

Abstract
Epitope-based vaccines provide a new strategy for prophylactic and therapeutic application of pathogen-specific immunity. A critical requirement of this strategy is the identification and selection of T-cell epitopes that act as vaccine targets. This study describes current methodologies for the selection process, with dengue virus as a model system. A combination of publicly available bioinformatics algorithms and computational tools are used to screen and select antigen sequences as potential T-cell epitopes of supertype HLA alleles. The selected sequences are tested for biological function by their activation of T-cells of HLA transgenic mice and of pathogen infected subjects. This approach provides an experimental basis for the design of pathogen specific, T-cell epitope-based vaccines that are targeted to majority of the genetic variants of the pathogen, and are effective for a broad range of differences in human leukocyte antigens among the global human population.
Keywords: T-cell epitopes, epitope-based vaccines, bioinformatics, pathogens, immune system, entropy, conserved sequences, immunological hotspots, altered-ligand effect, supertypes
1. Introduction
New developments in immunoinformatics and other computational methodologies, combined with the broad versatility in the design and synthesis of genetic (DNA) vaccines, underlay new strategies for the novel design of antigen-specific, epitope-based vaccines against the many pathogens that currently have proven refractive to conventional vaccine therapy [1,2]. Early clinical trials of epitope-based vaccines for human immunodeficiency virus (HIV), malaria and tuberculosis have produced promising results [3,4], supporting the protective and therapeutic uses of these vaccines. T-cell epitopes, important for cytolytic and regulatory responses to pathogens [5–7], are necessary elements of these vaccines. The rational selection of protein antigen sequences that function as T-cell epitopes in vaccine formulations is therefore crucial for successful application of this vaccination strategy [2,8].
This selection of pathogen antigen sequences to be included in epitope-based vaccines must address several determinative issues. The goal is to identify relevant T-cell epitopes, both HLA class I and II, that are both effective and sufficient in vaccine protection against pathogen challenge. A major question is the degree of protection that can be achieved without the concomitant administration of neutralizing antibody epitopes. Vaccines must also protect a broad spectrum of human population against as wide a variety of pathogenic strains as possible; this presents further challenges. Many pathogens exhibit high mutation rates, with selection of new genetic variants that are resistant to an existing immune response to earlier pathogen subtypes, or may subvert the immune response by the altered peptide ligand phenomena [9–11]. It is therefore important to choose epitopes derived from conserved peptide sequences. Also, the extreme polymorphism that characterizes human leukocyte antigens (HLAs) restricts the proportion of the human population that will respond to a particular antigen [8,12]. Thus, it is advantageous to select promiscuous T-cell epitopes that bind to several alleles of HLA supertypes for maximal population coverage [13]. The focus is on a bioinformatics-based approach as a means to enhance the optimal selection of potential targets of immune response that can then be validated by experiments that test the biological function of these antigen sequences in immune-system based assays.
In this report, we describe a combined immunoinformatics and molecular strategy for vaccine development. Based upon the growing number of bioinformatics tools and antigen sequences available in public databases [14] for identifying pathogen peptides, the in silico prediction of T-cell epitopes can greatly reduce the list of candidate epitopes. Such a shortlist is then the starting point for molecular experiments that can validate the vaccine targets based on the biological function of the selected antigen sequences.
2. Methodology and Results
2.1 Data collection and preparation
Predictions about future mutations are derived from past evolutionary history. It is therefore important to collect sequences that are as representative as possible of the genetic variants of the pathogen, over extended periods of time and broad geographical ranges. Ideally, all available protein sequences pertaining to the pathogen should be collected from major public databases, such as the NCBI Entrez protein database (www.ncbi.nlm.nih.gov/entrez). Since public databases often contain errors, discrepancies and duplicate entries, a data cleaning process is needed to correct such anomalies [15]. For example, annotation errors and discrepancies in 17 dengue virus records were identified and corrected prior to analysis [16]. While several methods are available, we found the ABK structural rule-based approach [17] well suited to this type of task, allowing fully annotated sets of over 40,000 influenza protein sequences to be cleaned and independently verified in two weeks.
