

Abstract
ComS is an anti-adapter protein that binds to MecA, displacing the competence transcription factor ComK. This protects ComK from degradation by the ClpCP protease and turns on the switch leading to bistable gene expression. Here we identify the motifs on ComK and ComS that mediate binding to MecA, and show that they contain similar core sequences (FMLYPK and IILYPR respectively), located near the C- and N-termini of the respective proteins. A 17 residue peptide from ComK including this sequence, has the same affinity for MecA as full length ComK and a peptide containing this sequence is sufficient to target green fluorescent protein for degradation in vivo. Cross-linking and competition experiments demonstrate that ComK- and ComS-derived peptides bind to the same region of MecA. We propose a model in which the anti-adapter protein ComS acts by direct competition to protect ComK from degradation.
Regulated proteolysis is an important mechanism in both prokaryotic and eukaryotic cells, particularly because the degradation of regulator molecules may be used to control signaling cascades and metabolic or developmental pathways. Examples of bacterial development controlled in part by proteolysis, include sporulation and competence in Bacillus subtilis (Pan et al., 2001; Turgay et al., 1998) and the cell cycle of Caulobacter crescentus (Skerker and Laub, 2004). In addition to these specific regulatory mechanisms, the removal of damaged or incomplete proteins is a universally important component of the stress response (reviewed in Hengge and Bukau, 2003; Jenal and Hengge-Aronis, 2003; Sauer et al., 2004).
In these, as well as other systems, the relevant proteases are often compartmental, energy-dependent, proteolysis machines, such as the eukaryotic and prokaryotic proteasomes and the Clp family of prokaryotic proteases (Butler et al., 2006; Gottesman, 2003). The ClpX, A and C ATPase proteins form hexamers that unfold substrate proteins and translocate them to the lumen of a structure, formed by two heptameric rings of the ClpP serine protease, where degradation ensues. Because proteolysis is confined to the interior of the complex, it can be targeted to a restricted number of potential substrates, requiring that substrate recognition occur at a stage prior to degradation. One intensively studied recognition system involves the 11 residue SsrA sequence tag, which is added to proteins when translation is interrupted (Keiler et al., 1996; reviewed in Sauer et al., 2004). This sequence serves for direct recognition by the ClpXP and ClpAP proteases of Escherichia coli, and illustrates the concept that recognition may target a short unstructured peptide sequence in the substrate (Flynn et al., 2003; Gonzalez et al., 2000; Hoskins et al., 2000; Laachouch et al., 1996; Levchenko et al., 1997; Ryan et al., 2002). Other systems involve the participation of so-called adapter proteins, which may be required for recognition or may modulate the interaction between the protease and the substrate (Ades, 2004; Dougan et al., 2002; Sauer et al., 2004). Adapter proteins can modify substrate preferences, thereby enabling a single protease to regulate several processes.
Competence in B. subtilis is a genetically programmed state permitting cells to be genetically transformed by environmental DNA (reviewed in Chen et al., 2005; Chen and Dubnau, 2003; Dubnau and Lovett Jr., 2001). The synthesis of proteins that mediate the internalization of DNA is controlled by the transcriptional activator ComK (van Sinderen et al., 1995) which is expressed in a bistable manner (Maamar and Dubnau, 2005; Smits et al., 2005). During exponential growth, ComK is degraded by the ClpCP protease, and is targeted to the protease by the adapter protein MecA (Turgay et al., 1998; Turgay et al., 1997). ClpC is an AAA+ (ATPasesassociated with a variety of activities) ATPase, related to ClpA and ClpX (Msadek et al., 1994). A hallmark of competence development is the stabilization of ComK by the small protein ComS, which is produced in response to quorum sensing. ComS binds to MecA, releasing ComK and protecting it from degradation. Both ComK and ComS interact primarily with the N-terminal domain of MecA whereas ClpC binds to its C-terminal domain (Persuh et al., 1999).
An unusual feature of this system is that both MecA and ComS can be degraded by ClpCP (Turgay et al., 1998). The degradation of ComS requires the presence of MecA, which therefore acts as an adapter for both ComS and ComK. It is possible that both ComS and ComK bind to MecA using related recognition signals. Thus, they might compete for binding to MecA, thereby explaining how ComS protects ComK from degradation during the development of competence. In this study, we show that this is indeed the case. A short peptide motif is identified on ComK that is sufficient for binding to MecA and for targeting ComK and the unrelated green fluorescent protein (GFP) for degradation. A similar peptide from ComS mediates interaction with MecA and we show that the ComK- and ComS-derived peptides compete for binding to MecA in vitro and bind to the same or an overlapping site on MecA.
Results
If ComS and ComK compete for a common site on MecA, we might expect them to possess similar sequence features. In fact, a cluster of residues near the C-terminus of ComK (residues 168-193, FMLYPK) resembles a cluster near the N-terminus of ComS (residues 12-17, IILYPR). Consistent with the possible involvement of these sequences in binding to MecA is the report from Ogura et al (1999) that individual alanine substitutions at the IILYPR motif interfered with ComS function in vivo, and the replacement of the ComS motif by IALYPR decreased binding to MecA in vitro. A preliminary experiment carried out with surface plasmon resonance (SPR), showed that the C-terminal half of ComK (S96-Y192) containing the FMLYPK sequence bound to MecA, while the N-terminal half of ComK (M1-P93) showed no binding to MecA (not shown).
ComK residues required for degradation and for transcriptional activation of comG
A mecA knockout strain fails to target ComK for degradation by ClpCP, resulting in ComK accumulation (Turgay et al., 1998). Thus, changes in comK that prevent binding to MecA would be expected to stabilize the mutant protein. We replaced the FMLYPK motif of ComK by FAAAAK. The mutant protein accumulated to high levels, and this accumulation was unaffected by mutational inactivation of mecA or comS (not shown). To investigate this ComK motif and flanking residues in more detail, residues 159-192 of ComK were individually replaced by alanine. Two methods were used to determine the effects of these mutations. In the first, a comG-lacZ reporter was used, since comG is dependent on ComK for its transcription. A potential difficulty with this approach is that mutations compromising both ComK degradation and the ability of ComK to act as a transcription factor may be subject to misinterpretation, since these two effects will be manifested respectively as increased and decreased β-galactosidase activity. In a second system, designed to obviate this problem, the mutant comK genes were placed under control of the xylose induced Pxyl promoter inserted at the ectopic amyE locus (Hahn et al., 1996) and the wild-type comK gene was inactivated. In these constructs, the auto-regulatory loop enabling ComK to activate the transcription of its own gene is interrupted, so that the accumulation of ComK is completely independent of the transcriptional activity of ComK.
