mediates the reaction. For example, if the oxidizing species is an intermediate formed on the main catalytic pathway, or even in a side reaction, an increase in ABTS oxidation rate suggests that the presence of the glutamine side chain can increase the lifetime of this species. Alternatively, the charge on the carboxylate moiety of Glu-162 may also play an important role in the conformational change that converts OxDC from an “open” to a “closed” form [8]. In this case, it is possible that the mutant enzyme remains in an “open” form permitting the dye molecule to access the Mn-binding site and any radical intermediates that are formed during enzyme-catalyzed cleavage of the oxalate C-C bond.
When the E162Q OxDC mutant was incubated with oxalate solution in the presence of peroxidase, the rate of ABTS oxidation increased (Table 3), suggesting that the mutant enzyme could catalyze the oxidation of oxalate. So as to rule out any unanticipated effects on enzyme structure or chemistry associated with the presence of ABTS, however, we re-examined the catalytic behavior of the E162Q OxDC mutant using an alternative assay in which oxalate oxidase activity was directly established by measuring the stoichiometry of formate production and oxalate consumption. In these experiments, we observed that (i) the rate of E162Q-catalyzed oxidation of oxalate showed an initial burst before dropping to zero after 50 min, and (ii) the total amount of oxalate converted to two molecules of CO2 by this “side-reaction” was approximately 2.5 mM (Figure 4). Because the concentration of dioxygen dissolved in buffer at 23 °C under air at 1 atm pressure is approximately 270 μM [27], however, the reason for this cessation in the oxidase activity of this OxDC mutant is not immediately obvious, especially as the decarboxylase activity persists for up to 200 min. The addition of 5 mM formate to the reaction mixture did not appear to affect the oxidase activity of the E162Q OxDC mutant, however, ruling out the possibility that this reaction product was suppressing the oxidation reaction. Moreover, as these assays were performed at pH 4.2, it also seems unlikely that bicarbonate ion was adversely impacting the oxidase reaction. Since hydrogen peroxide does not inhibit OxDC-catalyzed decarboxylation (data not shown), it is possible that cessation of the oxidase activity might arise from a side reaction that leaves Mn in an incorrect oxidation state for catalysis.
Fig. 4. Progress curves showing the time-dependence of the oxalate oxidase (■) and oxalate decarboxylase (□) activities of the E162Q OxDC mutant.

In these experiments, the initial oxalate concentration was 20 mM (see Materials and Methods). The extent to which oxalate was consumed by the decarboxylase activity was assessed by measuring formate, while oxidase activity was given by the expression: [oxalate oxidized] = 20 – [formate] – [residual oxalate].
The appearance of enhanced oxalate oxidase activity in the E162Q OxDC mutant, however, is consistent with the notion that the N-terminal Mn-binding site is catalytically active. The enhancement of oxidative activity in the absence of the Glu-162 carboxylate would then be consistent with our hypothesis in which the functional role of the Glu-162 side chain is to control protonation of the formate radical anion so as to prevent a second electron transfer to Mn(III)-bound superoxide thereby generating hydroperoxide anion and another CO2. On this point, we note that an asparagine residue is positioned in the active site of oxalate oxidase in place of the glutamate seen in OxDC (Figure 2), reinforcing the idea that protein environment can modulate the reactivity of the Mn(II) center. In contrast to the behavior of the E162Q OxDC mutant, the analogous E333Q variant did not exhibit any ABTS oxidative activity that was significantly increased relative to that of wild type OxDC in the presence or absence of peroxidase.
These findings on the catalytic activities of the E162Q OxDC mutant have important implications for more general questions concerning the extent to which local protein environment can modulate the intrinsic chemical reactivity of transition metal centers [28–30]. X-ray crystallographic [7, 8, 13, 31] and sequence alignment [14] studies suggest that oxalate oxidase (OxOx) [32] is evolutionarily related to OxDC [2]. OxOx is another manganese-dependent member of the cupin superfamily [9, 10], which oxidizes oxalate to two molecules of CO2 with the concomitant reduction of dioxygen to hydrogen peroxide (Equation 2). OxDC and OxOx therefore employ the same substrate (oxalate) and contain manganese centers in which the metal ion is coordinated by identical ligands (Figure 2) but catalyze different chemical transformations. Computational studies on model structures for the metal center in OxDC [18] suggest that the intrinsic reactivity of the metal is oxidative, as observed in the reaction catalyzed by oxalate oxidase [33] Hence, Mn-catalyzed decarboxylation rather than oxidation of oxalate is presumably controlled by the proton transfers mediated by the glutamate side chain (Glu-162 and perhaps Glu-333) in the OxDC active site(s) [18]. In such a model, the inability to re-protonate the Mn-bound formate radical anion might permit an additional electron transfer reaction to metal-bound superoxide anion and the formation of an additional molecule of CO2. Support for this hypothesis is 18 provided by the fact that the cognate residue to Glu-162 in the OxOx active site, Asn-85 (Figure 2), cannot participate in proton transfer even though it can interact with the Mn-bound substrate and reaction intermediates, at least as judged from a recent crystal structure of the OxOx/glycolate complex [31]. The change in catalytic behavior that can be achieved by the replacement of a single protein residue in the E162Q OxDC mutant therefore reveals the importance of small functional group changes to the evolution of new activities, as required by current models [34, 35].