2.2 Identification of conserved sequences
The identification of conserved sequences is an initial step to overcome pathogen genomic variation that in some cases is extensive, such as HIV, influenza A viruses and dengue viruses. Multiple sequence alignments of pathogen proteins are examined by a consensus-sequence based approach [18] for the selection of sequences conserved in the large majority of variants. For pathogens with multiple groups (clades, serotypes or subtypes), pan-group consensus sequences are obtained by aligning consensus sequences derived from each of the different groups (Fig. 1), rather than by analyzing pan-group alignments that combine sequences from all groups. This prevents over-represented groups from biasing the derived consensus sequence. Identification of conserved alignment sites is based on the representation (frequency) of the consensus residue among all sequences in the alignment. Depending on the variability exhibited by different pathogen groups, the cut-off intra-group representation for conserved sequences may be set between 50% and 100%. For example, in our dengue virus analysis we only selected conserved sites common across the four serotypes, exhibiting at least 80% representation in each of the four serotypes (Fig. 2). For immunological applications, a minimum conserved sequence length of nine amino acids is required because this represents the typical length of peptides that bind to HLA molecules [19].
Fig. 1.
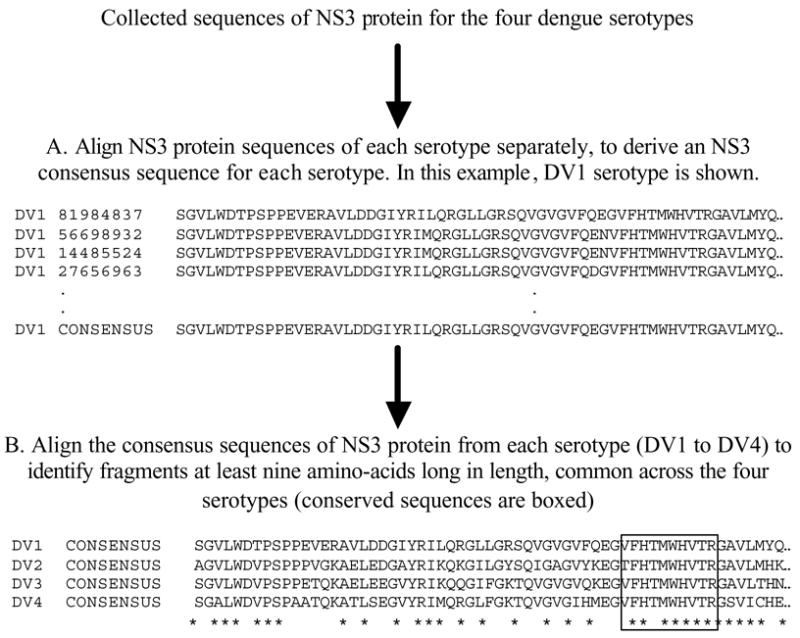
Steps involved in determining sequence fragments conserved across the four serotypes for NS3 protein using a consensus-sequence-based approach. A) The consensus sequence for NS3 protein is derived for each serotype (DV1–4) from their respective multiple sequence alignment. Each residue in the consensus sequence represents the predominant residue at that position in the corresponding multiple sequence alignment. B) The four consensus sequences of NS3 protein (one from each serotype) are aligned to reveal sequence fragments that are at least nine amino acids long and identical across the four consensus sequences.
Fig. 2.

Dengue pan-serotype conserved sequences of the NS3 protein and their intra-serotype percentage representation (conservation). The amino acid positions are numbered according to the aligned sequences of dengue proteins from all four serotypes.
2.3 Entropy-based analysis of conserved sequence variability
Consensus-based methods consider each alignment site independently. However, vaccine targets are short peptides, typically 9-mers, whose combinatorial composition can produce great diversity even when adjacent sites have highly conserved residues. A more robust method based on information entropy [20] can measure the degree of variability of peptides of any length, and infer their evolutionary stability. Entropy, H, representing the variability of nonamer peptides (9-mers) centered at any given alignment site, is computed from the probability, pa of each nonamer peptide a occurring at that site:
Peptides centered at any given position partially overlap peptides centered at neighbouring positions. Low entropy characterizes stable peptides, and an entropy value of 0 indicates a 100% conserved nonamer. Entropy rises with increasing variability of a site, and is affected both by the number of variants at that site, and also by their respective frequency. The ABK-AVANA antigenic variability analyzer tool (O.M. et al., manuscript in preparation) can perform peptide entropy analysis. Fig. 3 shows intra- and pan-serotype peptide entropy plots for dengue virus NS3 protein. The data shows that each of the four serotypes has distinct patterns of highly conserved and variable regions. Thus, the pan-serotype low entropy regions were restricted to discrete short regions, which corresponded to the conserved sequences selected by consensus-sequences method.