Individual alanine replacements of the FMLYPK residues resulted in significant increases in comG-lacZ expression (Fig. 1). Interestingly, mutations in the extreme C-terminus (T177-Y192) lowered the expression of comG-lacZ, as did mutations H159A-R161A, suggesting that these residues may be important for the transcriptional activity of ComK. We directly assessed the accumulation of mutant forms by Western blot analysis of ComK expressed under xylose-inducible control. Fig. 2A reveals that substitution of residues 168-173, which result in high comG-lacZ expression, also cause the accumulation of ComK, presumably due to the failure of the mutant proteins to be degraded. Alanine substitutions of residues 164-167 do not result in detectable increases in the level of ComK (not shown). Taken together, the data shown in Figs. 1 and 2 indicate that ComK residues 168-173 are involved in mediating the degradation of ComK and are not essential for its activity as a transcription factor on the comG promoter.
Fig. 1.
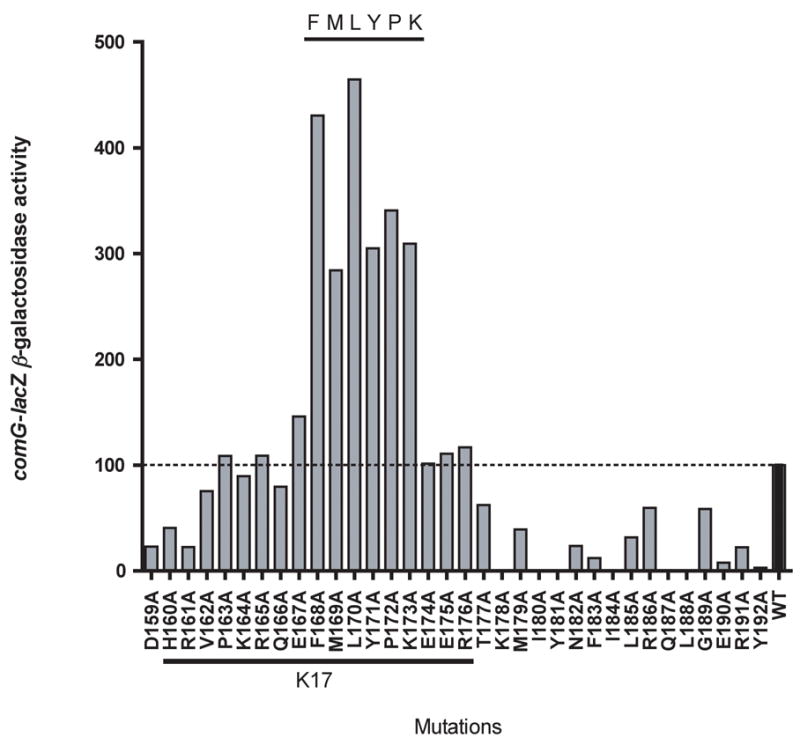
Alanine substitution mutations of the C-terminal part of ComK and their effects on the expression of comG-lacZ two hours after entering stationary phase. The sequence of the last 34 residues is presented on the X-axis and the effects of alanine replacements are shown by vertical bars. The dotted line represents the wild type level (100%) of comG-lacZ expression. comG-lacZ β-galactosidase activity is presented as a percentage of ComG-lacZ activity (measured in Miller units) of a strain producing wild type ComK. The sequence of the K17 peptide is underlined.
Fig. 2.
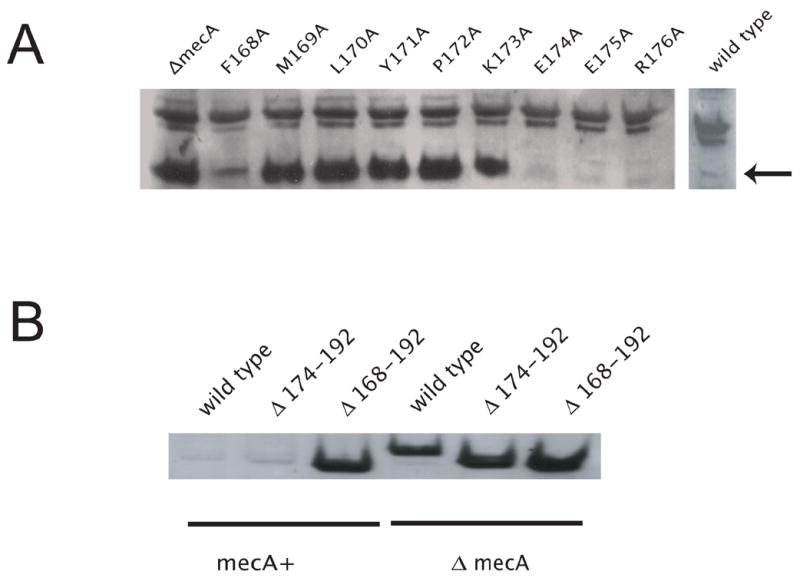
The effects of comK mutations on stability of ComK. A. ComK mutants in the FMLYPK region stabilize ComK in vivo. ComK was detected by Western blot analysis using anti-ComK antibody. Point mutants of ComK were grown in competence medium containing 2% xylose and expressed from PxylA. The copy of comK in its native locus was disrupted. Samples were collected three hours after entering stationary phase. The arrow shows the position of ComK. B. The deletion of the recognition region causes accumulation of ComK in the presence of MecA. The expression of ComK and of its truncated derivatives was induced by 2% xylose early in the vegetative phase of growth in competence medium. The copy of comK in its native locus was disrupted. Samples were collected 1 hour after induction and processed as described previously (Hahn et al., 2005). The gel-loads were normalized according to their protein concentrations. Immunoblot analysis was performed using anti-ComK antibody.