Steady-state 13C and 18O isotope effects
Based on their steady-state properties, heavy atom 13C and 18O kinetic isotope effects (IEs) were determined for the decarboxylase reaction catalyzed by the R92K, E162Q, R270K and E333D OxDC mutants, using the methodology developed for studies on the recombinant, wild type enzyme (Table 4) [4]. Similar studies were not carried out on the E162D OxDC mutant because we anticipated the observed 13C and 18O KIEs would be very similar to those of wild type OxDC based on the relative high activity of the mutant enzyme. IEs represent a powerful approach to probe hypotheses about functional roles of putative active site residues because they report only on active forms of OxDC, i.e. those enzyme molecules in which Mn is correctly incorporated. On the other hand, and somewhat unfortunately for efforts to assess the catalytic function of Glu-162, the competing oxidation and decarboxylation reactions catalyzed by the E162Q OxDC mutant under steady-state conditions preclude interpretation of the observed heavy-atom IEs because the amount of labeled CO2 that is formed by oxalate oxidation rather than decarboxylation cannot be determined accurately. As a result, heavy-atom IEs were not measured for the E162Q OxDC mutant.
Table 4.
13C and 18O isotope effects on the reaction catalyzed by selected Mn-binding site mutants of Bacillus subtilis OxDC.a
| OxDC | 13(V/K) (%) | 18(V/K) (%)b | |||
|---|---|---|---|---|---|
| CO2 | Formate | CO2 | Formate | ||
| WT OxDCc | 4.2 (BHEP)d | 0.5 ± 0.1 | 1.5 ± 0.1 | −0.2 ± 0.2 | 1.1 ± 0.2 |
| WT OxDC | 5.7 (Piperazine) | 0.8 ± 0.1 | 1.9 ± 0.1 | −0.7 ± 0.1 | 1.0 ± 0.1 |
| R92K | 4.2 (BHEP) | 0.4 ± 0.1 | 1.0 ± 0.1 | −0.5 ± 0.1 | 0.7 ± 0.1 |
| R92K | 5.7 (Piperazine) | 1.1 ± 0.1 | 1.9 ± 0.1 | −0.4 ± 0.2 | 0.8 ± 0.2 |
| R270K | 4.2 (BHEP) | 0.5 ± 0.1 | 1.4 ± 0.1 | 0.0 ± 0.2 | 0.4 ± 0.2 |
| R270K | 5.7 (Piperazine) | 1.5 ± 0.1 | 2.0 ± 0.1 | −0.5 ± 0.2 | 0.0 ± 0.2 |
| E162Q | 4.2 (BHEP) | 0.9 ± 0.1 | 1.5 ± 0.1 | −1.4 ± 0.2 | 0.9 ± 0.1 |
| E333D | 4.2 (BHEP) | 0.8 ± 0.1 | 1.5 ± 0.1 | −0.8 ± 0.1 | 0.7 ± 0.1 |
| E333D | 5.7 (Piperazine) | 1.3 ± 0.1 | 2.0 ± 0.1 | −0.8 ± 0.1 | 1.5 ± 0.1 |
Values are reported for 27–43 mM oxalate at 23 °C.
Isotope effect is given per 2 oxygen atoms.
Measurements for wild type OxDC have been reported previously [4] and are included here for ease of comparison.
BHEP: 1,4-bis-(2-hydroxyethyl)-piperazine.
