

Abstract
We have characterized the adsorption of bovine fibrinogen onto the biomedical polymer polyethylene terephthalate (PET) by performing mass spectrometric mapping with a lysine-reactive biotin label. After digestion with trypsin, MALDI-TOF mass spectrometry was used to detect peptides from biotinylated bovine fibrinogen, with the goal of identifying lysines that were more accessible for reaction with the chemical label after adsorption. Peptides within domains that are believed to contribute to heparin binding, leukocyte activation, and platelet adhesion were found to be biotin labeled only after bovine fibrinogen adsorbed to the PET surface. Additionally, the accessibility of lysine residues throughout the entire molecule was observed to increase as the concentration of the adsorbing bovine fibrinogen solution decreased, suggesting that the proximity of biologically active motifs to hydrophilic residues leads to their exposure. The surface area per adsorbed bovine fibrinogen molecule was quantified on PET using optical waveguide lightmode spectroscopy (OWLS), which revealed higher surface densities for bovine fibrinogen adsorbed from higher concentration solutions. By measuring changes in both the identity and conformation of proteins that adsorb from complex mixtures such as blood or plasma, this technique may have applications in fundamental studies of protein adsorption and may allow for more accurate predictions of the biocompatibility of materials.
Keywords: Protein Adsorption, Fibrinogen, Biocompatibility, Poly(ethylene terephthalate)
1. Introduction
Upon exposure to blood, implanted materials adsorb a thin layer of proteins onto their surfaces [1–3]. The rate of adsorption and the chemical properties of the surface influence protein conformations and surface coverage as the adsorbed proteins denature and unfold at this solid/liquid interface. Some adsorbed proteins expose previously hidden binding domains that participate in inflammatory, thrombotic, and immunologic responses [4–8]. The postadsorptive conformations of fibrinogen have been investigated on surfaces of varying surface energy [9, 10] and these transformations expose sites responsible for platelet activation and aggregation [11–14]. Here, we introduce mass spectrometric mapping to probe local conformational changes within bovine fibrinogen (bFg) following adsorption to the biomedical polymer polyethylene terephthalate (PET).
Fibrinogen is a 340 kDa dimeric glycoprotein composed of Aα, Bβ, and γ chains containing globular and α-helical coiled-coil secondary structures held together by 29 disulfide bonds. The structure is trinodular, composed of one inner and two outer globular domains linked by regions of coiled-coils [15]. The inner domain (E domain) contains the amino termini of the Aα, Bβ, and γ chains [16], while the two outer domains (D domains) consist of the carboxyl termini of the Bβ, and γ chains [17]. Extending from the D domains, beginning near Aα224 in bFg, are the mobile and highly flexible αC domains that associate non-covalently with the E domain [18]. Fibrinogen possesses several cell adhesion and protein interaction domains in all three of its major chains, including RGD integrin binding motifs in the Aα chain [19], a heparin [20] and cadherin [21] binding domain in the Bβ chain, and a dodecapeptide essential for platelet aggregation in the γ chain [22].
When proteins are reacted with a chemical label prior to enzymatic digestion, labeled peptides with increased masses can be identified by mass spectrometry. Since the rate of reaction of protein functional groups is dependent on their degree of solution exposure [23], mass spectrometry can be used to map changes in protein conformation by varying the experimental conditions and tracking the accessibility of specific residues to chemical modification. Mass spectrometric mapping has been previously utilized to discern protein-protein interactions and protein conformations in solution [24–26]. Arginine, lysine, tryptophan, and tyrosine are the most common targets of amino acid modification [24, 26, 27]. Since lysines occur frequently throughout proteins, are charged and have a high degree of solution exposure, amine-specific N-hydroxysuccinimide (NHS) ester labels have been used in various applications, including isotope-tagging for relative absolute quantification (ITRAQ) [28, 29]. The availability of water-soluble derivatives of NHS-esters provides an advantage over most other reagents used for protein labeling, in that the reaction can be performed under physiologic conditions [30].
Techniques such as antibody labeling [11], atomic force microscopy (AFM) [9, 31–35], circular dichroism [36], sum frequency generation (SFG) [37], total internal reflectance fluorescence (TIRF) [34], and Fourier transform infrared spectroscopy (FTIR) [35, 38, 39] have demonstrated structural changes in fibrinogen after adsorption onto surfaces. Each of these techniques has advantages for elucidating protein structural changes, but generally provides information at the domain-level. Antibody binding can be used to correlate the exposure of cell interaction motifs to post-adsorptive changes in fibrinogen conformation [11]. While antibody binding verifies the exposure of previously inaccessible domains of fibrinogen, it is dependent on the availability of specific antibodies and can only reveal changes in protein conformation in areas of the protein complementary to these antibodies. AFM has provided detailed information on the overall postadsorptive structure of fibrinogen by revealing the dimensions of the fibrinogen D and E domains on various surfaces. However, AFM is not well suited for discerning transitions in dense monolayers of protein. Detailed studies on the global changes in fibrinogen secondary structure after adsorption have been performed using SFG, TIRF, and FTIR, also yielding information at the domain-level. We believe that mass spectrometric mapping may contribute to the study of adsorbed proteins by providing residue-specific information on surface-dependent conformational changes under physiologically relevant experimental conditions.
We have developed a proteomics-based strategy to analyze the adsorption of bFg onto PET, which supplements our earlier proteomic studies on the adsorption of serum and plasma proteins on biomaterials [40]. The conformational changes that occur upon fibrinogen adsorption were examined by comparing the mass spectra of bFg that was biotin-labeled in solution to bFg that was labeled after adsorption to the surfaces of PET particles. We hypothesized that there was a tendency for sections of bFg to be exposed to solution during the adsorption process, and that peptides at these locations would be biotinylated at higher rates. We targeted primary amines with a chemical label due to their reactive properties and presence at 107 sites throughout the bFg molecule, which could be analyzed with respect to their location in three dimensional space due to the availability of the nearly complete bFg crystal structure. The reactions were performed under physiological conditions with an NHS-ester label. These residue-specific mass spectrometric mapping experiments correlated well with optical waveguide lightmode spectroscopy (OWLS) results, both of which demonstrated concentration-dependent changes in fibrinogen spreading on PET surfaces. Our data support previous findings that large scale changes in fibrinogen conformation occur upon adsorption from dilute solutions, evidenced by enhanced exposure of hydrophilic lysine groups throughout the protein. Furthermore, postadsorptive increases in lysine exposure were detected for several residues localized within biologically relevant fibrinogen motifs. Thus this method may be useful in localizing adsorption-induced conformational changes to specific sites within proteins with a residue-level resolution.
2. Materials/Methods
2.1 Spin Coating
Unless otherwise noted, all reagents were purchased from Sigma. Glass OWLS chips coated with a Si/Ti/O2 waveguide layer (48 × 16 × 0.5 mm, MicroVacuum Ltd.) and silicon wafers (48 × 16 × 0.5 mm) were cleaned by immersion in piranha solution (3:1 H2SO4 and H2O2) for 5 min followed by rinsing in DI water. Polyethylene terephthalate (PET, 0.05 mm thick, McMaster Carr, Chicago, IL) sheets were dissolved in chloroform with 5% (w/v) trifluoroacetic acid (TFA) to make 0.8% solutions of PET. The PET solution (100 μL) was applied to the surfaces of either the wafers or OWLS chips and spun at 4500 RPM for 50 s with a model KW-4A spin coater (CHEMAT Technology). PET layer thicknesses on the silicon wafers were confirmed with a variable angle Stokes ellipsometer (Gaertner Scientific Corporation). X-ray photoelectron spectroscopy (XPS, Kratos Analytical Axis 165 spectrometer, University of Missouri Rolla) was performed to verify the composition of the layers.
2.2 OWLS Analysis
PET coated waveguide chips were placed inside the flow chamber of a MicroVacuum OWLS 110 optical waveguide lightmode spectrometer. The adsorption of bFg from 1, 0.5 and 0.1 mg/mL solutions in phosphate buffered saline (PBS; 0.2 g/L KCl, 0.2 g/L KH2PO4, 8 g/L NaCl, 1.15 g/L anhydrous Na2HPO4, pH 7.4) was monitored with a time step of 30 sec. PBS was flowed over the surface of the chips for at least 1 h, or until a baseline with a transverse magnetic mode refractive index variation less that 1 × 10−6 was obtained. Solutions of bFg (0.1, 0.5, or 1 mg/mL) were allowed to flow over the surface for 4 h at 0.15 mL/min. The surface was then washed with PBS at 0.15 mL/min until a steady baseline was again reached. The thickness and density of the adsorbed layers were determined for each time point using MicroVacuum BioSense software.
2.3 Analysis of OWLS Results
The density and the change in density with time (dM/dt) were fit to a random sequential adsorption (RSA) model of the protein adsorption process [41], using MathWorks MATLAB version 6.1. The OWLS data was fit to the equation:
| (1) |
where θ is the fractional surface coverage, Ka is the deposition rate constant, Cb is the bulk solution protein concentration, and φ is a surface coverage dependent function that represents exclusion effects due to previously adsorbed proteins. The fractional surface coverage is defined by:
| (2) |
where M is the adsorbed surface density, m is the mass of a single fibrinogen molecule, and a is the surface area covered by one fibrinogen molecule. The dM/dt data were fit to the RSA equation previously derived using statistical mechanics of hard sphere adsorption to a two dimensional surface [42]:
| (3) |
The maximum θ, or jamming limit, resulting from the adsorption process for spheres is 0.547 [43]. By substituting this maximum coverage into equation (2), the protein surface density at the jamming limit, M∞, was calculated for each adsorption experiment. M∞ was used to directly convert OWLS surface density measurements into fractional surface coverages. Statistical significance (p-value < 0.05) between the fibrinogen concentrations was determined by ANOVA followed by a Scheffe post-hoc test.