Although individual alanine replacements of the residues downstream from E173 have no marked effects on ComK stability, it is possible that the recognition sequence must be located internally, i.e. that a length of protein must be present downstream for appropriate interaction with MecA or ClpC to occur. To further explore the role of the C-terminal region in degradation, we prepared truncated versions of ComK lacking residues F168-Y192 (recognition sequence absent) or E174-Y192 (with the recognition sequence intact). These constructs were placed under control of the Pxyl promoter and inserted at amyE in a background containing a comK loss-of-function mutation and a comG-lacZ reporter. Western blot analysis showed that while full length ComK and ComK∆ (174-192) were degraded similarly in a MecA-dependent fashion, ComK∆ (168-192) accumulated in both the mecA and wild-type backgrounds, presumably due to the absence of the recognition sequence (Fig. 2B). In the two truncated constructs, β-galactosidase activity from the comG-lacZ reporter was extremely low (not shown), consistent with a role for C-terminal residues of ComK in transcriptional activation.
Based on these results, we can conclude that residues within the region F168-R173 of ComK are critical for its degradation. Although this degradation signal is normally internal, this position is not obligatory, since the 19 residues downstream from position 173 are not needed for degradation. The most parsimonious explanation for the roles of residues 164-173 is that they are required for the binding of ComK to MecA.
Residues H160-R176 are sufficient for binding to MecA
To identify the ComK residues involved in binding to MecA, we immobilized synthetic peptides derived from the ComK sequence by primary amine chemistry, and measured binding of soluble MecA-His by SPR. A 17-mer (K17) corresponding to residues 160-176 (CHRVPKRQEFMLYPKEER) includes all the residues important for degradation, as well as several additional residues to make the peptide more soluble. As controls, we used a peptide with the same composition but a scrambled sequence and peptides containing alanine substitutions (FMAYPK and FMAAAK, substitutions underlined) (A cysteine residue was added at the N-terminus of all peptides to facilitate future studies). In addition to the N-termini, the peptides have two K residues and can therefore bind to the chip surface in several orientations. MecA-His6 bound well to K17 but not to K17scramble or to the alanine substitution versions (Fig. 3 and not shown).
Fig. 3.
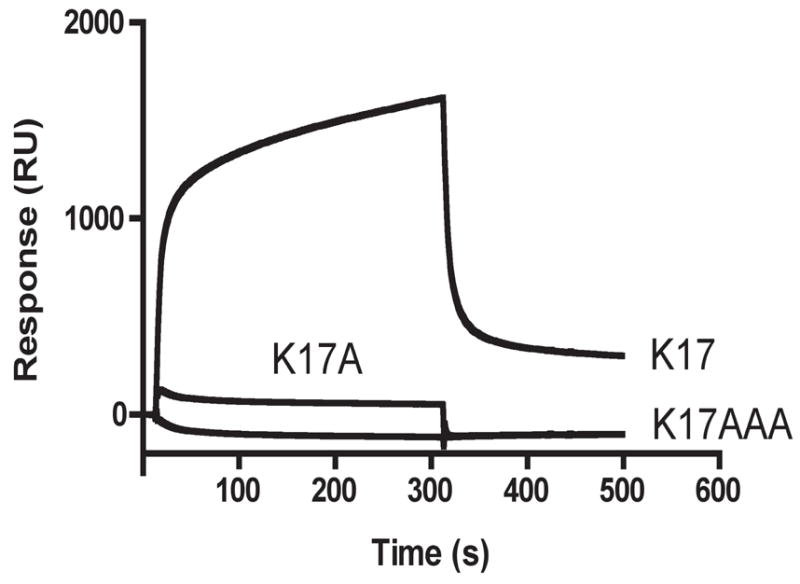
SPR analysis of the interactions between MecA and the K17 and alanine substituted peptides. MecA-His6 (1μM) was injected over 1100 RU of immobilized peptide. The response of the same analyte on a mock-coupled surface was subtracted from both curves. K17A and K17AAA refer to peptides in which the FMLYPK recognition sequence has been replaced by FMAYPK and FMAAAK respectively.
These experiments demonstrate that ComK residues H160-R176 are sufficient for specific binding to MecA. However, it is possible that additional ComK residues contribute to high affinity binding. To address this possibility, we compared the binding affinities of K17 and of full length ComK to MecA.
The affinities of ComK and K17 for MecA are similar
We compared the binding affinities of ComK and K17 to MecA in solution by SPR, using the Biacore instrument only to determine the concentrations of free MecA. This approach avoids potential difficulties associated with binding on surfaces, such as rebinding artifacts and transport limitations (Schuck, 1997). For these experiments, we immobilized a large amount (4800 RU) of K17 by primary amine chemistry. A control surface, treated with the coupling reagents in the absence of protein, was used as a negative control. Samples containing a constant concentration of MecA-His6 (1.3 μM) with increasing concentrations of K17 were incubated, injected over the chip surface and the signals at the 75-second time point were recorded. To construct a calibration curve, different concentrations of MecA-His6 were injected over the same surface and signals were recorded at the 75-second time point. Using these data to construct a standard curve, the concentration of free MecA-His6 could be determined for each concentration of K17 (Fig. 4A) and the affinity constant could be calculated as described in Methods. The analogous experiment was carried out using full length ComK in place of the K17 peptide (Fig. 4B). The KD for ComK binding to MecA determined from these data was 1.3 μM and that for K17 binding to MecA was 1.2 μM. This agreement suggests that the thermodynamically significant contacts between ComK and MecA involve the residues in K17. Additionally, the ability of added full-length ComK to compete for binding of MecA to K17 on the chip suggests that this peptide binds to the same site on MecA as full length ComK. Clearly, the residues in K17 are not only important for the degradation of ComK in vivo (Figs. 2 and 3) but are fully sufficient for the binding of ComK to MecA. Attempts to compare the MecA binding affinities of ComS and S17 using SPR were confounded by the tendency of ComS to aggregate on the surface of the flow cell. However, the addition of either ComS or S17 to a solution containing MecA and ClpC, stimulated the ClpC ATPase to the same extent, suggesting that the important residues of ComS for MecA binding are contained within S17 (data not shown).
Fig. 4.