Fig. 3.
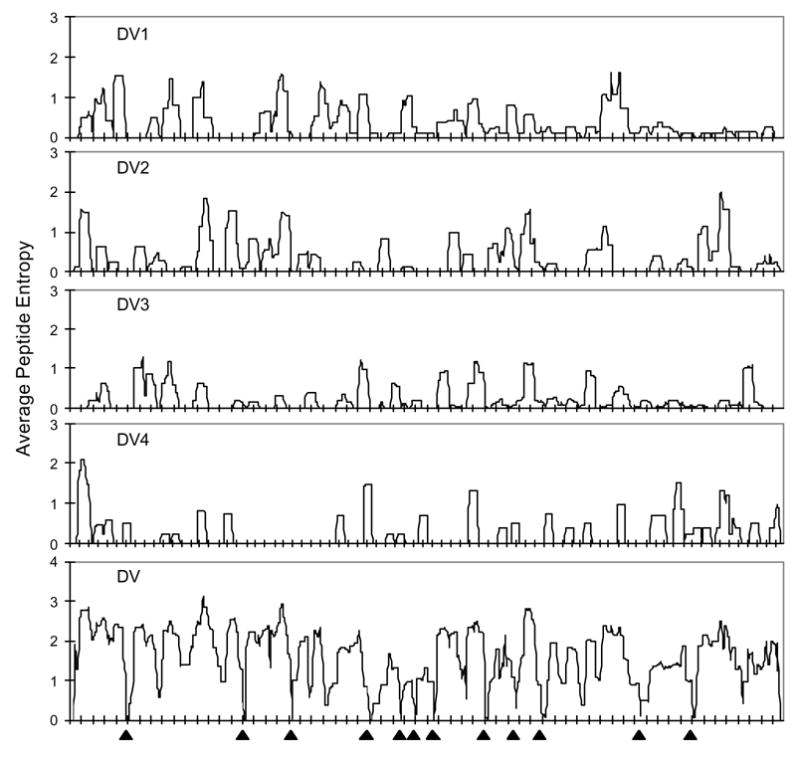
Peptide entropy plots for intra- and pan-serotype alignments of dengue virus (DV) NS3 protein (intra-serotype: DV1, DV2, DV3, DV4; pan-serotype: DV). The average peptide entropy value at each position is based on the frequency of nonamer peptide variants present at that position in the protein’s alignment. All 12 identified pan-serotype conserved sequences of NS3 protein were found to be localized in the pan-serotype conserved antigenic regions of the protein (▲), with values ranging from 0 to 0.4, indicating the high probability that these sequences will remain conserved in the future.
2.4 Functional and structural correlates of the conserved sequences
It is generally recognized that conserved protein sequences represent important functional domains [21], for which mutations would be detrimental to the survival of the pathogen. The functions of conserved sequences can be elucidated by databases that comprise data on protein families, domains and functional sites, such as the Pfam database [22] (www.sanger.ac.uk/Software/Pfam). Mapping the location of a conserved sequence on the 3-D structure of the protein may also provide relevant information (Fig. 4). Many such 3-D structures are available in the PDB database [23] (www.pdb.org).
Fig. 4.

Molecular location of dengue NS3 pan-serotype conserved sequences (148GLYGNGVVT156 and 189LTIMDLHPG197) on the protein’s 3-D structure. A) A major portion of 148GLYGNGVVT156 conserved sequence (in red) is localized in the buried regions of the 3-D structure. B) Most of the 189LTIMDLHPG197 conserved sequence (in red) is localized in the exposed region of the 3-D structure. This suggests that the conserved sequence 148GLYGNGVVT156 is less likely to mutate compared to 189LTIMDLHPG197, though both share identical level of intra-serotype percentage representation.