2.4 PET Particle Fabrication
PET solutions (1%) were obtained by dissolving polymer sheets in dichloromethane with 5% (v/v) TFA. Each sample was filtered with a 0.5 μm PTFE filter (Millipore) and poured into 1 L of DI water while stirring. Emulsions were stirred overnight and PET particles were removed by filtration. The particles were washed in ethanol and DI water and stored under vacuum until use. The surface area per gram of particles was determined for three separate fabrications using BET (Micrometrics Brunauer Emmett Teller model Gemini 2375).
2.5 Biotinylation
PET particles were placed in 1.5 mL silicone-coated polypropylene Eppendorf tubes and washed 3X in ethanol and 3X in PBS. Disposable polystyrene columns (Fisher Scientific) with 2 mL bed volumes were filled with the PET particles (150 mg) in PBS and allowed to settle for 30 min. PBS (3 mL) was added to the columns for washing, followed by 2 mL of 1, 0.5, or 0.1 mg/mL bFg in PBS. The PET surfaces were incubated with the bFg solutions for 2 h and then washed with 5 mL of PBS. The PBS washes were collected in 1 mL samples for analysis with a GENESYS 10 spectrophotometer (Spectronic) at 280 nm to monitor the decrease in protein concentration and to ensure that the non-adsorbed bFg solution was completely removed. A 10 mg/mL solution of Sulfo-NHS-LC-Biotin (Pierce) in PBS was added to the column and incubated for 1 h to allow reaction with the adsorbed bFg. PBS (5 mL) was then flowed through the column to remove excess NHS solution. The adsorbed protein was eluted from the column with 500 μL of an elution buffer (4% SDS, 40 mM Tris, 8M urea, pH 8.5). The elution buffer was allowed to incubate in the column overnight. Protein was collected by acetone precipitation and dried in an Eppendorf Vacufuge concentrator. Adsorption experiments were performed in triplicate for each concentration.
SDS-PAGE was used to monitor the extent of labeling in biotinylated samples. To maintain a similar degree of labeling between solution-labeled and surface-labeled samples, 0.5 mg/mL of bFg was biotinylated in solution with 1 mg/mL of Sulfo-NHS-LC-Biotin for 1 h at room temperature. The labeled bFg was concentrated and desalted by acetone precipitation. The solution-labeled samples were not exposed to PET surfaces nor the elution buffer. Solution-labeling experiments were performed in triplicate.
Each surface-labeled and solution-labeled fibrinogen precipitation was resuspended in DI water and separated into 3 samples. The samples were reduced with 100 mM DTT, alkylated with 100 mM iodoacetamide and separated by SDS-PAGE using Tris-HCl linear gradient 8–16% gels (Pierce). Fibrinogen Aα, Bβ, and γ chains were stained with SYPRO Ruby stain and excised from the gels. In-gel trypsin digestions were performed overnight for 16 h at 37°C. The peptides were purified for MALDI-TOF analysis using ZipTips (Fisher Scientific) with elution by 15 mg/mL α-cyano-4-hydroxycinnamic acid matrix solution in 0.1% TFA/50% acetonitrile.
2.6 MALDI-TOF Analysis
Mass spectrometry was performed with an Applied Biosystems Voyager System 4254 MALDI-TOF in positive-ion reflectron mode to produce centroided spectra. Each bFg sample was divided into three separate aliquots prior to in-gel trypsin digestion and thus each sample was analyzed with MALDI-TOF three times (see workflow diagram, Fig. 4). The three spectra obtained for each sample were normalized to each other using internal bFg peptide standards. Peptides common to each trypsin digest that did not contain lysines were selected as standards. Trypsin-digested bFg peptide masses were predicted using the ExPASy proteomics server [44]. Peptides with up to three missed cleavages ranging in size from 700 – 4000 Da were found in MALDI-TOF spectra with in-house Perl scripts that identified peaks consistent with tryptic bFg peptides. A tolerance of ± 0.1% was utilized with a maximum upper deviation of 2 Da and a maximum lower deviation of 1 Da was utilized. Biotin-labeled peptides were identified by the following criteria: 1) the peptide contained at least one missed cleavage, 2) the peptide mass increased by the size of the Sulfo-NHS-LC-Biotin label (+339.452 Da), and, 3) the peptide contained at least one lysine residue that was not located at the carboxyl terminus, since trypsin cleavage after labeled lysines should be unfavorable. If two separate peaks corresponded to different peptides that contained the same labeled lysine location due to incomplete trypsin digestion, the normalized intensities were summed. The summed normalized intensities of peptides containing each lysine residue were averaged across the three trypsin digests. These average lysine intensities were then averaged across three separate experiments for each of the tested conditions. Non-parametric Mann-Whitney pairwise comparisons were used to identify statistically significant (p-value < 0.05) changes in intensity. Labeled bFg locations were visualized using PDB file 1DEQ [45] with Visual Molecular Dynamics version 1.8.3 [46]. A sequence comparison between human and bovine fibrinogen was performed using CLUSTAL W version 1.83 [47].
Fig. 4.
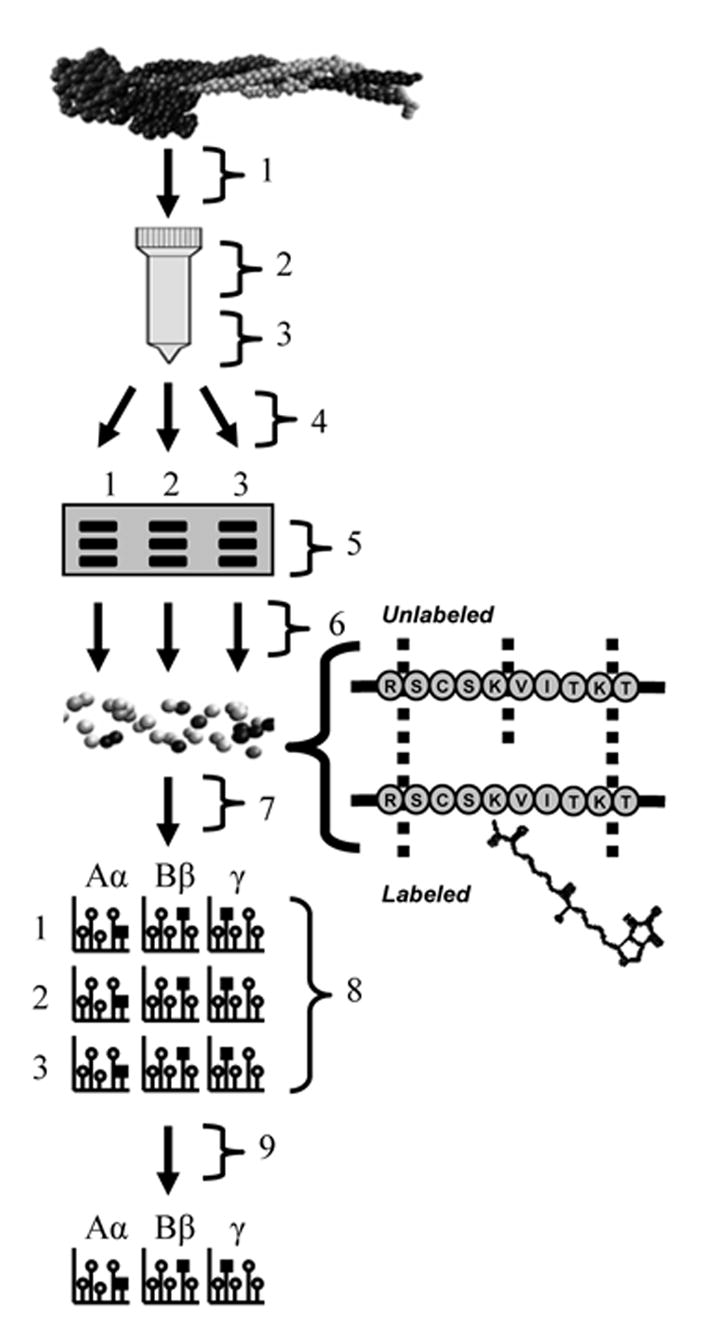
Workflow diagram for bovine fibrinogen (bFg) surface adsorption experiments. Diagram is labeled as follows: 1) Adsorption of bFg onto PET beads packed in a column, from 0.1, 0.5, or 1 mg/mL solutions. Each concentration was tested in triplicate, 2) Incubation of adsorbed bFg with Sulfo-NHS-LC-Biotin for 1 h, 3) Elution of adsorbed bFg with 4% SDS, 100 mM DTT and 8 M urea elution buffer. Elutions were separated into three samples for separate trypsin digestion, 4) bFg purification by acetone precipitation, 5) bFg chain separation by SDS-PAGE, 6) In-gel trypsin digestion, 7) Peptide purification with Zip-tips, 8) MALDI-TOF analysis, (each PET column yielded three separate samples, and bFg Aα, Bβ, and γ chains were analyzed for each sample), 9) Intensities of peptides containing labeled lysine residues were averaged across the three separate digests.