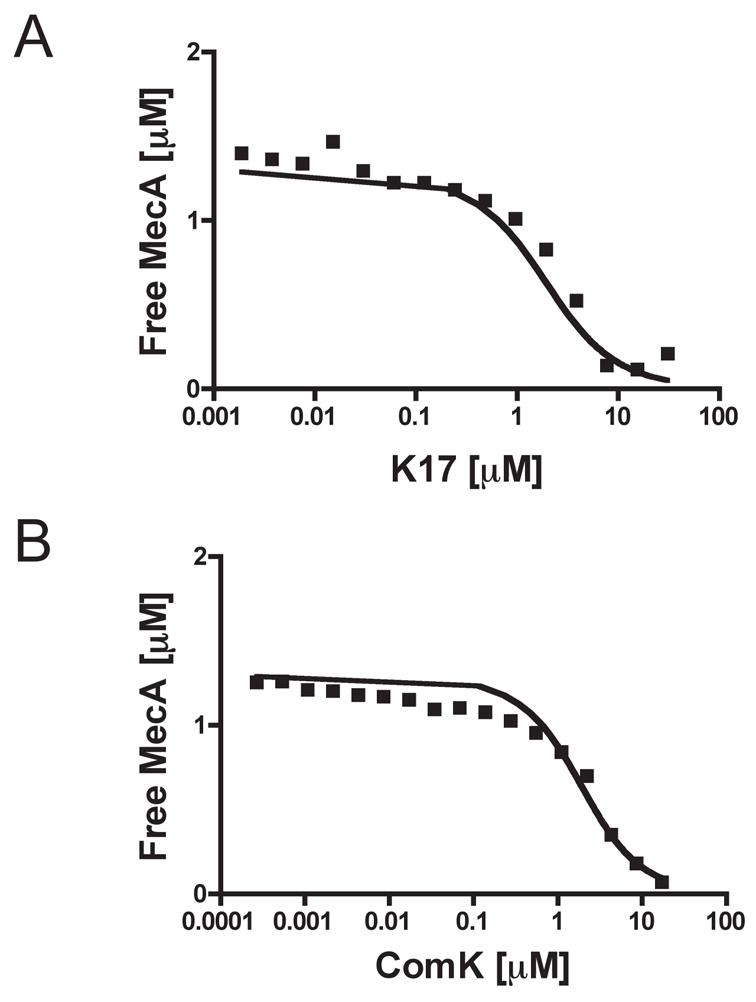
Binding affinities of ComK and K17 for MecA. The binding affinities were determined using a solution affinity method (see text and Experimental procedures). The KD determined for MecA-His6 binding to K17 is 1.2 μM (A) and that for MecA-His6 binding to ComK is 1.3 μM (B). All the plotted concentrations are in units of μM.
Residues H160-R176 target GFP for degradation in vivo
To investigate the sufficiency of the K17 sequence to target a protein for degradation, we fused the K17 sequence to the C-terminus of GFP and placed the fusion under control of Pxyl inserted at amyE. Additional fusions of GFP to full length ComK and to R176-Y192 were constructed (Fig. 5A). Immunoblot analysis with anti-GFP antibodies showed that GFP-ComK and GFP-H160-Y176 were largely degraded in the wild-type background, but were protected from degradation in a mecA background (Fig. 5A). The R176-Y192 fusion and GFP control were unaffected by the presence of MecA and appeared to be stable. These results were confirmed by fluorescence microscopy (Fig. S1) and similar results were obtained when the clpC gene was inactivated instead of mecA (not shown). The GFP-K17 fusion was not only degraded in vivo by ClpCP by a MecA-dependent mechanism, but was also protected from degradation in the presence of ComS (Fig. 5B). We conclude that the residues H160-R176 are sufficient to target GFP for degradation in vivo, and that the ClpC ATPase is therefore able to unfold the stable GFP molecule.
Fig. 5.
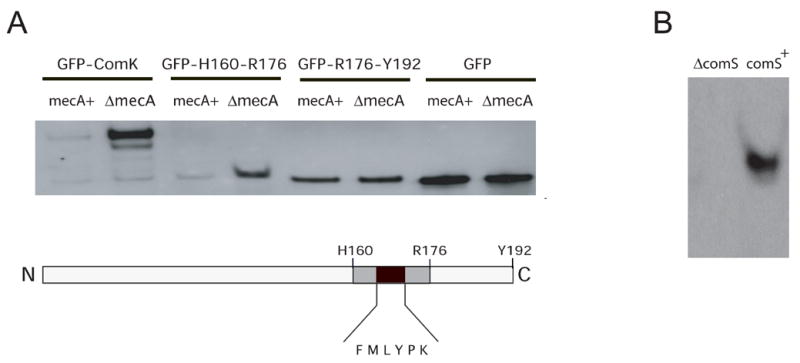
K17 targets GFP for degradation and is protected by ComS. A. Immunoblotting with anti-GFP antibody shows that GFP fused to ComK and to the K17 region are readily degraded in vivo by a MecA-dependent mechanism, while the GFP fusion to the downstream sequence H176-192 and un-fused GFP are not affected by inactivation of mecA. The expression of GFP fusions was induced by adding 2% xylose in the mid-exponential phase of growth. Exponential cells were used to avoid the expression of comS. Samples were collected after 30 minutes. The constructs tested are diagrammed. B. To demonstrate that GFP-K17 is protected in vivo by ComS, the same GFP-K17 construct was introduced into strains carrying a plasmid that over-expresses comS (comS+) and into one in which comS was inactivated by mutation (comS). These strains were grown to T2 for analysis. In panels A and B, the gel-loads were normalized according to their protein concentrations.
Binding of a ComS peptide to MecA; competition by K17
We next determined whether the portion of ComS containing the hexapeptide IILYPR is sufficient to mediate binding to MecA, and whether this binding would be competed by K17. As noted previously, alanine substitutions within this sequence were reported to decrease ComS binding to MecA (Ogura et al., 1999). We prepared synthetic peptides, K17BB and S17BB. The first is identical to K17, but contains a p-benzoyl-L-phenylalanine (BPA) photo cross-linking moiety at its N-terminus and a biotin molecule attached to its C-terminus. S17BB consists of ComS residues G5-E21 (GKHLISSIILYPRPSGE), and is similarly modified by the addition of BPA and biotin.