2.5. Distribution of conserved sequences in nature
Potential vaccine targets should be analyzed for specificity to the target pathogen. In vaccine design, epitopes common to other pathogens could either be useful by inducing cross-protection, or detrimental by inducing altered-ligand effect [9–11]. Identified conserved sequences should therefore be submitted to a BLAST search against all protein sequences at NCBI, excluding the target pathogen. If the sequences are found in other pathogens, the extent of their representation should be analyzed. For example, many dengue virus conserved sequences are found widely present in other Flaviviruses.
2.6 Characterization of candidate promiscuous T-cell epitopes
2.6.1 Algorithms for prediction of HLA binding peptides
Dedicated algorithms based on distinct prediction models are used to locate putative promiscuous T-cell epitopes for HLA class I or II supertypes within conserved sequences. Computational epitope prediction systems, such as NetCTL [24] (www.cbs.dtu.dk/services/NetCTL), MULTIPRED [25](research.i2r.a-star.edu.sg/multipred) and TEPITOPE [26] have been proven to be effective in accurately mapping T-cell epitopes. When selecting peptides for experimental validation, putative epitopes predicted by multiple models are chosen, since consensus predictions from a combination of models have been shown to be more accurate than individual model predictions [24,27].
In addition to being promiscuous with respect to multiple alleles of an HLA supertype, some putative T-cell epitopes exhibit multiple-supertype promiscuity. This additional form of promiscuity has been observed in several viruses, such as dengue [28] and HIV [3]. T-cell epitopes specific to multiple HLA supertypes are advantageous for vaccine design because they effectively increase the numbers of epitopes to which an individual can respond, and provide much more extensive coverage of the population [3].
2.6.2 Immunological hotspots
Putative promiscuous T-cell epitopes may be localized in clusters, as reported in studies of HIV-1 [29–32] and the outer membrane of Chlamydia trachomatis [33], among others [34,35]. The clusters are also ideal for developing epitope-based vaccines because they contain multiple promiscuous epitopes. MULTIPRED [25] can be used to predict immunological hotspots.
2.6.3 HLA distribution analysis
The percentage of individuals in the population predicted to respond to the putative conserved promiscuous T-cell epitopes is predicted by the population coverage analysis tool of the Immune Epitope Database [36] (www.immuneepitope.org/tools/population). The tool provides allele frequencies for 78 populations grouped into 11 different geographical areas.
2.7 Probability of altered-ligand effect
The genotypic differences between primary and secondary pathogens, or between the vaccine and challenge infection, constitute a critical consideration for protective and, in some cases, pathologic immunity [11]. Because of intra- and inter-group sequence variability, most T-cell epitope sequences may contain single or multiple amino acid differences within and between the groups. Variants of the putative promiscuous T-cell epitopes are identified among the reported sequences in the pathogen groups, and their representation within the group and across groups is observed. Variants of a putative epitope at a given alignment position comprise all nonamers at that site that possess at least one amino acid difference. Putative epitopes with no or low variant representation (~100% conserved) are potentially advantageous in avoiding altered peptide ligands.
2.8 Experimental Validation
2.8.1 Survey of reported human T-cell epitopes in the conserved sequences
Predictions of T-cell epitopes of the conserved sequences can in many cases be conformed (commonly without identification of the specific allele, however) by reports of experimentally confirmed T-cell epitopes. Therefore, search against both extant literature and the Immune Epitope Database (www.immuneepitope.org) is performed for reported human T-cell epitopes (both class I and II) that fully or partially overlap with identified conserved sequences. For example, eight reported human NS3 T-cell epitopes of dengue virus corresponded to the predicted promiscuous T-cell epitopes in the NS3 conserved sequences (Table 1).
Table 1.
Reported human T-cell epitopes in dengue virus NS3 pan-serotype conserved sequences.
| Protein | Pan-serotype sequence | Reported T-cell epitopes
|
|---|---|---|
| Reference(s) | ||
| NS3 | 46FHTMWHVTRG55 | [39] |
| 148GLYGNGVVT156 | [39,40] | |
| 189LTIMDLHPG197 | [41] | |
| 256EIVDLMCHATFT267 | [39,42,43] | |
| 313IFMTATPPG321 | [39] | |
| 357GKTVWFVPSIK367 | [44,45] | |
| 383VIQLSRKTFD392 | [39] | |
| 406VVTTDISEMGANF418 | [39] | |
| 537LMRRGDLPVWL547 | [39] |
The amino acid positions are numbered according to the aligned sequences of dengue proteins from all four serotypes.