2.7 Liquid Chromatography Electrospray-Ionization Ion Trap Tandem Mass Spectrometry
The reliability of labeled peptide detection was confirmed by processing some solution-labeled samples with liquid chromatography electrospray-ionization ion trap tandem mass spectrometry (LC-ESI-MS/MS). After biotinylation, PBS solutions of bFg were buffer exchanged using microcentrifuge filters (NMWL 10,000, PLGC cellulosic membrane, Millipore Corp.) into 25 mM ammonium bicarbonate and digested with sequence grade trypsin overnight at 37°C. The ammonium bicarbonate was evaporated in a Vacufuge and samples were stored at −20°C. Tryptic peptides were dissolved in 25 μL of 5% acetonitrile/0.02% N-heptafluorobutyric acid (HFBA). The peptide solutions (4 μL) were injected into a nano-HPLC system (Agilent 1100 capillary pump) connected to a Picoview nanoelectrospray head (New Objective, Woburn, MA) attached to an ion trap mass spectrometer (LCQ DECA, ThermoFinnigan, San Jose, CA). The peptides in the mixture were separated at a flow rate of 300 nL/min. A gradient of 5% acetonitrile/0.02% HFBA to 80% acetonitrile/0.02% HFBA was run over 60 min, with peptides separated on a 75 μm × 5 cm C18 Biobasic column (New Objective). The LCQ-DECA was operated in positive ion mode using a capillary voltage of 12 V, a capillary temperature of 150°C, and a spray voltage of 2.2 kV. Each experiment was immediately followed by a blank injection to ensure that proteins were not carried over from previous injections. Data were collected in full MS/MS mode and biotin-labeled peptides were identified using an in-house program as described previously [40].
3. Results
3.1 Characterization of PET Films
The spin coating of PET layers was initially characterized on silicon wafers. Ellipsometry showed consistent thicknesses of between 45 and 50 nm across the surfaces of the silicon wafers (Fig. 1). XPS analysis verified the composition of these layers (Table 1). Peaks at 283.5 eV for carbon and 530.5 eV for oxygen revealed their respective atomic compositions to be approximately 71.0% and 24.8%, which corresponded well with the expected amounts for a PET layer of 72% for carbon and 28% for oxygen. OWLS waveguide chips were coated using the protocol developed for the silicon wafers and the changes in thickness were verified by OWLS to be between 50 and 75 nm.
Fig. 1.
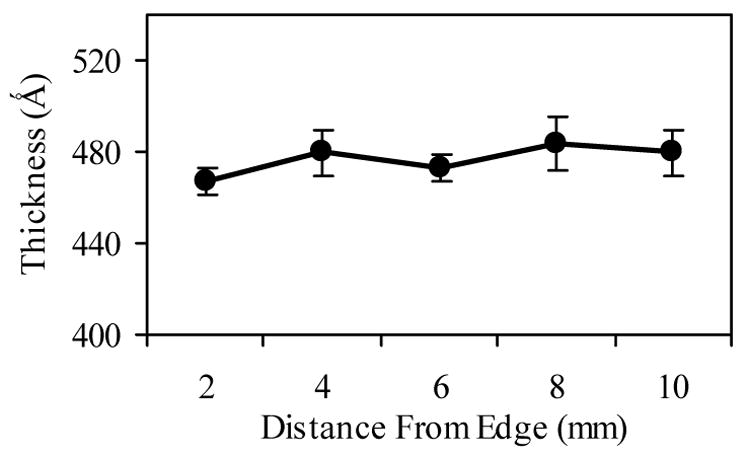
Ellipsometry measurements of polyethylene terephthalate (PET) layer thickness and uniformity on silicon wafers. PET coatings were applied to surfaces using spin coating, and ellipsometry was used to determine the layer thickness at 2 mm increments across a wafer. Error bars represent standard deviations of four samples.
Table 1.
XPS analysis of PET coated silicon wafers.
| Peak | Position BE (eV) | Atomic Conc. % |
|---|---|---|
| O | 530.5 | 24.80 |
| Ti | 464 | 0.02 |
| N | 397 | 0.19 |
| C | 283 | 71.48 |
| S | 167 | 0.12 |
| Si | 100.5 | 3.05 |
| Cl | 99.5 | 0.00 |
3.2 OWLS Analysis of Fibrinogen Adsorption onto PET
A random sequential adsorption model was used to determine the average surface area of individual bFg molecules on PET-coated OWLS chips at the jamming limit (Table 2). Changes in protein surface density over time for the bulk solution concentrations of 0.1, 0.5 and 1 mg/mL were non-linear least-squares fit to equation (3) by adjusting the a/m and K parameters. Statistically significant differences in surface density were found when comparing the 1 mg/mL samples to either the 0.5 mg/mL or the 0.1 mg/mL samples. The 0.5 mg/mL and 0.1 mg/mL samples were not significantly different.
Table 2.
Summary of bFg m/a values estimated from OWLS and AFM experiments. OWLS analysis of PET coated waveguide chips was performed under flowing bFg solutions in PBS. A random sequential adsorption model was used to determine the m/a from measured changes in surface density during the adsorption process. Statistically significant differences were determined by ANOVA with a Scheffe post-hoc test. OWLS samples that are significantly different (p-value < 0.05) from the 1 mg/mL samples are marked with an (*). AFM Calculations were made using fibrinogen volume and height measurements obtained from Sit et al [9].
| Instrument | Material | Bovine fibrinogen Concentration | m/a (ng/cm2) |
|---|---|---|---|
| OWLS | PET | 1 mg/ml | 275.48 ± 4.34 |
| PET | 0.5 mg/ml | *129.68 ± 51.73 | |
| PET | 0.1 mg/ml | *119.63 ± 22.82 | |
| AFM | Mica | 50 ng/ml | 162.31 ± 19.28 |
| APTES | 50 ng/ml | 124 ± 17.96 | |
| OTS | 50 ng/ml | 79.82 ± 13.19 |
3.3 MALDI-TOF Sequence Coverage
The bFg peptides identified in the MALDI-TOF spectra of the solution-labeled samples revealed the sequence coverages of the Aα, Bβ, and γ chains to be 69%, 68%, and 61%, respectively (Fig. 2). Within these regions, 81 of the 107 lysine locations in bFg were identified. Due to the lack of lysines present in certain sections (e.g. a 128 amino acid span between Aα272–400), several large segments of bFg were neither available for MALDI-TOF detection after trypsin digestion nor accessible for labeling with biotin.
Fig. 2.
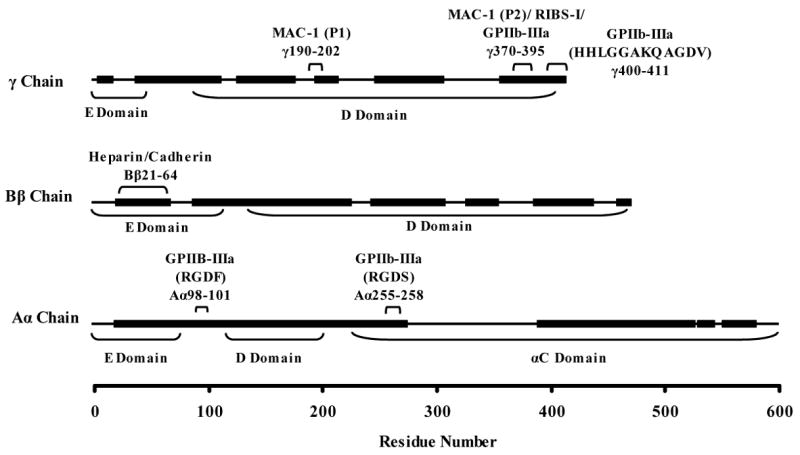
Sequence coverage obtained for Aα, Bβ, and γ chain in-gel trypsin digests of biotinylated bovine fibrinogen. Residues that were found in peptides recovered after the trypsin digests are represented with thick lines, while residues that were not found are represented with thin lines. Locations of important binding motifs are indicated. Sequence coverages of 69%, 68%, and 61% were obtained for the Aα, Bβ, and γ chains, respectively.
To determine whether the sequences spanned by the recovered peptides included areas of the protein that were complementary to binding motifs previously studied in human fibrinogen, the sequences of human and bovine fibrinogen were compared using multiple sequence alignments with CLUSTAL W. Although only approximately 66% of the entire bFg molecule was observable by MALDI-TOF, identified regions included several locations of interest, which are labeled in Fig. 2. In the Aα chain, an RGDF platelet binding domain for αIIbβ3 integrins at Aα98–101 was found to be complementary to Aα95–98 in human fibrinogen and was found in the MALDI-TOF sequence coverage. A region containing an RGGS motif at Aα541–544 in bFg that was not found in the sequence coverage had high sequence complementarity to a region in human fibrinogen containing an RGDS (Aα572–575). An RGDS motif is present in bFg at Aα255–258 and was within the sequence coverage, but an RGDS is not present in human fibrinogen at the complementary site. The Bβ chain of human fibrinogen contains domains linked to cadherin binding (Bβ15–42) and heparin binding (Bβ15–57), which were respectively located in bFg at Bβ21–49 and Bβ21–64. Both sequences were identified in bFg by MALDI-TOF after trypsin digestion. The binding domains of the γ chain are highly conserved between species. The MAC-1 leukocyte binding sites at γ190–202 and γ377–395, the RIBS-I epitope at γ373–385, the αIIbβ3 platelet-binding site within γ370–385, and the platelet-binding dodecapeptide at γ400–411 were in similar locations for human and bovine fibrinogen. The biologically-active γ chain binding domains were within the sequence coverage except for the MAC-1 leukocyte motif.
3.4 PET Particle Fabrication
SEM analysis revealed mostly spherical particles with diameters ranging from 1–200 μm that tended to aggregate in PBS solution (Fig. 3). Higher percentages of spherical particles were obtained when the surface tension was adjusted with surfactant during formation, but this step was not performed to eliminate the possibility of unwanted influences of surfactant on protein adsorption. BET analysis determined the surface area of the particles to be 20.1 ± 2.9 cm2/mg.
Fig. 3.