S17BB was tested for binding to MecA-His6 by SPR, immobilizing S17BB on a streptavidin-coated matrix. MecA-His6 bound well to this surface but no binding to an empty surface was detected (not shown). When either peptide was incubated with MecA-His6, and UV irradiated, they readily cross-linked to the MecA protein. Cross-linking of K17BB to MecA-His6 was reduced when ComS was added as a competitor, consistent with the hypothesis that they compete for the same site on MecA (Fig. 6A). Cross-linking of S17BB was also reduced by the addition of ComS, as would be expected, confirming that the S17BB peptide is binding to the correct site on MecA (Fig. 6B). Finally, cross-linking of the S17BB peptide to MecA, was reduced by the addition of K17, but not by K17scramble (Fig. 6C and D), consistent with the specific binding of the K17 and S17 peptides to the same or an overlapping site on MecA. These experiments demonstrate that the predicted binding sequence of ComS does bind to MecA, that this binding is specific and can be reduced by the addition of the binding peptide derived from ComK. These data identify related recognition motifs on ComS and ComK and support a model in which ComS functions by competing with ComK for a common site on MecA.
Fig. 6.
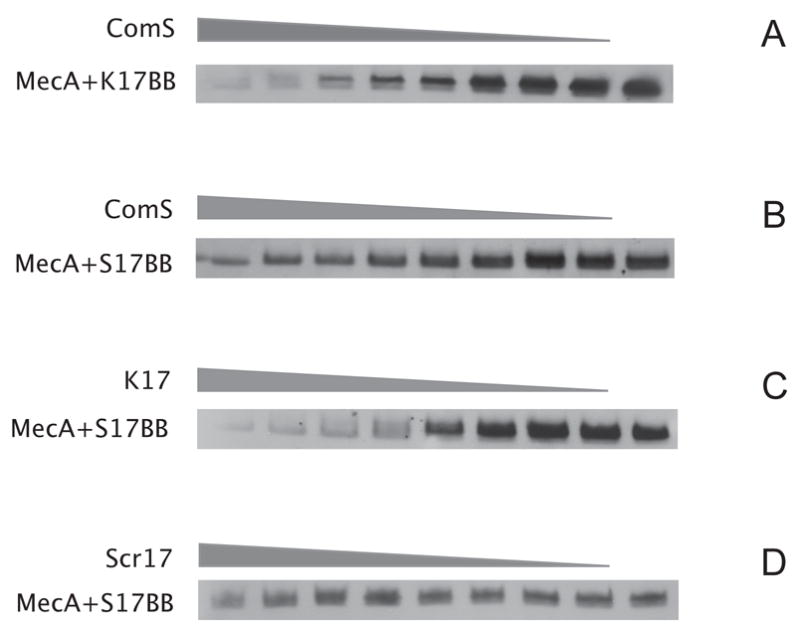
Photo cross-linking of K17 BB and S17 BB to MecA-His6 shows competition between K17 and S17. Cross-linking reactions were prepared as described in Material and Methods. Samples containing MecA-His6 (7μM final concentration), K17BB or S17BB (11 μM final concentration) and K17, K17scramble or synthetic ComS, were cross-linked, separated by Tris-tricine SDS polyacrylamide gels followed by immunoblot analysis with anti-biotin antibodies. The grey triangles show relative concentrations of competing peptides and proteins. A. Cross-linking of MecA-His6 to K17BB in the presence of varying synthetic ComS. ComS was added to the reaction in the range 0–200 μM, in two-fold increments. B. Cross-linking of MecA-His6 to S17BB in the presence of varying synthetic ComS. ComS was added to the reaction in the range 0–200 μM, in two-fold increments. C. Cross-linking of MecA-His6 to S17BB in the presence of varying K17. K17 was added to the reaction in the range 0–200 μM, in two-fold increments. D. Cross-linking of MecA-His6 to S17BB in the presence of varying K17scramble. K17scramble was added to the reaction in the range 0–200 μM, in two-fold increments.
R17BB and S17BB cross-link to nearby binding sites on MecA
We determined whether K17BB and S17BB cross-link to nearby sites on MecA. The peptides were cross-linked to MecA-His6, as described above, and the cross-linked complexes were isolated by nickel affinity chromatography to remove un-cross-linked peptides. The complexes were then cleaved with AspN, a protease that cuts at the N-terminal side of aspartates. (the S17 and K17 peptides lack aspartates). The cross-linked fragments were captured by binding to streptavidin-coated magnetic beads via their biotin moieties and the eluted material was resolved by SDS-PAGE. Cross-linked fragments of 5–6 kD were detected by Western blotting with anti-biotin monoclonal antibody and were missing from AspN-cleaved MecA, not previously cross-linked. These bands were blotted to a PVDF membrane and subjected to sequencing by Edman degradation. The S17 and K17 peptide sequences were not detected, because they are N-terminally cross-linked to MecA and thereby blocked for the Edman reaction. With both peptides, an identical MecA sequence was detected: DVISLSKLNVNGSKTT, corresponding to 16 residues at the N-terminal portion of a predicted MecA-derived AspN cleavage product of 28 residues. With the addition of the BPA and biotin moieties and the 17 residues of K17 or S17 to this segment of MecA, the total molecular weight of the cross-linked fragments should be 6026 and 5649 respectively, in agreement with their sizes determined by SDS-PAGE. We conclude that the K17BB and S17BB peptides, when bound to MecA, are similarly oriented, with their N-terminal BPA moieties positioned near this 28-residue AspN fragment of MecA. These data provide further support for the conclusion that ComS directly competes with ComK for binding to the same or an overlapping site on MecA.
Discussion
Recognition of substrates by MecA
The principal result of this study is that two proteins, each dependent on MecA for degradation by ClpCP, are recognized using similar short peptide motifs. Six ComK residues (from K168 to K173) are required for optimal binding to MecA and residues 160-176 can mediate not only binding, but also the degradation of GFP in vivo when fused to the C-terminus of that protein. The ComK residues identified by these experiments are apparently the only ones that make significant contacts with MecA, at least for initial binding, since the affinities of K17 and of full length ComK for MecA are similar. It is noteworthy that in ComS, the recognition motif is near the N-terminus of the protein and in ComK it is near the C-terminus. Additionally, the sequence downstream from the ComK motif can be deleted without effect on degradation. Thus, the recognition motif does not need to be positioned at a particular location, within the primary sequence to mediate degradation.
The results from competition and cross-linking experiments support the idea that ComS and ComK bind to a common or overlapping site on MecA. It is interesting that although ComK and ComS both bind to the N-terminal domain of MecA (Persuh et al., 1999), the 28-residue AspN fragment to which K17BB and S17BB became cross-linked is located within the C-terminal domain of MecA. Our previous work established that the N-terminal domain of MecA binds ComK and ComS strongly, while the C-terminal domain binds ClpC (Persuh et al., 1999). However, that report also showed weak binding of MBP-ComK and ComS with the C-terminal domain. Taken together, the available evidence suggests that the binding sites for ComK and ComS include residues from both domains of MecA and that these sites may be formed at the interface of the two domains. Recent mutational analysis of MecA is consistent with this hypothesis (M. DeFrancesca and D.D., unpublished).