2.8.2 Experimental measurements to validate predictions
Experimental measurements for validation of computational predictions are necessary for accurate interpretation of results. Such measurements currently include HLA binding assays [37], immunization of HLA transgenic mice and ELISpot assay for peptide-specific T-cell activation [38] and of pathogen infected human subjects. We performed functional assessment of the dengue virus NS1 conserved sequences: four were predicted to contain HLA-DR epitopes and three of these four were confirmed by ELISpot assay with T-cell activation peptides that closely mimic the conserved sequences (Table 2). An additional two that were also ELISpot positive were not predicted to bind to DR molecules. In summary, of seven conserved NS1 sequences, five contained HLA-DR T-cell epitopes and at least three are promiscuous for multiple HLA-DR alleles. The predictive models are helpful in selecting antigen sequences for additional study of immune responses, especially for sequences predicted by multiple algorithms.
Table 2.
IFN-gamma ELISpot responses of CD4-depleted splenocytes from HLA transgenic mice immunized with peptides overlapping dengue virus NS1 pan-serotype conserved sequences.
| Pan-serotype sequence | Predicted DR-2, -3, -4 | ELISpot positive HLA transgenic mouse | ELISpot activation peptide |
|---|---|---|---|
| 12ELKCGSGIF20 | DR-2 | DR-2 | 13LKCGSGIFVTNEVHT27 |
| 25VHTWTEQYKFQ35 | DR-4 | DR-3 and -4 | 25VHTWTEQYKFQADSP39 |
| 193AVHADMGYWIES204 | DR-2 and -3 | None | 193AVHADMGYWIESQKN207 |
| 229HTLWSNGVLES239 | DR-3 and -4 | DR-3 and -4 | 229HTLWSNGVLESDMII243 |
| 266GPWHLGKLE274 | None | DR-3 and -4 | 265AGPWHLGKLELDFNY279 |
| 294RGPSLRTTT302 | None | DR-4 | 293TRGPSLRTTTVSGKL307 |
| 325GEDGCWYGMEIRP337 | None | None | 325GEDGCWYGMEIRPIS339 |
The amino acid positions are numbered according to the aligned sequences of dengue proteins from all four serotypes. Prediction for DR alleles was performed by use of MULTIPRED [25], TEPITOPE [26] and ARB [46]. The ELISpot assays were performed for DR-2, DR-3 and DR-4 transgenic mice. ELISpot activation peptides are the actual peptides used to test the ELISpot.
3. Conclusion
The bioinformatics approach presented in this paper proved generic as it was successfully applied to several viruses, such as dengue virus (A.M.K. et al., manuscript in preparation), influenza (A.T.H. et al., manuscript in preparation) and HIV (K.N.S et al., manuscript in preparation). Thus, the approach can be used as a template for the analysis of other pathogens, providing a novel and generalized approach to the formulation of epitope-based vaccines that are effective against broad diversity of pathogens and applicable to the human population at large. This new methodology enables the systematic screening of pathogen data which would otherwise be impossible to carry out experimentally, due to too many pathogen sequences (high viral diversity) and variations in immune system among individuals (extensive polymorphism of HLA). It therefore significantly reduces the efforts and cost of experimentation, while providing for systematic screening.
Acknowledgments
This project has been funded in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, USA, under Grant No. 5 U19 AI56541 and Contract No. HHSN2662-00400085C.