Scanning electron microscopy (SEM) of fabricated PET particles. a) An individual PET particle at 2000x magnification. b) SEM image of an aggregate of PET particles at 300x magnification. Particle sizes ranged from 1 to 200 μm.
3.5 Biotinylation of Surface Adsorbed Fibrinogen
The biotinylation protocol used for labeling and analyzing bFg after adsorption to PET surfaces is outlined in Fig. 4. Plastic disposable chromatography columns were packed with 150 mg of PET particles to provide approximately 3000 cm2 of surface area. Based on OWLS measurements of adsorbed bFg surface density, this surface area was estimated to adsorb between 0.3 – 0.9 mg of bFg as the solution concentration increased from 0.1 mg/mL to 1mg/mL. Decreased reaction between surface adsorbed bFg and the biotin label was expected when compared to the reaction of bFg in solution with the biotin label. Therefore, a 10-fold higher concentration of Sulfo-NHS-LC-Biotin was used for the surface adsorption experiments to maintain similar levels of biotin labeling between surface and solution samples (Fig. 5a). SDS-PAGE of bFg eluted from the PET columns revealed differences in molecular weights between labeled and unlabeled bFg chains. The changes in mass of solution-labeled and surface-labeled samples were compared to verify similar levels of biotin incorporation. The size of each band was determined via the linear relationship between the mobility of proteins in SDS-PAGE gels and the logarithms of their molecular weights [48]. Using the molecular weight standards to produce a standard curve, the Aα, Bβ, and γ chains were found to correlate well with their expected molecular weights of 63.5 kDa, 56 kDa, and 47 kDa (Fig 5b). The changes in molecular weight of the bFg chains in the solution-labeled samples corresponded to the binding of approximately 68.4% ± 17.0%, 53.8% ± 11.0%, and 52.1% ± 17.1% of the lysines in each respective chain. For the surface-labeled samples, the respective changes in molecular weight of the Aα, Bβ, and γ chains corresponded to the reaction of approximately 68.7% ± 12.8%, 50.6% ± 14.0%, and 41.6% ± 21.5% of the lysines. An ANOVA revealed no statistically significant differences in the extent of biotinylation between the solution-labeled and surface-labeled fibrinogen chains.
Fig. 5.
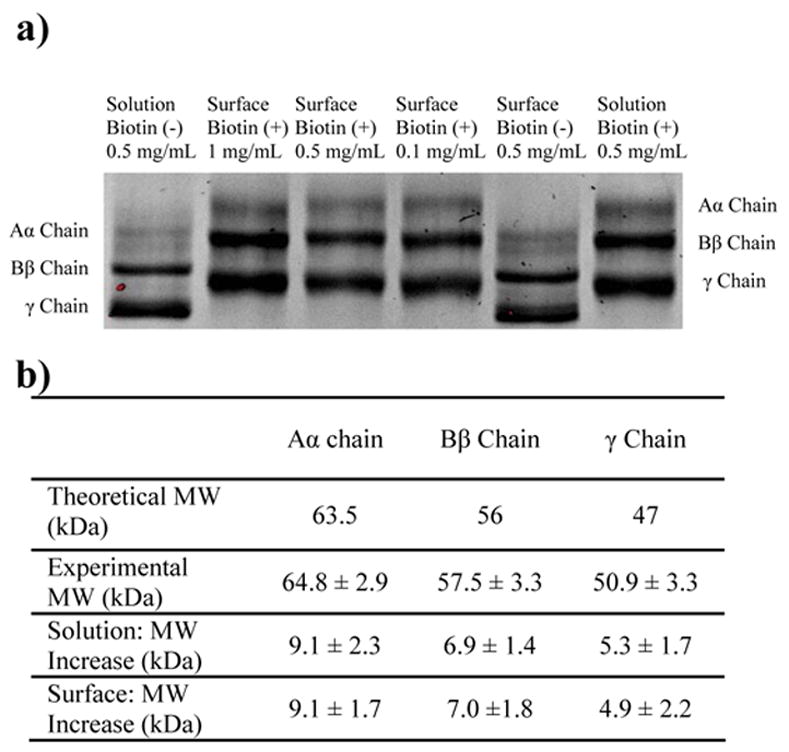
SDS-PAGE gel of bFg before and after reaction with the Sulfo-NHS-LC-Biotin label. The concentration of the label was adjusted to achieve the same degree of labeling for both bFg in solution and bFg adsorbed to PET. a) Aα, Bβ, and γ chains were separated by SDS-PAGE and stained with SYPRO Ruby stain to compare the increases in molecular weight due to labeling. Lanes are labeled as follows: Unlabeled bFg in solution (lane 1); labeled bFg eluted from the surfaces of PET particles after adsorption from 1 mg/mL (lane 2), 0.5 mg/mL (lane 3), and 0.1 mg/mL (lane 4); unlabeled bFg eluted from PET surfaces (lane 5); and bFg labeled in solution (lane 6). b) The increases in molecular weight due to labeling were determined from the changes in migration the distances of the protein bands.
3.6 Identification of Biotinylated Residues
After excision from SDS-PAGE gels and digestion with trypsin, the labeled and unlabeled fibrinogen chains were analyzed with MALDI-TOF and fibrinogen peptides were identified based on the expected molecular weights of tryptic fragments. Fig. 6 demonstrates the detection of labeled and unlabeled peptides for fibrinogen biotinylated in solution and fibrinogen biotinylated after adsorption onto a PET surface. Trypsin should not cleave after lysines that have been labeled, and thus all Sulfo-NHS-LC-Biotin labeled peptides contained at least one missed cleavage. As a result, the average peptide mass increased as the number of biotinylated peptides increased, and the majority of labeled peptides had molecular weights above 2000 Da.
Fig. 6.
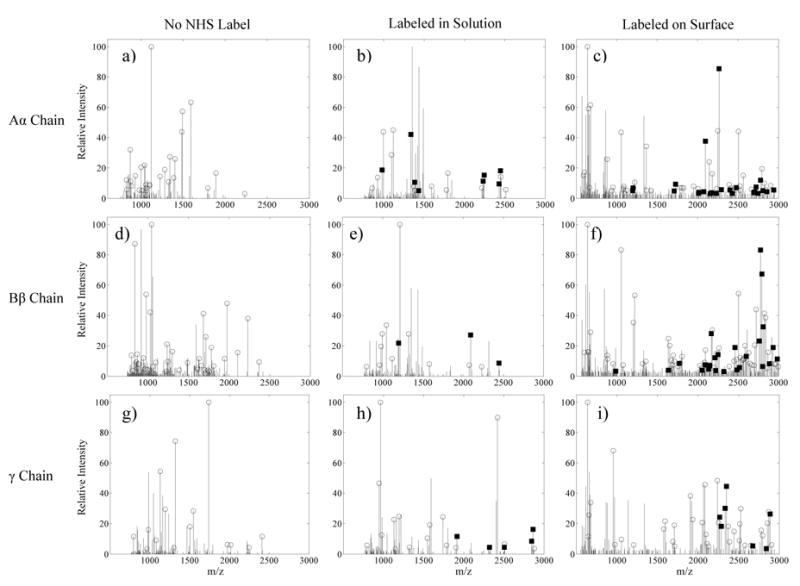
Centroided MALDI spectra corresponding to bFg trypsin digest fragment masses from the 0.5 mg/mL adsorption experiments. Open circles indicate the identification of masses that correspond to unlabeled bFg trypsin fragments. Black squares mark peaks that match an expected peptide after the Sulfo-NHS-LC-Biotin label was added to the mass. Spectra are shown for: a) Unlabeled bFg Aα chain, b) Aα chain labeled with biotin, c) Aα chain labeled with biotin after adsorption onto PET, d) Unlabeled Bβ chain, e) Bβ chain labeled with biotin, f) Bβ chain labeled with biotin after adsorption onto PET, g) Unlabeled γ chain, h) γ chain labeled with biotin, i) γ chain labeled with biotin after adsorption onto PET.
The ability to discern labeled from unlabeled peptides based on peptide molecular weight was verified with LC-ESI-MS/MS sequence analysis after an in-solution trypsin digestion. Table 3 lists the peptide sequences that were detected and the locations of the labeled lysines within them. These lysine locations and peptide masses were used to verify solution-labeled peptides that were identified with MALDI-TOF (Fig. 7 (✓)). A total of 19 of the 81 lysine residues spanned by the MALDI-TOF sequence coverage were identified as biotin-labeled, and 11 of these lysines were also detected as labeled by LC-ESI-MS/MS.
Table 3.