MecA clearly plays other roles, unrelated to competence. MecA overproduction inhibits sporulation (Kong et al., 1993), while its inactivation results in over-expression of the eps operon (M. Albano and D.D., unpublished). However, additional substrates for MecA have not been described. Spore formation and eps expression are not affected by mutation of comS or comK, suggesting that additional substrate binding sites may be present on MecA.
A search of the predicted protein sequences encoded by the B. subtilis genome, has revealed one gene product containing a sequence closely similar to the ComS recognition sequence identified in this study. YxeI, annotated as resembling a widespread family of penicillin amidases, contains the sequence VILYPR. No additional sequence identical to that of the ComK recognition sequence or differing from it by only a single residue was detected. YxeI is clearly a candidate for a MecA/ClpCP regulated protein.
Mechanism of ComK degradation
Because the H160-R176 sequence of ComK is sufficient to target GFP in vivo for degradation by ClpCP, we can exclude models in which the initial binding to MecA mediated by this sequence is followed by the obligatory interaction of additional ComK residues with MecA or ClpC, unless a sequence competent for this second interaction is fortuitously present in GFP. Such a two-step mechanism has been demonstrated in the case of RpoS degradation, mediated by the adapter RssB (Studemann et al., 2003).
Secondary structure predictions for ComK using Predict Protein (http://www.predictprotein.org) suggest that the MecA binding residues are flanked by α-helices (Fig. S2). Our results imply that most of the predicted helical residues are not needed for binding of ComK to MecA, because mutation of these residues has no detectable effect on ComK stability (Fig. 2). These results together with those of Susanna et al (Susanna et al., 2006) demonstrate that the putative helical residues are needed for transcriptional activity and therefore possibly for DNA binding. If the predicted helices, which flank the recognition sequence for MecA interact with DNA, the simultaneous binding of ComK to MecA and to DNA might be sterically prevented. Consequently, the degradation of ComK would be secondary to the inhibition of its activity by MecA binding and the MecA-mediated ComK activity switch would be quickly responsive to increases in the concentration of ComS. In fact, it is known that ComK cannot bind simultaneously to MecA and to the comK promoter (Turgay et al., 1997).
The role of ComS in the release of ComK
This work provides strong support for the idea that ComS competes with ComK for MecA binding, thereby protecting this protein from degradation. First, the original impetus for examining the C-terminal region of ComK came from the similarity between its hexapeptde sequence FMLYPK and the IILYPR sequence in ComS. The work of Ogura et al (1999) had shown that the first five of these residues in ComS were required for its function, and particularly for binding to MecA. Our mutational data confirms that the equivalent ComK residues are likewise needed for MecA binding. The cross-linking data shows that the BPA moieties at the N-termini of the ComS- and ComK-derived 17-mers contact the same 28 residue fragment of MecA, consistent with them occupying the same or at least overlapping binding sites. Furthermore, the K17BB and S17BB peptides compete for binding to MecA (Fig. 6). Since these peptides appear to establish all the important contacts of their parent proteins with MecA (Fig. 4 and unpublished results) this competition must reflect the binding properties of ComK and ComS, strengthening the competition model.
Our data is subject to an alternative, but in our view less likely, explanation. The two peptides and hence ComS and ComK might bind to nearby sites on MecA. These binding sites would be non-overlapping, involving different residues in the MecA N-terminal domain, but extending to nearby regions within the MecA C-terminal domain. According to this model, MecA exists in two mutually exclusive conformational states. In the ComS-bound state, the affinity for ComK is low. In the ComK-bound state, the reverse is true. This more complex hypothesis ignores the similarity in the recognition sequences of ComK and ComS.
Anti-adapters and the regulation of protein stability
The system that seems most similar to the present one in terms of its regulatory logic is the degradation of RpoS (σS), a master regulator of the stress response by ClpXP in E. coli. This degradation requires the adapter protein RssB, which targets RpoS for degradation. Recently, the 89-residue IraP protein has been shown to bind to RssB, releasing RpoS, thereby protecting this sigma factor from degradation (Bougdour et al., 2006). Although the design of the RpoS and ComK systems appear similar, the proteins involved are not related in their sequences, except for ClpCP and ClpXP. IraP thus joins ComS as the latest member of what has been termed the “anti-adapter” family of proteins (Bougdour et al., 2006). It is not known if IraP acts by competition as we suggest here for ComS.
Alignment of ComK sequences
ComK-like genes have been sequenced from a number of Gram-positive bacteria. Fig. S2 shows a multiple alignment with ComK sequences from six bacterial species closely related to B. subtilis. Although much of the protein is quite conserved, the C-terminal region, downstream from residue H160 in the B. subtilis sequence, is highly divergent, except in B. licheniformis. B. licheniformis, like B. subtilis, encodes MecA and is the only bacterium of the 28 that is known to be competent for transformation. Perhaps the binding sequence for MecA has been retained or acquired only when interaction with MecA is needed, and that comK expression is regulated differently in the other bacteria. Although comK is widespread among low GC Gram positive bacteria, mecA is even more widespread, existing in organisms (Lactobacillus, Enterococcus, Desulfitobacterium, Leuconstoc) that appear to lack a comK ortholog (not shown). This supports the contention that MecA can play a variety of roles, unrelated to the regulation of ComK stability.
Experimental procedures
Strains and strain construction
The strains used in this work and their construction are described in Supplementary Material. Site-directed mutagenesis was performed with a QuikChange Mutagenesis kit (Stratagene). All the constructs obtained by PCR were confirmed by sequencing.
Peptide synthesis and protein purification
All the peptides used in this study except for ComS, were chemically synthesized by the Molecular Resource Facility at UMDNJ Newark. The purity and correctness of the products were verified by mass spectrometry. ComS peptide was a gift from Peter Zuber. MBP-ComK fusion proteins and MecA-His6 were purified as previously described (Turgay et al., 1997). Peptide and protein concentrations were measured by spectrophotometry in 6 M guanidium hydrochloride buffered by 50 mM phosphate buffer, pH 6.5 at 280 nm. Extinction coefficients were calculated from http://au.expasy.org/tools/protparam.html.