Abbreviations
- HIV
human immunodeficiency virus
- HLA
human leukocyte antigen
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sette A, Fikes J. Epitope-based vaccines: an update on epitope identification, vaccine design and delivery. Curr Opin Immunol. 2003;15:461–470. doi: 10.1016/s0952-7915(03)00083-9. [DOI] [PubMed] [Google Scholar]
- 2.Sette A, Livingston B, McKinney D, Appella E, Fikes J, Sidney J, Newman M, Chesnut R. The development of multi-epitope vaccines: epitope identification, vaccine design and clinical evaluation. Biologicals. 2001;29:271–276. doi: 10.1006/biol.2001.0297. [DOI] [PubMed] [Google Scholar]
- 3.Wilson CC, McKinney D, Anders M, MaWhinney S, Forster J, Crimi C, Southwood S, Sette A, Chesnut R, Newman MJ, Livingston BD. Development of a DNA vaccine designed to induce cytotoxic T lymphocyte responses to multiple conserved epitopes in HIV-1. J Immunol. 2003;171:5611–5623. doi: 10.4049/jimmunol.171.10.5611. [DOI] [PubMed] [Google Scholar]
- 4.Robinson HL, Amara RR. T cell vaccines for microbial infections. Nat Med. 2005;11:S25–32. doi: 10.1038/nm1212. [DOI] [PubMed] [Google Scholar]
- 5.Zinkernagel RM, Hengartner H. On immunity against infections and vaccines: credo 2004. Scand J Immunol. 2004;60:9–13. doi: 10.1111/j.0300-9475.2004.01460.x. [DOI] [PubMed] [Google Scholar]
- 6.Esser MT, Marchese RD, Kierstead LS, Tussey LG, Wang F, Chirmule N, Washabaugh MW. Memory T cells and vaccines. Vaccine. 2003;21:419–430. doi: 10.1016/s0264-410x(02)00407-3. [DOI] [PubMed] [Google Scholar]
- 7.Pulendran B, Ahmed R. Translating innate immunity into immunological memory: implications for vaccine development. Cell. 2006;124:849–863. doi: 10.1016/j.cell.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 8.Brusic V, August JT. The changing field of vaccine development in the genomics era. Pharmacogenomics. 2004;5:597–600. doi: 10.1517/14622416.5.6.597. [DOI] [PubMed] [Google Scholar]
- 9.Sloan-Lancaster J, Allen PM. Altered peptide ligand-induced partial T cell activation: molecular mechanisms and role in T cell biology. Annu Rev Immunol. 1996;14:1–27. doi: 10.1146/annurev.immunol.14.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Evavold BD, Sloan-Lancaster J, Allen PM. Tickling the TCR: selective T-cell functions stimulated by altered peptide ligands. Immunol Today. 1993;14:602–609. doi: 10.1016/0167-5699(93)90200-5. [DOI] [PubMed] [Google Scholar]
- 11.Rothman AL. Dengue: defining protective versus pathologic immunity. J Clin Invest. 2004;113:946–951. doi: 10.1172/JCI21512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ovsyannikova IG, Jacobson RM, Poland GA. Variation in vaccine response in normal populations. Pharmacogenomics. 2004;5:417–427. doi: 10.1517/14622416.5.4.417. [DOI] [PubMed] [Google Scholar]
- 13.Sette A, Sidney J. Nine major HLA class I supertypes account for the vast preponderance of HLA-A and -B polymorphism. Immunogenetics. 1999;50:201–212. doi: 10.1007/s002510050594. [DOI] [PubMed] [Google Scholar]
- 14.Brusic V, Bajic VB, Petrovsky N. Computational methods for prediction of T-cell epitopes--a framework for modelling, testing, and applications. Methods. 2004;34:436–443. doi: 10.1016/j.ymeth.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Srinivasan KN, Gopalakrishnakone P, Tan PT, Chew KC, Cheng B, Kini RM, Koh JL, Seah SH, Brusic V. SCORPION, a molecular database of scorpion toxins. Toxicon. 2002;40:23–31. doi: 10.1016/s0041-0101(01)00182-9. [DOI] [PubMed] [Google Scholar]
- 16.Khan AM, Heiny AT, Lee KX, Srinivasan KN, Tan TW, August BV JT. Large-scale analysis of antigenic diversity of T-cell epitopes in dengue virus. BMC Bioinformatics. 2006;7:S4. doi: 10.1186/1471-2105-7-S5-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miotto O, Tan TW, Brusic V. Extraction by Example: Induction of Structural Rules for the Analysis of Molecular Sequence Data from Heterogeneous Sources. In: Gallagher M, Hogan J, Maire F, editors. Lecture Notes in Computer Science. Vol. 3578. Springer; Berlin: 2005. pp. 398–405. [Google Scholar]