Biotin-labeled bFg peptides identified by liquid chromatography electrospray-ionization ion trap tandem mass spectrometry. bFg was labeled with biotin while in a 0.5 mg/mL solution. Collision-induced dissociation fragmentation spectra of tryptic bFg peptides were compared to the predicted spectra of tryptic peptides from a database of 1×106 proteins. Only peptides that matched bFg peptides with p-values < 1×10−7 were considered significant. Labeled lysine locations are displayed in lowercase italics.
| Peptide Sequence | Lysine Location | Mol. Wt. (Da) | p-value of match |
|---|---|---|---|
| MkGLIDEVDQDFTSR | Aα55 | 2093.63 | 1×10−15 |
| GDFAkANNNDNTFK | Aα103 | 1895.43 | 2.45×10−12 |
| ANNNDNTFkQISEDLR | Aα112 | 2218.23 | 6.66×10−11 |
| kVIEQVQR | Aα128 | 1339.54 | 2.32×10−09 |
| LEVDIDIkIR | Aα160 | 1554.08 | 8.49×10−12 |
| ALEHkVDLEDYK | Aα175 | 1799.48 | 4.14×10−12 |
| NQQkQLEQVIAINLLPSR | Aα186 | 2432.49 | 2.6×10−14 |
| EFkSQLQEAPLEWK | Aα222 | 2072.62 | 1.21×10−12 |
| ALLEMQQTkMVLETFGGDGHAR | Aα242 | 2772.7 | 1.98×10−10 |
| SCSkVITkTVTNADGR | Aα438 | 2415.95 | 6.73×10−10 |
| VITkTVTNADGR | Aα442 | 1614.47 | 2.21×10−12 |
| TETTkEVVK | Aα455 | 1374.64 | 2.69×10−09 |
| Dk DDFFTR | Aα491 | 1383.43 | 5.06×10−09 |
| kPPDADGCLHADPDLGVLCPTGCK | Bβ65 | 2933.07 | 1.91×10−11 |
| kSIEDLR | Bβ102 | 1200.23 | 5.38×10−10 |
| VYCDMkTEK | Bβ250 | 1513.58 | 6.69×10−11 |
| VYCDMkTEkGGWTVIQNR | Bβ250, Bβ253 | 2865.09 | 3.8×10−09 |
| TEkGGWTVIQNR | Bβ253 | 1727.93 | 7.66×10−13 |
| QGFGNIATNAEGkK | Bβ290 | 1774.73 | 4.34×10−08 |
| ISQLTNMGPTkLLIEMEDWK | Bβ316 | 2687.71 | 3.64×10−13 |
| DNDGWkTTDPR | Bβ393 | 1644.27 | 4.96×10−11 |
| kQCSkEDGGGWWYNR | Bβ399, Bβ403 | 2550.37 | 5.22×10−15 |
| NSkSMMEEIMK | γ87 | 1667.27 | 3.57×10−11 |
| DkVVQLEANCQEPCQDTVk IHDVTGR | γ127, γ144 | 3719.16 | 1.62×10−08 |
| DCQDVANkGAK | γ159 | 1544.92 | 8.48×10−13 |
| LDGSLDFkK | γ205 | 1362.57 | 2.57×10−10 |
| LAIGEGQQHQLGGAkQAGDV | γ406 | 2316.76 | 3.77×10−09 |
Fig. 7.

Normalized MALDI-TOF spectral peaks corresponding to biotinylated lysine residues within bFg Aα, Bβ, and γ chains, labeled while in a 0.5 mg/mL solution. A cumulative sum of the intensities of all the labeled peptides that contained the listed lysine residues was determined, normalized relative to the highest intensity obtained, and averaged across three separate experiments. Error bars represent one standard deviation. (✓) mark peaks that were also found to be labeled by the more accurate LC-ESI-MS/MS sequence analysis. The MALDI-TOF sequence coverages for each chain are displayed below each graph. Labeled lysine locations are in italics.
The lysine residues that were labeled after bFg was adsorbed onto PET from 1, 0.5, and 0.1 mg/mL solutions are displayed in Fig. 8. The exposure of lysine residues was higher for the 0.5 mg/mL and 0.1 mg/mL samples when compared to the 1 mg/mL samples. Both the degree of label incorporation and the surface coverage of the MALDI-TOF spectra increased after adsorption onto PET (Table 4). A statistical comparison between solution-labeled and the 1 mg/mL surface-labeled samples revealed a significant decrease in the relative intensity of Aα128 and a significant increase in exposure of Aα438 and Aα442 following adsorption. Several biotin-labeled locations found for the 1 mg/mL surface sample were within the sequence coverage of the solution-labeled samples, yet were not found labeled in solution. Overall, 7 labeled lysines were found in the Aα chain only in the adsorbed protein, along with 10 in the Bβ chain, and 9 in the γ chain. Two of theses residues were Bβ60, which is located within a heparin/cadherin binding domain, and γ196, which is located within a MAC-1 binding motif.
Fig. 8.
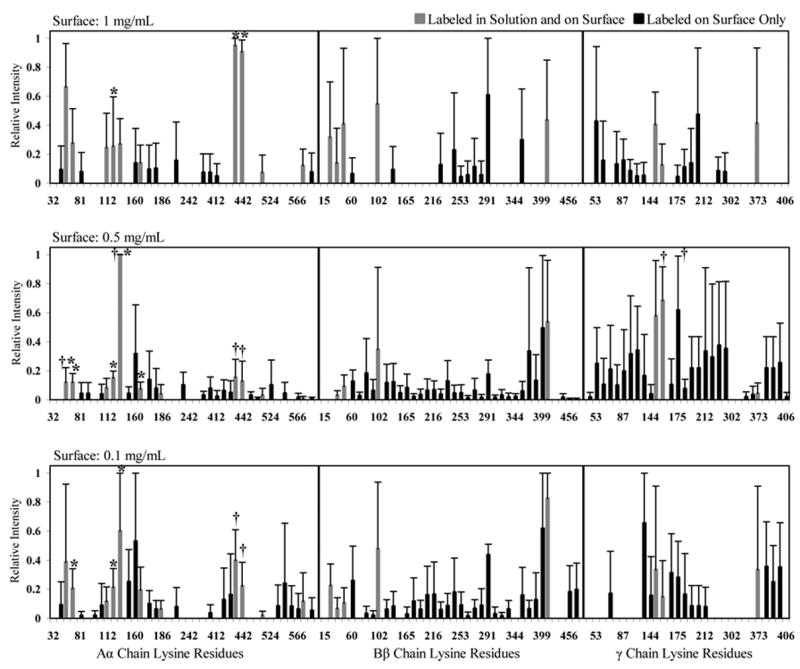
Normalized MALDI-TOF spectral peak intensities of lysine residues within bFg Aα, Bβ, and γ chains that were biotinylated after adsorption to a PET surface from 1, 0.5 and 0.1 mg/mL solutions. Error bars represent one standard deviation. Locations that were labeled both in solution and after adsorption to a PET surface are represented by grey bars. Locations that were labeled only after adsorption to a PET surface are represented by black bars. Pairwise Mann-Whitney comparisons were used to determine statistically significant differences in intensity (p-value <0.05) between solution-labeled samples and surface-labeled samples (*) and between the high concentration (1 mg/mL) surface-labeled samples and the low concentration (0.5 mg/mL, 0.1 mg/mL) surface-labeled samples (†).
Table 4.
Sequence coverage and percentage of labeled lysines observed after MALDI-TOF analysis of tryptic bFg peptides. Peptides originated from bFg that was labeled with Sulfo-NHS-LC-Biotin while in solution or after adsorption onto PET from different solution concentrations.
| Sequence Coverage | Lysines Labeled in Aα Chain | Lysines Labeled in Bβ Chain | Lysines Labeled in γ Chain | |
|---|---|---|---|---|
| Solution | 66% | 28% | 13% | 10% |
| 1 mg/mL | 74% | 51% | 39% | 53% |
| 0.5 mg/mL | 89% | 77% | 92% | 93% |
| 0.1 mg/mL | 84% | 69% | 84% | 50% |
After adsorption from 0.5 mg/mL solutions, there were 17 biotinylated locations each in the Aα and γ chains and 21 locations in the Bβ chain that were within the sequence coverage of the solution samples, but were not labeled while in solution. When the 0.5 mg/mL samples were compared to the solution samples, statistically significant decreases in intensity were found for Aα55, Aα71, Aα128 and Aα165, while Aα141 increased in intensity. In addition to Bβ60 and γ196, a multitude of previously inaccessible lysine residues were found labeled, including γ380, γ381, γ385, and γ406 which all contained binding motifs essential for platelet adhesion.
For adsorption from 0.1 mg/mL solutions, statistically significant decreases in lysine exposure were seen for Aα71, and Aα128, and an increase in ranked intensity was observed for Aα141. As seen in the 0.5 mg/mL samples, previously inaccessible locations were labeled in all three chains, with notable exposures of γ380, γ381, and γ385. Increased labeling in the Aα chain was mainly localized at the carboxyl terminus, while residues throughout the Bβ chain increased in exposure. Within these locations, a total of 12 residues in the Aα chain, 18 in the Bβ chain, and 9 in the γ chain were found to have reacted with biotin exclusively in the surface-labeled samples.
Protein visualization software was utilized to display the three-dimensional locations of biotinylation sites (Fig. 9). Lysines labeled while in solution were found primarily within the coiled coils of the Aα chain and the globular domains of the Bβ and γ chains. Several locations in the αC domain and in the coiled coil regions of the Bβ chain were biotin labeled as well, but these residues were not present in the bFg crystal structure. After adsorption, lysines throughout both the globular and coiled regions of the Bβ and γ chains were labeled, including several locations within the protein interior that are not normally solution-exposed.
Fig. 9.
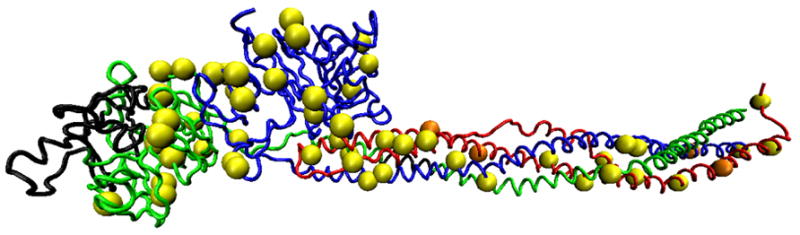
3D image of bFg displaying the locations of labeled lysines after adsorption to a PET surface from a 0.1 mg/mL solution. The image is labeled as follows: Aα chain, red; Bβ chain, blue; γ chain, green; residues not included in sequence coverage, black; labeled lysine residues, yellow spheres; lysine residues with statistically significant changes in relative intensity after adsorption to PET surface, orange spheres. Image was generated using the bFg PDB file 1DEQ and Visual Molecular Dynamics version 1.8.3.