In the case of the R92K, R270K and E333D OxDC mutants, no dramatic changes were observed for the heavy-atom IEs, suggesting that these site-specific changes had affected the relative rates of individual steps in the catalytic mechanism rather than the overall kinetic mechanism of the enzyme. Unfortunately, any detailed interpretation of the IEs determined for the R270K and E333D OxDC mutants is precluded by the fact that these mutations in the C-terminal Mn-binding site may modify enzyme structure. On the other hand, because of the high level of Mn incorporation for the R92K OxDC mutant, and the fact that highly pure enzyme could be obtained using phenyl-Sepharose chromatography, we undertook a quantitative analysis of the experimental IEs for this OxDC mutant using the model developed in our previous studies of OxDC (Scheme 1E) [4]. In this analysis, the following equation describes the effects of isotopic substitution:
where k1-k5 are rate constants (Scheme 1E), xKeq3 is the equilibrium IE for the formation of the intermediate species containing the putative oxalate radical anion, xk5 is the IE on decarboxylation, and x = 13 or 18 for a 13C or 18O IE, respectively. The use of this equation to obtain estimates of k3/k2 and k5/k4 is presented fully in the supporting information for this paper. 13C or 18O fractionation factors computed from the values of 13Keq3 and 18Keq3, respectively, (Table 5) yielded a C-O bond order of 1.22 for the Mn-bound carbonyl group in the transition state for decarboxylation of the radical anion intermediate (Scheme 1B). This value is greater than that observed for the reaction catalyzed by wild type OxDC (1.16), suggesting that the C-O bond is less polarized during the R92K-catalyzed decarboxylation (at least as computed on the basis of our kinetic model). In turn, the k5/k4 ratio is decreased because decarboxylation is slowed down in the R92K OxDC mutant relative to wild type enzyme, implying that polarization of this C-O bond may also destabilize the transition state associated with proton-coupled electron transfer. Given that the mutation of arginine to lysine likely maintains the charge of the side chain, the change in bond polarization is probably associated with the fact that the ammonium ion in lysine is located farther from the Mn-bound substrate. This change in distance could then affect the electrostatic field felt by the carboxyl moiety bound to the metal and also weaken any hydrogen bonding interaction that might be needed to stabilize the transition state for decarboxylation. 20 Whatever the molecular details, removal of this charged side chain in the R92A OxDC mutant yields an enzyme with activity that is considerably lower than that of R92K even though the extent of Mn incorporation is similar.
Table 5.
Quantitative interpretation of 13C and 18O IEs using the standard kinetic model (Scheme 1E).
| 13Keq3 | 18Keq3 | k3/k2 | k5/k4 | 13C FFa | Bond Orderb | Calc’d./Exptl.13C IE (%)c | (V/K)/[E]ok1 | [E]ok1 (%) | |
|---|---|---|---|---|---|---|---|---|---|
| OxDCd | 1.021 | 1.016 | 0.7 | 4.0 | 0.963 | 1.16 (1.14) | 1.2/1.5 | 0.375 | 100 |
| R92K | 1.017 | 1.011 | 2.4 | 2.6 | 0.968 | 1.22 (1.22) | 0.7/1.0 | 0.636 | 1 |
Fractionation factor for 13C at pH 4.2 calculated from the standard kinetic model for the OxDC-catalyzed reaction as described previously [4].
Bond orders are based on the results of BEBOVIB calculations [36] as reported previously [4]. Values in parentheses are the bond orders calculated on the basis of 18O fractionation factors (not shown here). nd – not computed.
13C IEs are calculated for the reaction at pH 4.2 using values of 13Keq3, 13k3, 13k5, k3/k2, k5/k4 computed from observations at pH 5.7. Experimental IEs are those observed at pH 4.2.
Measurements for wild type OxDC have been reported previously [4] and are included here for ease of comparison.
Evidence to suggest that Arg-92 may play multiple roles in enzyme function, however, is provided by manipulation of the steady-state equations describing our minimal mechanism of decarboxylation (Scheme 1E). Thus, the equation for V/K can be expressed in the following form:
and rearranging gives:
Using this form of the equation, we were able to calculate relative values of k1[E]0 for wild type OxDC and the R92K OxDC mutant because the V/K ratio (Table 2) and estimates of the relevant commitment factors (k3/k2 and k5/k4) (Table 5) were available. In addition, because (i) Mn was incorporated into wild type OxDC and the R92K mutant to a similar extent, and (ii) approximately equal concentrations of both enzymes were used in our IE experiments, the relative decrease in k1[E]0 calculated for the R92K OxDC mutant probably reflects an alteration in the rate of oxalate binding (k1). Hence, Arg-92 may not only function to polarize the Mn-bound oxalate radical anion, but also be involved in positioning oxalate in a catalytically productive orientation, or perhaps facilitating any conformational change in the mobile loop (residues 161–165) [8] to render this Mn-binding site inaccessible to solvent after substrate binding.