- 18.Novitsky V, Smith UR, Gilbert P, McLane MF, Chigwedere P, Williamson C, Ndung’u T, Klein I, Chang SY, Peter T, Thior I, Foley BT, Gaolekwe S, Rybak N, Gaseitsiwe S, Vannberg F, Marlink R, Lee TH, Essex M. Human immunodeficiency virus type 1 subtype C molecular phylogeny: consensus sequence for an AIDS vaccine design? J Virol. 2002;76:5435–5451. doi: 10.1128/JVI.76.11.5435-5451.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rammensee HG. Chemistry of peptides associated with MHC class I and class II molecules. Curr Opin Immunol. 1995;7:85–96. doi: 10.1016/0952-7915(95)80033-6. [DOI] [PubMed] [Google Scholar]
- 20.Shannon CE. A mathematical theory of communication. Bell System Technical Journal. 1948;27:379–423. 623–656. [Google Scholar]
- 21.Valdar WS. Scoring residue conservation. Proteins. 2002;48:227–241. doi: 10.1002/prot.10146. [DOI] [PubMed] [Google Scholar]
- 22.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, Studholme DJ, Yeats C, Eddy SR. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larsen MV, Lundegaard C, Lamberth K, Buus S, Brunak S, Lund O, Nielsen M. An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur J Immunol. 2005;35:2295–2303. doi: 10.1002/eji.200425811. [DOI] [PubMed] [Google Scholar]
- 25.Zhang GL, Khan AM, Srinivasan KN, August JT, Brusic V. MULTIPRED: a computational system for prediction of promiscuous HLA binding peptides. Nucleic Acids Res. 2005;33:W172–179. doi: 10.1093/nar/gki452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bian H, Hammer J. Discovery of promiscuous HLA-II-restricted T cell epitopes with TEPITOPE. Methods. 2004;34:468–475. doi: 10.1016/j.ymeth.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 27.Donnes P, Kohlbacher O. Integrated modeling of the major events in the MHC class I antigen processing pathway. Protein Sci. 2005;14:2132–2140. doi: 10.1110/ps.051352405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gagnon SJ, Zeng W, Kurane I, Ennis FA. Identification of two epitopes on the dengue 4 virus capsid protein recognized by a serotype-specific and a panel of serotype-cross-reactive human CD4+ cytotoxic T-lymphocyte clones. J Virol. 1996;70:141–147. doi: 10.1128/jvi.70.1.141-147.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shankar P, Fabry JA, Fong DM, Lieberman J. Three regions of HIV-1 gp160 contain clusters of immunodominant CTL epitopes. Immunol Lett. 1996;52:23–30. doi: 10.1016/0165-2478(96)02574-6. [DOI] [PubMed] [Google Scholar]
- 30.Surman S, Lockey TD, Slobod KS, Jones B, Riberdy JM, White SW, Doherty PC, Hurwitz JL. Localization of CD4+ T cell epitope hotspots to exposed strands of HIV envelope glycoprotein suggests structural influences on antigen processing. Proc Natl Acad Sci U S A. 2001;98:4587–4592. doi: 10.1073/pnas.071063898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown SA, Stambas J, Zhan X, Slobod KS, Coleclough C, Zirkel A, Surman S, White SW, Doherty PC, Hurwitz JL. Clustering of Th cell epitopes on exposed regions of HIV envelope despite defects in antibody activity. J Immunol. 2003;171:4140–4148. doi: 10.4049/jimmunol.171.8.4140. [DOI] [PubMed] [Google Scholar]
- 32.Berzofsky JA, Pendleton CD, Clerici M, Ahlers J, Lucey DR, Putney SD, Shearer GM. Construction of peptides encompassing multideterminant clusters of human immunodeficiency virus envelope to induce in vitro T cell responses in mice and humans of multiple MHC types. J Clin Invest. 1991;88:876–884. doi: 10.1172/JCI115389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim SK, DeMars R. Epitope clusters in the major outer membrane protein of Chlamydia trachomatis. Curr Opin Immunol. 2001;13:429–436. doi: 10.1016/s0952-7915(00)00237-5. [DOI] [PubMed] [Google Scholar]