Protein Interaction analysis
Real time biomolecular interaction analysis experiments were performed using a Biacore 2000 instrument at 25°C. To test the interaction between K17 and MecAHis6 we used CM5 sensor chips. 1500 RU of K17 and the mutant peptides were covalently immobilized to separate cells using amine-coupling chemistry. Solutions containing 1.4 μM MecAHis6 in HBS-EP buffer (0.01 M HEPES, pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.005% v/v surfactant P20) were passed over the sensor chip surface at a constant flow rate 20 μl/min. The response of the same solution on a mock-coupled surface was subtracted from both sensograms.
To measure affinity in solution, we immobilized 4800 RU of the K17 peptide and passed a mixture of MecAHis6 at a constant concentration of 1.3 μM and K17 or ComK at final concentrations in the range 0–35 μM. The chip surface was regenerated twice after each injection of analyte with 1M NaCl and 2.5% of the detergent P20 in HBS-EP. The data were analyzed as follows. The same chip surface, containing the K17 peptide, was used with known concentrations of MecA, to construct a standard curve of RU versus input MecA. This curve was used to measure the concentrations of unbound MecA and the data were analyzed with the equation:
Where, BT is the amount of unbound analyte, KD is the dissociation constant and AT is the concentration of the varied component (K17 or ComK).
To immobilize S17BB, we used the biotin moiety of the peptide to couple it to a sensor chip SA (with pre-immobilized streptavidin).
Cross-linking and cleavage reactions
Cleavage reactions with AspN are described in Supplementary Material. Edman sequencing of protein fractions was performed by Mary Ann Gawinowicz at the Columbia University Protein Core and DNA Sequencing Facility.
Crosslinking reactions were carried out in 96 well microtiter plates (Greiner, Cellstar). The final volume of a typical cross-linking reaction was 0.3 ml. MecA-His6((7 μM final concentration), a benzoylphenylalanine (Bpa) and biotin-labeled peptide (K17BB or S17BB, 11μM final concentration) and one of the competing peptides (ComS, K17, Kscramble, final concentration in the range of 200-0μM) were mixed in PBS in the dark and incubated for 10 minutes at room temperature. The samples were then exposed to UV light for 2 minutes in UV crosslinker (FB-UVXL-1000, Fisher Scientific) to trigger the cross-linking reaction. Following the cross-linking reaction, reducing SDS loading buffer was added and the samples were boiled for 5 minutes. Western blotting was performed by standard procedures, as follows.
Western blot analysis and microscopy
To assess the stability of ComK mutants expressed from the PxylA promoter, cells were grown in the presence of 2% xylose until three hours after the transition to stationary phase and lysates were prepared as described previously (Hahn et al., 2005). The lysates were resolved by electrophoresis in 16% Tris-tricine SDS-polyacrylamide gels, transferred to nitrocellulose membrane (Biorad), and probed with anti-ComK antibody (Kong and Dubnau, 1994).
For the GFP degradation assays, cultures in mid-logarithmic phase were induced with 2% xylose. After 30 minutes, cells were harvested and samples for Western blot analysis were prepared and examined as described above. Anti-GFP primary antibody was used according to Rudner and Losick (Rudner and Losick, 2002).
Supplementary Material
Acknowledgments
We thank D. Wah, I. Chen, M. Persuh, J. Hahn and M. Albano for helpful discussion and valuable comments and I. Chen and Jeanie Dubnau for critical reading of the manuscript. We also thank B. Lang from Biacore Inc. for advice concerning interaction analysis using the Biacore instrument, D. Rudner for the gift of GFP antibody, P. Zuber for a kind gift of ComS and Leendert W. Hamoen for technical assistance with the purification of ComK. This work was supported by National Institutes of Health grant GM057720.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ades SE. Proteolysis: Adaptor, adaptor, catch me a catch. Curr Biol. 2004;14:R924–926. doi: 10.1016/j.cub.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Bougdour A, Wickner S, Gottesman S. Modulating RssB activity: IraP, a novel regulator of sigma(S) stability in Escherichia coli. Genes Dev. 2006;20:884–897. doi: 10.1101/gad.1400306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler SM, Festa RA, Pearce MJ, Darwin KH. Self-compartmentalized bacterial proteases and pathogenesis. Mol Microbiol. 2006;60:553–562. doi: 10.1111/j.1365-2958.2006.05128.x. [DOI] [PubMed] [Google Scholar]
- Chen I, Christie PJ, Dubnau D. The ins and outs of DNA transfer in bacteria. Science. 2005;310:1456–1460. doi: 10.1126/science.1114021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I, Dubnau D. DNA transport during transformation. Front Biosci. 2003;8:s544–556. doi: 10.2741/1047. [DOI] [PubMed] [Google Scholar]
- Dougan DA, Mogk A, Zeth K, Turgay K, Bukau B. AAA+ proteins and substrate recognition, it all depends on their partner in crime. FEBS Lett. 2002;529:6–10. doi: 10.1016/s0014-5793(02)03179-4. [DOI] [PubMed] [Google Scholar]
- Dubnau D, Lovett CM., Jr . Transformation and Recombination. In: Hoch JA, Losick R, Sonenshein AL, editors. Bacillus subtilis and Its Relatives: From Genes to Cells. Washington, DC: American Society for Microbiology; 2001. pp. 453–471. [Google Scholar]
- Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol Cell. 2003;11:671–683. doi: 10.1016/s1097-2765(03)00060-1. [DOI] [PubMed] [Google Scholar]
- Gonzalez M, Rasulova F, Maurizi MR, Woodgate R. Subunit-specific degradation of the UmuD/D′ heterodimer by the ClpXP protease: the role of trans recognition in UmuD′ stability. Embo J. 2000;19:5251–5258. doi: 10.1093/emboj/19.19.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman S. Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol. 2003;19:565–587. doi: 10.1146/annurev.cellbio.19.110701.153228. [DOI] [PubMed] [Google Scholar]