CONCLUSIONS
The results of these studies provide a clear picture of the technical difficulties associated with prior site-directed mutagenesis experiments, which might explain the conflicting conclusions in the literature regarding the location of the catalytic site(s) in OxDC. Our data are consistent with the hypothesis that the N-terminal Mn-binding site catalyzes the transformation of oxalate to formate and CO2, as proposed previously [8], and our proposal for the functional role of Arg-92 [4]. Hence, replacement of this residue by lysine lowers the specific activity of the enzyme, and lowers the amount of C-O polarization in the Mn-bound C-O moiety of the substrate that eventually becomes formate (Scheme 1B). The replacement of Arg-92 by lysine also appears to affect the rate of substrate binding (k1).
Our observations on the E162D and E162Q OxDC mutants also suggest that this residue functions as a general base in the catalytic mechanism of decarboxylation. Hence, although replacement of the glutamate by a chemically similar, but shorter, aspartate residue lowers enzyme activity, this may result from lowered Mn incorporation rather than significant perturbations to proton transfer steps. The importance of controlling proton transfers within the active site is also shown by the gain in oxidase activity that is observed for the E162Q OxDC mutant. On the other hand, because changes to the residues in the C-terminal domain impact quaternary structure and the level of Mn incorporation in the recombinant enzyme, it is difficult to interpret the outcome of steady-state kinetic measurements and obtain clear evidence against the notion that the C-terminal Mn-binding site also possesses catalytic activity. The use of alternate experimental strategies will therefore likely be required to determine if the C-terminal site also has catalytic activity.
Supplementary Material
ABBREVIATIONS
- ABTS
2,2′-azinobis-(3-ethylbenzthiazoline-6-sulphonic acid)
- DEAE
diethylaminoethanol
- EPR
electron paramagnetic resonance
- FDH
formate dehydrogenase
- HRP
horseradish peroxidase
- ICP-MS
inductively coupled plasma mass spectroscopy
- o-PDA
ortho-phenylenediamine
- OxDC
oxalate decarboxylase
- OxOx
oxalate oxidase
Footnotes
This work was supported by National Institutes of Health grants R01 GM18938 (W.W.C.) and R01 DK61666 (N.G.J.R.)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shimazono H, Haiyashi O. J Biol Chem. 1957;227:151–159. [PubMed] [Google Scholar]
- 2.Svedružić D, Jónsson S, Toyota CG, Reinhardt LA, Ricagno S, Lindqvist Y, Richards NGJ. Arch Biochem Biophys. 2005;433:176–192. doi: 10.1016/j.abb.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 3.Begley TP, Ealick SE. Curr Op Chem Biol. 2004;8:508–515. doi: 10.1016/j.cbpa.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Reinhardt LA, Svedruzic D, Chang CH, Cleland WW, Richards NGJ. J Am Chem Soc. 2003;125:1244–1252. doi: 10.1021/ja0286977. [DOI] [PubMed] [Google Scholar]
- 5.Tanner A, Bornemann S. J Bacteriol. 2000;182:5271–5273S. doi: 10.1128/jb.182.18.5271-5273.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanner A, Bowater L, Fairhurst SA, Bornemann S. J Biol Chem. 2001;276:43627–43634. doi: 10.1074/jbc.M107202200. [DOI] [PubMed] [Google Scholar]
- 7.Anand R, Dorrestein PC, Kinsland C, Begley TP, Ealick SE. Biochemistry. 2002;41:7659–7669. doi: 10.1021/bi0200965. [DOI] [PubMed] [Google Scholar]
- 8.Just VJ, Stevenson CEM, Bowater L, Tanner A, Lawson DM, Bornemann S. J Biol Chem. 2004;279:19867–19874. doi: 10.1074/jbc.M313820200. [DOI] [PubMed] [Google Scholar]