- 34.Gupta V, Tabiin TM, Sun K, Chandrasekaran A, Anwar A, Yang K, Chikhlikar P, Salmon J, Brusic V, Marques ET, Kellathur SN, August TJ. SARS coronavirus nucleocapsid immunodominant T-cell epitope cluster is common to both exogenous recombinant and endogenous DNA-encoded immunogens. Virology. 2006;347:127–139. doi: 10.1016/j.virol.2005.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srinivasan KN, Zhang GL, Khan AM, August JT, Brusic V. Prediction of class I T-cell epitopes: evidence of presence of immunological hot spots inside antigens. Bioinformatics. 2004;20(Suppl 1):I297–I302. doi: 10.1093/bioinformatics/bth943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peters B, Sidney J, Bourne P, Bui HH, Buus S, Doh G, Fleri W, Kronenberg M, Kubo R, Lund O, Nemazee D, Ponomarenko JV, Sathiamurthy M, Schoenberger SP, Stewart S, Surko P, Way S, Wilson S, Sette A. The design and implementation of the immune epitope database and analysis resource. Immunogenetics. 2005;57:326–336. doi: 10.1007/s00251-005-0803-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sidney J, Southwood S, Oseroff C, del Guercio MF, Grey HM, Sette A. Measurement of MHC/peptide interactions by gel filtration. Current Protocols in Immunology. 1998:18.13.11–18.13.19. doi: 10.1002/0471142735.im1803s31. [DOI] [PubMed] [Google Scholar]
- 38.Rosloniec EF, Brand DD, Myers LK, Whittington KB, Gumanovskaya M, Zaller DM, Woods A, Altmann DM, Stuart JM, Kang AH. An HLA-DR1 transgene confers susceptibility to collagen-induced arthritis elicited with human type II collagen. J Exp Med. 1997;185:1113–1122. doi: 10.1084/jem.185.6.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simmons CP, Dong T, Chau NV, Dung NT, Chau TN, Thaole TT, Dung NT, Hien TT, Rowland-Jones S, Farrar J. Early T-cell responses to dengue virus epitopes in Vietnamese adults with secondary dengue virus infections. J Virol. 2005;79:5665–5675. doi: 10.1128/JVI.79.9.5665-5675.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurane I, Okamoto Y, Dai LC, Zeng LL, Brinton MA, Ennis FA. Flavivirus-cross-reactive, HLA-DR15-restricted epitope on NS3 recognized by human CD4+ CD8− cytotoxic T lymphocyte clones. J Gen Virol. 1995;76(Pt 9):2243–2249. doi: 10.1099/0022-1317-76-9-2243. [DOI] [PubMed] [Google Scholar]
- 41.Mangada MM, Rothman AL. Altered cytokine responses of dengue-specific CD4+ T cells to heterologous serotypes. J Immunol. 2005;175:2676–2683. doi: 10.4049/jimmunol.175.4.2676. [DOI] [PubMed] [Google Scholar]
- 42.Kurane I, Dai LC, Livingston PG, Reed E, Ennis FA. Definition of an HLA-DPw2-restricted epitope on NS3, recognized by a dengue virus serotype-cross-reactive human CD4+ CD8− cytotoxic T-cell clone. J Virol. 1993;67:6285–6288. doi: 10.1128/jvi.67.10.6285-6288.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okamoto Y, Kurane I, Leporati AM, Ennis FA. Definition of the region on NS3 which contains multiple epitopes recognized by dengue virus serotype-cross-reactive and flavivirus-cross-reactive, HLA-DPw2-restricted CD4+ T cell clones. J Gen Virol. 1998;79(Pt 4):697–704. doi: 10.1099/0022-1317-79-4-697. [DOI] [PubMed] [Google Scholar]
- 44.Zeng L, Kurane I, Okamoto Y, Ennis FA, Brinton MA. Identification of amino acids involved in recognition by dengue virus NS3-specific, HLA-DR15-restricted cytotoxic CD4+ T-cell clones. J Virol. 1996;70:3108–3117. doi: 10.1128/jvi.70.5.3108-3117.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loke H, Bethell DB, Phuong CX, Dung M, Schneider J, White NJ, Day NP, Farrar J, Hill AV. Strong HLA class I--restricted T cell responses in dengue hemorrhagic fever: a double-edged sword? J Infect Dis. 2001;184:1369–1373. doi: 10.1086/324320. [DOI] [PubMed] [Google Scholar]
- 46.Bui HH, Sidney J, Peters B, Sathiamurthy M, Sinichi A, Purton KA, Mothe BR, Chisari FV, Watkins DI, Sette A. Automated generation and evaluation of specific MHC binding predictive tools: ARB matrix applications. Immunogenetics. 2005;57:304–314. doi: 10.1007/s00251-005-0798-y. [DOI] [PubMed] [Google Scholar]