4. Discussion
Conformational changes can occur in proteins after adsorption to biomaterial surfaces. These changes can influence protein surface coverage with time and expose binding sites that are responsible for eliciting biological responses [4–8]. Since fibrinogen adsorbed on biomaterials directs numerous cellular and thrombotic responses [49], a better understanding of fibrinogen conformational changes at the molecular level may provide information useful in the development of devices with enhanced biocompatibility. We are developing methods to allow a more complete analysis of the fibrinogen structure-function relationship at solid/liquid interfaces by using mass spectrometric mapping to probe changes in the exposure of individual residues. These methods may be beneficial for studying conformation changes in proteins adsorbed from complex mixtures, such as blood or plasma.
Prior to performing the chemical labeling experiments, we used optical waveguide lightmode spectroscopy (OWLS) to measure the extent of bFg spreading on PET as a function of solution protein concentration. OWLS has the ability to measure the surface density of protein layers in situ with a 30 s time-step, which permits the determination of parameters in kinetic models of molecular interactions with surfaces [41, 50]. Since our interest lies in the interactions of proteins at the solid/liquid interface between blood and biomaterial surfaces, we modified the surfaces of OWLS waveguide chips by spin coating to study the frequently used biomaterial PET.
OWLS results for bFg adsorbed onto PET demonstrated the postadsorptive spreading of the protein in a concentration-dependent manner, consistent with previous results on other surfaces [41, 51]. During protein adsorption, the rate of protein unfolding appears to be on the order of the time required to approach the RSA jamming limit on surfaces [41]. Proteins thus will not have adequate time to reach their maximum spread area when adsorbed from highly-concentrated solutions, due to the filling of adjacent spaces required for major conformational changes [52]. Statistical analysis of OWLS results showed that the 0.5 and 0.1 mg/mL samples were not different from each other, while both were significantly different from the 1 mg/mL samples. The RSA model does not account for the kinetics of spreading, diffusion or hydrodynamic effects on the adsorption process. However, diffusion and hydrodynamic influences have been shown to be counteractive [53]. A model that incorporates multiple states of protein denaturation may improve the measurement of protein unfolding kinetics on surfaces, but would require the curve-fitting of additional parameters to the OWLS surface density measurements, which may introduce additional inaccuracies.
Atomic force microscopy (AFM) has previously provided a detailed picture of fibrinogen’s post-adsorptive conformations. We compared our OWLS results with published AFM experiments, estimating fibrinogen m/a values from reported volume measurements [9] (Table 2). The m/a estimates were calculated based on dimensions of fibrinogen obtained by Sit et al, who used AFM to study the structure of fibrinogen adsorbed onto hydrophobic (octadecyltrichlorosilane, OTS), negatively charged (mica), and positively charged (3-aminopropyltriethoxysilane, APTES) surfaces. Their results were obtained under experimental conditions that differed from ours, allowing spreading to occur only over a 1 h period, while our studies allowed 4 h of spreading. We monitored adsorption only on PET, a surface with a higher surface energy than OTS and lower surface energy than mica and APTES [54]. Moreover, Sit et al. used a much lower fibrinogen concentration (50 ng/mL), so that the jamming limit would not be reached. The OTS surface resulted in a lower m/a in the AFM experiments than the PET used in our OWLS experiments, which was expected due to the higher surface energy of PET and the more concentrated fibrinogen solutions that we tested. PET in the presence of 0.5 and 0.1 mg/mL fibrinogen solutions showed larger spread areas than the hydrophilic mica surfaces exposed to a 50 ng/mL bulk concentration. Wertz et al. showed that the majority of postadsorptive conformational changes occurred within the first 15 minutes after adsorption [51], and thus the measurements of Sit et al. may represent the maximum obtainable surface coverage for individual fibrinogen molecules on the surfaces tested. Assuming that bFg has a rod-shaped geometry with dimensions of 6.0 × 6.0 × 45 nm, the theoretical unspread end-on and side-on close packing densities for adsorbed bFg at complete surface coverage are 1.57 and 0.21 μg/cm2 [55]. Thus, even at the highest concentration tested, bFg adsorption levels were in the vicinity of a side-on molecular packing arrangement. These adsorption densities were even lower than the 0.4 – 0.5 μg/cm2 achieved on low energy methylated surfaces [55–57], which may reflect extensive spreading of fibrinogen on the PET surface.
Our OWLS experiments revealed increases in the average area per bFg molecule as the solution concentration decreased from 1 mg/mL to 0.1 mg/mL, indicating that different degrees of conformational change occurred between these concentrations. By proteomic methods, we were then able to determine the specific residues within bFg that were susceptible to biotinylation after adsorption to the surfaces of PET particles. These small PET particles were fabricated with high surface areas using emulsion chemistry to increase the amount of adsorbed protein for the labeling experiments. Our hypothesis was that if a predisposition existed for certain biologically active domains within bFg to be exposed upon adsorption, then lysine residues found near these sites would have a higher tendency to react with the chemical label and could be detected by changes in MALDI-TOF peak intensities. Based on the OWLS results, we expected to observe differences in labeling between higher concentration, 1 mg/mL, and lower concentration, 0.5 and 0.1 mg/mL, samples. While only a few statistically significant changes in intensities were found, large differences in the number of biotin labeled lysines were observed. These qualitative results suggest that conformational changes influenced by the energetically more favorable interactions of hydrophilic residues with the solution over the PET surface may be driving the exposure of biologically active motifs. This is evidenced by the drastic increase in accessibility of lysines within the highly interspecies conserved γ chain, which coincides with the exposure of several cell interaction motifs essential to thrombosis and inflammation. Overall, our results are consistent with previous experiments examining the interactions of proteins with low energy surfaces, but at a higher level of resolution and revealing additional complexity.
Although SDS-PAGE showed equivalent increases in molecular weight between solution-labeled and surface-labeled samples, mass spectrometric analysis may have revealed considerable differences in the distribution of the label throughout the bFg molecule. MALDI-TOF has a strong bias for higher concentration peptides [58], and, consequently, many solution-labeled peptides originating from sterically-hindered locations may have been overshadowed by highly labeled locations. However, the nonlinear nature of MALDI-TOF intensity may have amplified the differences in labeling between solution and surface biotinylated samples. LC-ESI-MS/MS demonstrated the presence of several labeled peptides that were never detected in the MALDI-TOF spectra of solution-labeled samples. More quantitative mass spectrometric methods will be required to definitively show that such differences between surface and solution labeled fibrinogen exist.
Increases in lysine exposure within the Aα chain appeared to occur most prominently in the αC domain. Previous immunochemical studies of the human fibrinogen Aα chain performed by Cierniewski et al. revealed that in solution, the majority of the immunogenic surface resided between Aα1–238, with moderate immunogenicity between 239–476 and minimal antibody binding at the C-terminus between Aα518–584 [59]. Since the inaccessible areas of the αC domain were readily bound by antibody after plasmin cleavage, they concluded that this region was integrated into the native fibrinogen molecule in a manner that influenced epitope exposure. Mass spectrometric mapping of the bFg Aα chain displayed similar distributions of accessibility to the biotin label. Of the 11 lysines biotinylated to a high degree in the Aα chain, only one location was found labeled near the carboxyl terminus in the solution-labeled samples. The surface-labeled samples showed that post-adsorptive conformational changes readily exposed these minimally accessible carboxyl-terminal domains, possibly due to decreased interaction between the αC domain and the E domain. Binding of an antibody that recognizes human Aα572–575 has been correlated with platelet adhesion to adsorbed human fibrinogen [60]. A region within the bFg αC domain containing an RGGS site (Aα541–544) has high sequence homology with a region in the human fibrinogen sequence that contains an RGDS motif (Aα572–575). A lysine (Aα549) near this site was shown to increase in exposure after adsorption to PET from the lower concentration solutions. Similarly, a lysine (Aα103) in close proximity to the Aα98–101 RGDF site in the bovine fibrinogen, which is synonymous to RGDF Aα95–98 in human fibrinogen, was found to increase in exposure upon adsorption to PET as the solution concentration decreased. While biotinylated Aα103 was detected in the LC-ESI-MS/MS analysis of solution-labeled samples, Aα549 was only found to be biotinylated in the surface-labeled samples by MALDI-TOF.
Although similar percentages of labeled lysines was found in the γ chain when comparing the high concentration and low concentration adsorption experiments (Table 4), the labeling in the low concentration samples shifted towards the carboxyl terminus. In the 0.5 and the 0.1 mg/mL samples, residues were labeled within the sequence γ370–383, which has been shown to have an essential role in platelet adhesion [61]. Sites located in the γ377–395 sequence, thought to bind the αMβ2/Mac-1 leukocyte integrin and participate in inflammation, were also found labeled [62]. LC-ESI-MS/MS analysis revealed that γ406 within the platelet adhesion dodecapeptide (400–411) was accessible while in solution. This lysine was not found labeled in MALDI-TOF analysis of solution samples nor the 1 and 0.1 mg/mL surface-labeled samples. However, this residue was detected as labeled after adsorption to PET from the 0.5 mg/mL solutions [13]. These results suggest that the peptide containing this lysine either does not ionize well in MALDI-TOF or is present at very low amounts.
It has been shown that nonactivated platelets bind less to human fibrinogen fragments containing only γ400–411, only Aα95–98, or without Aα572–575 [63]. These previous findings are supported by our data that suggest a post-adsorptive exposure of the locations in bFg that are synonymous to these platelet binding human fibrinogen sites. Furthermore, a lysine near the bovine complement to Aα572–575 in human fibrinogen was only found exposed after adsorption, while a lysine near the bovine complement to Aα95–98 in human fibrinogen was accessible both in solution and on the surface. Thus our data also supports previously performed experiments that suggest that Aα572–575 may be more specific to post-adsorptive platelet binding than Aα95–98.