- 9.Dunwell JM, Purvis A, Khuri S. Phytochem. 2004;65:7–17. doi: 10.1016/j.phytochem.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 10.Dunwell JM, Khuri S, Gane PJ. Microbiol Mol Biol Rev. 2000;64:153–179. doi: 10.1128/mmbr.64.1.153-179.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muthusamy M, Burrell MR, Thorneley RNF, Bornemann S. Biochemistry. 2006;45:10667–10673. doi: 10.1021/bi060460q. [DOI] [PubMed] [Google Scholar]
- 12.Chang CH, Svedruzic D, Ozarowski A, Walker L, Yeagle G, Britt RD, Angerhofer A, Richards NGJ. J Biol Chem. 2004;279:52840–52849. doi: 10.1074/jbc.M402345200. [DOI] [PubMed] [Google Scholar]
- 13.Woo EJ, Dunwell JM, Goodenough PW, Marvier AC, Pickersgill RW. Nat Struct Biol. 2000;7:1036–1040. doi: 10.1038/80954. [DOI] [PubMed] [Google Scholar]
- 14.Escutia MR, Bowater L, Edwards A, Bottrill AR, Burrell MR, Polanco R, Vicuña R, Bornemann S. Appl Environ Microbiol. 2005;71:3608–3616. doi: 10.1128/AEM.71.7.3608-3616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cleland WW. Methods Enzymol. 1995;249:341–373. doi: 10.1016/0076-6879(95)49041-8. [DOI] [PubMed] [Google Scholar]
- 16.Cleland WW. Adv Enzymol Relat Areas Mol Biol. 1977;45:273–387. doi: 10.1002/9780470122907.ch4. [DOI] [PubMed] [Google Scholar]
- 17.Northrop DB. Annu Rev Biochem. 1981;50:103–131. doi: 10.1146/annurev.bi.50.070181.000535. [DOI] [PubMed] [Google Scholar]
- 18.Chang CH, Richards NGJ. J Chem Theor Comput. 2005;1:994–1007. doi: 10.1021/ct050063d. [DOI] [PubMed] [Google Scholar]
- 19.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 20.Schutte H, Flossdorf J, Sahm H, Kula MR. Eur J Biochem. 1976;62:151–160. doi: 10.1111/j.1432-1033.1976.tb10108.x. [DOI] [PubMed] [Google Scholar]
- 21.Cleland WW. Methods Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 22.Requena L, Bornemann S. Biochem J. 1999;343:185–190. [PMC free article] [PubMed] [Google Scholar]
- 23.Holmquist B. Methods Enzymol. 1988;158:6–14. doi: 10.1016/0076-6879(88)58042-4. [DOI] [PubMed] [Google Scholar]
- 24.Olivares JA. Methods Enzymol. 1988;158:205–232. doi: 10.1016/0076-6879(88)58057-6. [DOI] [PubMed] [Google Scholar]
- 25.Northrop DB. In: Isotope Effects on Enzyme-Catalyzed Reactions. Cleland WW, O’Leary MH, Northrop DB, editors. University Park Press; Baltimore: 1977. pp. 122–148. [Google Scholar]
- 26.Dunwell JM, Culham A, Carter CE, Sosa-Aguirre CR, Goodenough PW. Trends Biochem Sci. 2001;26:740–746. doi: 10.1016/s0968-0004(01)01981-8. [DOI] [PubMed] [Google Scholar]
- 27.Lide DR, editor. CRC Handbook of Chemistry and Physics. 85. CRC Press; Boca Raton: 2004. [Google Scholar]
- 28.Emerson JP, Wagner ML, Reynolds MF, Que L, Sadowsky MJ, Wackett LP. J Biol Inorg Chem. 2005;10:751–760. doi: 10.1007/s00775-005-0017-1. [DOI] [PubMed] [Google Scholar]
- 29.Jackson TA, Brunold TC. Acc Chem Res. 2004;37:461–470. doi: 10.1021/ar030272h. [DOI] [PubMed] [Google Scholar]
- 30.Edwards RA, Whittaker MM, Whittaker JW, Baker EN, Jameson GB. Biochemistry. 2001;40:15–27. doi: 10.1021/bi0018943. [DOI] [PubMed] [Google Scholar]
- 31.Opaleye O, Rose RS, Whittaker MM, Woo EJ, Whittaker JW, Pickersgill RW. J Biol Chem. 2006;281:6428–6433. doi: 10.1074/jbc.M510256200. [DOI] [PubMed] [Google Scholar]
- 32.Whittaker MM, Whittaker JW. J Biol Inorg Chem. 2002;7:136–145. doi: 10.1007/s007750100281. [DOI] [PubMed] [Google Scholar]
- 33.Borowski T, Bassan A, Richards NGJ, Siegbahn PEM. J Chem Theor Comput. 2005;1:686–693. doi: 10.1021/ct050041r. [DOI] [PubMed] [Google Scholar]
- 34.Gerlt JA, Babbitt PC. Annu Rev Biochem. 2001;70:209–246. doi: 10.1146/annurev.biochem.70.1.209. [DOI] [PubMed] [Google Scholar]
- 35.Yoshikuni Y, Ferrin TE, Keasling JD. Nature (Lond) 2006;440:1078–1082. doi: 10.1038/nature04607. [DOI] [PubMed] [Google Scholar]
- 36.Berti PJ. Methods Enzymol. 1999;308:355–397. doi: 10.1016/s0076-6879(99)08016-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.