- Hahn J, Luttinger A, Dubnau D. Regulatory inputs for the synthesis of ComK, the competence transcription factor of Bacillus subtilis. Mol Microbiol. 1996;21:763–775. doi: 10.1046/j.1365-2958.1996.371407.x. [DOI] [PubMed] [Google Scholar]
- Hahn J, Maier B, Haijema BJ, Sheetz MP, Dubnau D. Transformation proteins and DNA uptake localize to the cell poles in Bacillus subtilis. Cell. 2005;122:1–13. doi: 10.1016/j.cell.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge R, Bukau B. Proteolysis in prokaryotes: protein quality control and regulatory principles. Mol Microbiol. 2003;49:1451–1462. doi: 10.1046/j.1365-2958.2003.03693.x. [DOI] [PubMed] [Google Scholar]
- Hoskins JR, Kim SY, Wickner S. Substrate recognition by the ClpA chaperone component of ClpAP protease. J Biol Chem. 2000;275:35361–35367. doi: 10.1074/jbc.M006288200. [DOI] [PubMed] [Google Scholar]
- Jenal U, Hengge-Aronis R. Regulation by proteolysis in bacterial cells. Curr Opin Microbiol. 2003;6:163–172. doi: 10.1016/s1369-5274(03)00029-8. [DOI] [PubMed] [Google Scholar]
- Keiler KC, Waller PR, Sauer RT. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science. 1996;271:990–993. doi: 10.1126/science.271.5251.990. [DOI] [PubMed] [Google Scholar]
- Kong L, Dubnau D. Regulation of competence-specific gene expression by Mec-mediated protein-protein interaction in Bacillus subtilis. Proc Natl Acad Sci USA. 1994;91:5793–5797. doi: 10.1073/pnas.91.13.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laachouch JE, Desmet L, Geuskens V, Grimaud R, Toussaint A. Bacteriophage Mu repressor as a target for the Escherichia coli ATP-dependent Clp Protease. Embo J. 1996;15:437–444. [PMC free article] [PubMed] [Google Scholar]
- Levchenko I, Yamauchi M, Baker TA. ClpX and MuB interact with overlapping regions of Mu transposase: implications for control of the transposition pathway. Genes Dev. 1997;11:1561–1572. doi: 10.1101/gad.11.12.1561. [DOI] [PubMed] [Google Scholar]
- Maamar H, Dubnau D. Bistability in the Bacillus subtilis K-state (competence) system requires a positive feedback loop. Mol Microbiol. 2005;56:615–624. doi: 10.1111/j.1365-2958.2005.04592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Msadek T, Kunst F, Rapoport G. MecB of Bacillus subtilis is a pleiotropic regulator of the ClpC ATPase family, controlling competence gene expression and survival at high temperature. Proc Natl Acad Sci USA. 1994;91:5788–5792. doi: 10.1073/pnas.91.13.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura M, Liu L, Lacelle M, Nakano MM, Zuber P. Mutational analysis of ComS: evidence for the interaction of ComS and MecA in the regulation of competence development in Bacillus subtilis. Mol Microbiol. 1999;32:799–812. doi: 10.1046/j.1365-2958.1999.01399.x. [DOI] [PubMed] [Google Scholar]
- Pan Q, Garsin DA, Losick R. Self-reinforcing activation of a cell-specific transcription factor by proteolysis of an anti-sigma factor in B. subtilis. Mol Cell. 2001;8:873–883. doi: 10.1016/s1097-2765(01)00362-8. [DOI] [PubMed] [Google Scholar]
- Persuh M, Turgay K, Mandic-Mulec I, Dubnau D. The N- and C-terminal domains of MecA recognize different partners in the competence molecular switch. Mol Microbiol. 1999;33:886–894. doi: 10.1046/j.1365-2958.1999.01544.x. [DOI] [PubMed] [Google Scholar]
- Rudner DZ, Losick R. A sporulation membrane protein tethers the pro-sigmaK processing enzyme to its inhibitor and dictates its subcellular localization. Genes Dev. 2002;16:1007–1018. doi: 10.1101/gad.977702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan KR, Judd EM, Shapiro L. The CtrA response regulator essential for Caulobacter crescentus cell-cycle progression requires a bipartite degradation signal for temporally controlled proteolysis. J Mol Biol. 2002;324:443–455. doi: 10.1016/s0022-2836(02)01042-2. [DOI] [PubMed] [Google Scholar]
- Sauer RT, Bolon DN, Burton BM, Burton RE, Flynn JM, Grant RA, Hersch GL, Joshi SA, Kenniston JA, Levchenko I, et al. Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell. 2004;119:9–18. doi: 10.1016/j.cell.2004.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerker JM, Laub MT. Cell-cycle progression and the generation of asymmetry in Caulobacter crescentus. Nat Rev Microbiol. 2004;2:325–337. doi: 10.1038/nrmicro864. [DOI] [PubMed] [Google Scholar]
- Smits WK, Eschevins CC, Susanna KA, Bron S, Kuipers OP, Hamoen LW. Stripping Bacillus: ComK auto-stimulation is responsible for the bistable response in competence development. Mol Microbiol. 2005;56:604–614. doi: 10.1111/j.1365-2958.2005.04488.x. [DOI] [PubMed] [Google Scholar]
- Studemann A, Noirclerc-Savoye M, Klauck E, Becker G, Schneider D, Hengge R. Sequential recognition of two distinct sites in sigma(S) by the proteolytic targeting factor RssB and ClpX. Embo J. 2003;22:4111–4120. doi: 10.1093/emboj/cdg411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susanna KA, Fusetti F, Thunnissen AM, Hamoen LW, Kuipers OP. Functional analysis of the competence transcription factor ComK of Bacillus subtilis by characterization of truncation variants. Microbiology. 2006;152:473–483. doi: 10.1099/mic.0.28357-0. [DOI] [PubMed] [Google Scholar]
- Turgay K, Hahn J, Burghoorn J, Dubnau D. Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J. 1998;17:6730–6738. doi: 10.1093/emboj/17.22.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgay K, Hamoen LW, Venema G, Dubnau D. Biochemical characterization of a molecular switch involving the heat shock protein ClpC, which controls the activity of ComK, the competence transcription factor of Bacillus subtilis. Genes Dev. 1997;11:119–128. doi: 10.1101/gad.11.1.119. [DOI] [PubMed] [Google Scholar]
- van Sinderen D, Luttinger A, Kong L, Dubnau D, Venema G, Hamoen L. comK encodes the competence transcription factor, the key regulatory protein for competence development in Bacillus subtilis. Mol Microbiol. 1995;15:455–462. doi: 10.1111/j.1365-2958.1995.tb02259.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.