We are currently investigating several potential changes to our chemical labeling protocol that could broaden its applications in the study of protein conformation at solid/liquid interfaces. First, MALDI-TOF was selected for our initial mass spectrometric mapping experiments because of the quickness and ease of sample preparation and analysis. Unlike MALDI-TOF, more sensitive electrospray ion-trap and Fourier transform mass spectrometry can take advantage of quantitative techniques such as stable-isotope labeling by amino acids in cell culture (SILAC, [64]) or ITRAQ [29], as well as provide vastly more reliable peptide identification. With mass spectrometry coupled to liquid chromatography, adsorption from multicomponent protein solutions or even whole blood could potentially be analyzed, which would have novel applications in the study of post-adsorptive processes such as the Vroman effect [65, 66]. Another area that could be considerably improved in our experiments is sequence coverage, since we were only able to observe approximately 66% of the fibrinogen molecule after in-gel trypsin digestion and purification of solution samples. Digesting samples with different enzymes should provide access to sections of the fibrinogen molecule that were not observed with trypsin digestion. Also, by using multiple labels that react with functional groups other than primary amines, a higher percentage of fibrinogen’s surface could be monitored for conformational changes.
5. Conclusion
In summary, we have applied OWLS and mass spectrometric mapping in the study of proteins at solid/liquid interfaces. We have shown that OWLS waveguide chips can be coated with PET and that substantial differences in protein spread area are found between bFg adsorbed at 1 and 0.1 mg/mL. Using chemical labeling and proteomic methods, we compared the differences in exposure of lysines between bFg in solution and bFg adsorbed to PET. Our results suggested extensive conformational changes in all three chains and specific increases in the exposure of biologically relevant residues in the Aα and γ chains during the unfolding process on PET. Based on our results, we are pursuing more comprehensive and quantitative adaptations of this methodology.
Acknowledgments
We acknowledge funding from the Center for Materials Innovation at Washington University in St. Louis, NIH training grant 5T32HL07916-05 (EAS), and partial support from NIH R01HL085364 (DLE). We are grateful to Dr. Seunghwan Lee at the Swiss Federal Institute of Technology Zurich for advice on preparing coated OWLS waveguides, to Dr. Nathan Baker at the Washington University School of Medicine Center for Computational Biology for helpful discussion on statistical analysis, and to Jeff Wight at the University of Missouri-Rolla for performing the XPS analysis.
Footnotes
No benefit of any kind will be received either directly or indirectly by the author(s).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baier RE, Dutton RC. Initial events in interactions of blood with a foreign surface. J Biomed Mater Res. 1969;3(1):191–206. doi: 10.1002/jbm.820030115. [DOI] [PubMed] [Google Scholar]
- 2.Andrade JD, Hlady V. Protein adsorption and materials biocompatibility: a tutorial review and suggested hypothesis. Adv Polym Sci. 1986:1–63. [Google Scholar]
- 3.Brash JL, Davidson VJ. Adsorption of glass and polyethylene from solutions of fibrinogen and albumin. Thromb Res. 1976;9(3):249–59. doi: 10.1016/0049-3848(76)90214-0. [DOI] [PubMed] [Google Scholar]
- 4.Zucker MB, Vroman L. Platelet adhesion induced by fibrinogen adsorbed onto glass. Proc Soc Exp Biol Med. 1969;131(2):318–20. doi: 10.3181/00379727-131-33866. [DOI] [PubMed] [Google Scholar]
- 5.Mohammad SF, Hardison MD, Glenn CH, Morton BD, Bolan JC, Mason RG. Adhesion of human blood platelets to glass and polymer surfaces. I. Studies with platelets in plasma. Haemostasis. 1974;3(5–6):257–70. doi: 10.1159/000214064. [DOI] [PubMed] [Google Scholar]
- 6.Shen M, Horbett TA. The effects of surface chemistry and adsorbed proteins on monocyte/macrophage adhesion to chemically modified polystyrene surfaces. J Biomed Mater Res. 2001;57(3):336–45. doi: 10.1002/1097-4636(20011205)57:3<336::aid-jbm1176>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 7.Kottke-Marchant K, Anderson JM, Miller KM, Marchant RE, Lazarus H. Vascular graft-associated complement activation and leukocyte adhesion in an artificial circulation. J Biomed Mater Res. 1987;21(3):379–97. doi: 10.1002/jbm.820210310. [DOI] [PubMed] [Google Scholar]
- 8.Anderson JM, Bonfield TL, Ziats NP. Protein adsorption and cellular adhesion and activation on biomedical polymers. Int J Artif Organs. 1990;13(6):375–82. [PubMed] [Google Scholar]
- 9.Sit PS, Marchant RE. Surface-dependent conformations of human fibrinogen observed by atomic force microscopy under aqueous conditions. Thrombosis and Haemostasis. 1999;82(3):1053–1060. [PubMed] [Google Scholar]
- 10.Tang L, Wu Y, Timmons RB. Fibrinogen adsorption and host tissue responses to plasma functionalized surfaces. J Biomed Mater Res. 1998;42(1):156–63. doi: 10.1002/(sici)1097-4636(199810)42:1<156::aid-jbm19>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 11.Shiba E, Lindon JN, Kushner L, Matsueda GR, Hawiger J, Kloczewiak M, et al. Antibody-detectable changes in fibrinogen adsorption affecting platelet activation on polymer surfaces. Am J Physiol. 1991;260(5 Pt 1):C965–74. doi: 10.1152/ajpcell.1991.260.5.C965. [DOI] [PubMed] [Google Scholar]
- 12.Farrell DH, Thiagarajan P, Chung DW, Davie EW. Role of fibrinogen alpha and gamma chain sites in platelet aggregation. Proc Natl Acad Sci U S A. 1992;89(22):10729–32. doi: 10.1073/pnas.89.22.10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bennett JS. Platelet-fibrinogen interactions. Ann N Y Acad Sci. 2001;936:340–54. doi: 10.1111/j.1749-6632.2001.tb03521.x. [DOI] [PubMed] [Google Scholar]
- 14.Liu QD, Rooney MM, Kasirer-Friede A, Brown E, Lord ST, Frojmovic MM. Role of the gamma chain Ala-Gly-Asp-Val and A alpha chain Arg-Gly-Asp-Ser sites of fibrinogen in coaggregation of platelets and fibrinogen-coated beads. Biochimica Et Biophysica Acta-Protein Structure and Molecular Enzymology. 1998;1385(1):33–42. doi: 10.1016/s0167-4838(98)00039-9. [DOI] [PubMed] [Google Scholar]
- 15.Hall CE, Slayter HS. The fibrinogen molecule: its size, shape, and mode of polymerization. J Biophys Biochem Cytol. 1959;5(1):11–6. doi: 10.1083/jcb.5.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madrazo J, Brown JH, Litvinovich S, Dominguez R, Yakovlev S, Medved L, et al. Crystal structure of the central region of bovine fibrinogen (E5 fragment) at 1.4-A resolution. Proc Natl Acad Sci U S A. 2001;98(21):11967–72. doi: 10.1073/pnas.211439798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spraggon G, Everse SJ, Doolittle RF. Crystal structures of fragment D from human fibrinogen and its crosslinked counterpart from fibrin (vol 389, pg 455, 1997) Nature. 1997;390(6657):315–315. doi: 10.1038/38947. [DOI] [PubMed] [Google Scholar]
- 18.Veklich YI, Gorkun OV, Medved LV, Nieuwenhuizen W, Weisel JW. Carboxyl-terminal portions of the alpha chains of fibrinogen and fibrin. Localization by electron microscopy and the effects of isolated alpha C fragments on polymerization. J Biol Chem. 1993;268(18):13577–85. [PubMed] [Google Scholar]
- 19.Andrieux A, Hudry-Clergeon G, Ryckewaert JJ, Chapel A, Ginsberg MH, Plow EF, et al. Amino acid sequences in fibrinogen mediating its interaction with its platelet receptor, GPIIbIIIa. J Biol Chem. 1989;264(16):9258–65. [PubMed] [Google Scholar]
- 20.Yakovlev S, Gorlatov S, Ingham K, Medved L. Interaction of fibrin(ogen) with heparin: further characterization and localization of the heparin-binding site. Biochemistry. 2003;42(25):7709–16. doi: 10.1021/bi0344073. [DOI] [PubMed] [Google Scholar]
- 21.Bach TL, Barsigian C, Yaen CH, Martinez J. Endothelial cell VE-cadherin functions as a receptor for the beta15–42 sequence of fibrin. J Biol Chem. 1998;273(46):30719–28. doi: 10.1074/jbc.273.46.30719. [DOI] [PubMed] [Google Scholar]
- 22.Kloczewiak M, Timmons S, Lukas TJ, Hawiger J. Platelet receptor recognition site on human fibrinogen. Synthesis and structure-function relationship of peptides corresponding to the carboxy-terminal segment of the gamma chain. Biochemistry. 1984;23(8):1767–74. doi: 10.1021/bi00303a028. [DOI] [PubMed] [Google Scholar]
- 23.Suckau D, Mak M, Przybylski M. Protein surface topology-probing by selective chemical modification and mass spectrometric peptide mapping. Proc Natl Acad Sci U S A. 1992;89(12):5630–4. doi: 10.1073/pnas.89.12.5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carven GJ, Stern LJ. Probing the ligand-induced conformational change in HLA-DR1 by selective chemical modification and mass spectrometric mapping. Biochemistry. 2005;44(42):13625–13637. doi: 10.1021/bi050972p. [DOI] [PubMed] [Google Scholar]
- 25.Robinson CV, Gross M, Eyles SJ, Ewbank JJ, Mayhew M, Hartl FU, et al. Conformation of GroEL-bound alpha-lactalbumin probed by mass spectrometry. Nature. 1994;372(6507):646–51. doi: 10.1038/372646a0. [DOI] [PubMed] [Google Scholar]
- 26.Bennett KL, Matthiesen T, Roepstorff P. Probing protein surface topology by chemical surface labeling, crosslinking, and mass spectrometry. Methods Mol Biol. 2000;146:113–31. doi: 10.1385/1-59259-045-4:113. [DOI] [PubMed] [Google Scholar]
- 27.Hager-Braun C, Tomer KB. Characterization of the tertiary structure of soluble CD4 bound to glycosylated full-length HIVgp120 by chemical modification of arginine residues and mass spectrometric analysis. Biochemistry. 2002;41(6):1759–66. doi: 10.1021/bi011626k. [DOI] [PubMed] [Google Scholar]
- 28.Roesli C, Elia G, Neri D. Two-dimensional mass spectrometric mapping. Curr Opin Chem Biol. 2006;10(1):35–41. doi: 10.1016/j.cbpa.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 29.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3(12):1154–69. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Staros JV. N-hydroxysulfosuccinimide active esters: bis(N-hydroxysulfosuccinimide) esters of two dicarboxylic acids are hydrophilic, membrane-impermeant, protein cross-linkers. Biochemistry. 1982;21(17):3950–5. doi: 10.1021/bi00260a008. [DOI] [PubMed] [Google Scholar]
- 31.Cacciafesta P, Humphris ADL, Jandt KD, Miles MJ. Human plasma fibrinogen adsorption on ultraflat titanium oxide surfaces studied with atomic force microscopy. Langmuir. 2000;16(21):8167–8175. [Google Scholar]
- 32.Holland NB, Marchant RE. Individual plasma proteins detected on rough biomaterials by phase imaging AFM. J Biomed Mater Res. 2000;51(3):307–15. doi: 10.1002/1097-4636(20000905)51:3<307::aid-jbm3>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 33.Lu XY, Huang Y, Qian WP, Tang ZM, Lu ZH. An effective method for quantitative evaluation of proteins adsorbed on biomaterial surfaces. J Biomed Mater Res A. 2003;66(3):722–7. doi: 10.1002/jbm.a.10014. [DOI] [PubMed] [Google Scholar]
- 34.Toscano A, Santore MM. Fibrinogen adsorption on three silica-based surfaces: conformation and kinetics. Langmuir. 2006;22(6):2588–97. doi: 10.1021/la051641g. [DOI] [PubMed] [Google Scholar]
- 35.Tunc S, Maitz MF, Steiner G, Vazquez L, Pham MT, Salzer R. In situ conformational analysis of fibrinogen adsorbed on Si surfaces. Colloids and Surfaces B: Biointerfaces. 2005;42(3–4):219–225. doi: 10.1016/j.colsurfb.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 36.Hylton DM, Shalaby SW, Latour RA., Jr Direct correlation between adsorption-induced changes in protein structure and platelet adhesion. J Biomed Mater Res A. 2005;73(3):349–58. doi: 10.1002/jbm.a.30295. [DOI] [PubMed] [Google Scholar]
- 37.Clarke ML, Wang J, Chen Z. Conformational changes of fibrinogen after adsorption. J Phys Chem B Condens Matter Mater Surf Interfaces Biophys. 2005;109(46):22027–35. doi: 10.1021/jp054456k. [DOI] [PubMed] [Google Scholar]
- 38.Buijs J, Norde W, Lichtenbelt JWT. Changes in the secondary structure of adsorbed IgG and F(ab')(2) studied by FTIR spectroscopy. Langmuir. 1996;12(6):1605–1613. [Google Scholar]
- 39.Lu DR, Park K. Effect of surface hydrophobicity on the conformational changes of adsorbed fibrinogen. Journal of Colloid and Interface Science. 1991;144(1):271–281. [Google Scholar]
- 40.Kim JK, Scott EA, Elbert DL. Proteomic analysis of protein adsorption: serum amyloid P adsorbs to materials and promotes leukocyte adhesion. J Biomed Mater Res A. 2005;75(1):199–209. doi: 10.1002/jbm.a.30424. [DOI] [PubMed] [Google Scholar]
- 41.Ramsden JJ. Concentration scaling of protein deposition kinetics. Physical Review Letters. 1993;71(2):295–298. doi: 10.1103/PhysRevLett.71.295. [DOI] [PubMed] [Google Scholar]
- 42.Schaaf P, Talbot J. Kinetics of random sequential adsorption. Physical Review Letters. 1989;62(2):175–178. doi: 10.1103/PhysRevLett.62.175. [DOI] [PubMed] [Google Scholar]
- 43.Hinrichsen EL, Feder J, Jossang T. Geometry of random sequential adsorption. J Stat Phys. 1986;44:793–827. [Google Scholar]
- 44.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31(13):3784–8. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown JH, Volkmann N, Jun G, Henschen-Edman AH, Cohen C. The crystal structure of modified bovine fibrinogen. Proc Natl Acad Sci U S A. 2000;97(1):85–90. doi: 10.1073/pnas.97.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–8. 27–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 47.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weber K, Osborn M. The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis. J Biol Chem. 1969;244(16):4406–12. [PubMed] [Google Scholar]
- 49.Weisel JW. Fibrinogen and fibrin. Adv Protein Chem. 2005;70:247–99. doi: 10.1016/S0065-3233(05)70008-5. [DOI] [PubMed] [Google Scholar]
- 50.Voros J, Ramsden JJ, Csucs G, Szendro I, De Paul SM, Textor M, et al. Optical grating coupler biosensors. Biomaterials. 2002;23(17):3699–710. doi: 10.1016/s0142-9612(02)00103-5. [DOI] [PubMed] [Google Scholar]
- 51.Wertz CF, Santore MM. Adsorption and relaxation kinetics of albumin and fibrinogen on hydrophobic surfaces: Single-species and competitive behavior. Langmuir. 1999;15(26):8884–8894. [Google Scholar]
- 52.Schaaf P, Talbot J. Surface Exclusion Effects in Adsorption Processes. Journal of Chemical Physics. 1989;91(7):4401–4409. [Google Scholar]
- 53.Bafaluy J, Senger B, Voegel J, Schaaf P. Effect of hydrodynamic interactions on the distribution of adhering Brownian particles. Physical Review Letters. 1993;70(5):623–626. doi: 10.1103/PhysRevLett.70.623. [DOI] [PubMed] [Google Scholar]
- 54.Baier RE, Shafrin EG, Zisman WA. Adhesion: mechanisms that assist or impede it. Science. 1968;162(860):1360–8. doi: 10.1126/science.162.3860.1360. [DOI] [PubMed] [Google Scholar]
- 55.Lassen B, Malmsten M. Structure of protein layers during competitive adsorption. Journal of Colloid and Interface Science. 1996;180(2):339–349. [Google Scholar]
- 56.Malmsten M. Ellipsometry Studies of Protein Layers Adsorbed at Hydrophobic Surfaces. Journal of Colloid and Interface Science. 1994;166(2):333–342. [Google Scholar]
- 57.Wojciechowski PW, Brash JL. Fibrinogen and albumin adsorption from human blood plasma and from buffer onto chemically functionalized silica substrates. Colloids and Surfaces B: Biointerfaces. 1993;1(2):107–117. [Google Scholar]
- 58.Knochenmuss R, Stortelder A, Breuker K, Zenobi R. Secondary ion-molecule reactions in matrix-assisted laser desorption/ionization. J Mass Spectrom. 2000;35(11):1237–45. doi: 10.1002/1096-9888(200011)35:11<1237::AID-JMS74>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 59.Cierniewski CS, Plow EF, Edgington TS. Conformation of the carboxy-terminal region of the A alpha chain of fibrinogen as elucidated by immunochemical analyses. Eur J Biochem. 1984;141(3):489–96. doi: 10.1111/j.1432-1033.1984.tb08219.x. [DOI] [PubMed] [Google Scholar]
- 60.Tsai WB, Grunkemeier JM, Horbett TA. Human plasma fibrinogen adsorption and platelet adhesion to polystyrene. J Biomed Mater Res. 1999;44(2):130–9. doi: 10.1002/(sici)1097-4636(199902)44:2<130::aid-jbm2>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 61.Podolnikova NP, Yakubenko VP, Volkov GL, Plow EF, Ugarova TP. Identification of a novel binding site for platelet integrins alpha(IIb)beta(3) (GPIIbIIIa) and alpha(5)beta(1) in the gamma C-domain of fibrinogen. Journal of Biological Chemistry. 2003;278(34):32251–32258. doi: 10.1074/jbc.M300410200. [DOI] [PubMed] [Google Scholar]
- 62.Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, et al. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest. 2004;113(11):1596–606. doi: 10.1172/JCI20741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Savage B, Ruggeri ZM. Selective recognition of adhesive sites in surface-bound fibrinogen by glycoprotein IIb-IIIa on nonactivated platelets. J Biol Chem. 1991;266(17):11227–33. [PubMed] [Google Scholar]
- 64.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–86. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 65.Slack SM, Horbett TA. Changes in the Strength of Fibrinogen Attachment to Solid-Surfaces - an Explanation of the Influence of Surface-Chemistry on the Vroman Effect. Journal of Colloid and Interface Science. 1989;133(1):148–165. [Google Scholar]
- 66.Vroman L, Adams AL. Identification of rapid changes at plasma-solid interfaces. J Biomed Mater Res. 1969;3(1):43–67. doi: 10.1002/jbm.820030106. [DOI] [PubMed] [Google Scholar]